

Abstract
In this work, we critically assess the ability of the all-atom enhanced sampling method accelerated molecular dynamics (aMD) to investigate conformational changes in proteins that typically occur on the millisecond time scale. We combine aMD with the inherent power of graphics processor units (GPUs) and apply the implementation to the bovine pancreatic trypsin inhibitor (BPTI). A 500 ns aMD simulation is compared to a previous millisecond unbiased brute force MD simulation carried out on BPTI, showing that the same conformational space is sampled by both approaches. To our knowledge, this represents the first implementation of aMD on GPUs and also the longest aMD simulation of a biomolecule run to date. Our implementation is available to the community in the latest release of the Amber software suite (v12), providing routine access to millisecond events sampled from dynamics simulations using off the shelf hardware.
Introduction
Conventional molecular dynamics allows one to access time scales on the order of tens to hundreds of nanoseconds; however, many biological processes of interest occur on longer time scales of up to milliseconds or more.1−6 Efforts to explore these long time scales have led to the development of several advanced sampling techniques such as conformational flooding,7,8 hyperdynamics,9,10 metadynamics,11−13 and the adaptive biasing force method.13−15 Inspired by Voter, accelerated molecular dynamics (aMD) is an additional enhanced conformational sampling method that provides access to events beyond those obtainable with conventional molecular dynamics (cMD).9 Here, we emphasize one of the great advantages of aMD, which is that no prior knowledge of the potential energy landscape needs to be known and, consequently, no reaction coordinate needs to be defined prior to running the simulation.
In addition to advances in algorithms to achieve dynamics on longer time scales, recent efforts by D. E. Shaw Research have focused on building a specialized computer, Anton, with the sole purpose of simulating protein dynamics.16 With this great engineering achievement, simulation time scales have been pushed into the range of hundreds of microseconds to milliseconds using unbiased brute force cMD.17,18 While time on an Anton machine has been generously granted to the scientific community, access is still limited, and many researchers are stuck waiting in queues on crowded super computers or local clusters. Recently, the advancement of computational science on conventional graphic processing units (GPU) has allowed researchers efficient and inexpensive access to microseconds of simulation time on just a single desktop computer.18−21 By combining the advanced sampling method of aMD and the inherent power of the GPU, we present the synthesis of a tool that allows researchers access to inexpensive efficient exploration of long time scale events. Here, we point out that a time scale cannot be directly determined from an aMD simulation, but we can correlate events which occur on long time scales from either experimental results or long conventional molecular dynamics simulations. However, future work using the conformational space explored by aMD as a seed for other methods, namely, Markov models,22 would allow one access to time dependent properties, such as NMR relaxation data, which was shown can be computed directly from the Anton millisecond trajectory.23
In its original form, the aMD method modifies the potential energy landscape by raising energy minima that lie below a defined threshold level, while leaving those areas lying above the threshold unmodified. As a result, barriers separating adjacent energy basins are effectively reduced, providing the simulation access to conformational space that cannot be easily accessed in a cMD simulation. Historically, aMD was first implemented by Hamelberg et al. within the framework of the sander module in the AMBER 7 package and used to study several small peptide and protein systems.24,25 Since the original implementation, there have been several notable variations of the aMD method,26−28 but no official implementation has been released. However, Wang et al. recently ported the aMD method to NAMD,29 and following this approach, we have ported aMD to the three main MD engines included in Amber 12:30 the CPU versions sander and pmemd and the GPU version pmemd.cuda. The performance enhancements for cMD on GPUs obtained with pmemd.cuda alone are remarkable and can be found on the Amber Web site (ambermd.org) and in the following publications.21,31 For this work, we have used our implementation of aMD in the pmemd.cuda MD engine and will refer to it simply as aMD.
Bovine pancreatic trypsin inhibitor (BPTI) is a small protein with 58 residues that has been extensively studied experimentally, being the first subject of NMR experiments to characterize individual hydration water molecules in proteins,32 and also the first protein to be simulated with molecular dynamics.33 D. E. Shaw Research reported in 2010 a remarkable 1.03 ms MD simulation of BPTI in explicit water.34 Using our aMD implementation in pmemd.cuda, we have performed a 500 ns aMD simulation of BPTI in explicit water, maintaining the same conditions as the simulation on Anton. In order to mimic the force field used in the Anton simulation, we modified the ff99SB-ILDN force field removing the modifications to leucine, aspartic acid, and asparagine, which we refer to as ff99SB-I.
Using the 1.03 ms simulation of BPTI provided by D. E. Shaw Research throughout our analysis, we have shown that both methodologies sample the same conformational space. Additionally, we performed a 500 ns cMD simulation to use as a measure of the amount of sampling attainable by conventional MD with the same computational effort. To the best of our knowledge, our aMD implementation is the first to support GPU acceleration, and the work presented here represents the longest single aMD simulation of a biomolecule run to date. Both Amber simulations were completed in two weeks on individual desktop computers containing single $500 GTX580 GPUs. Details of the simulations can be found in the Supporting Information.
Results
Structural Analysis
In the millisecond Anton simulation, five long-lived structural states were identified that persisted for 6–26 μs with deviations of up to 3.5 Å from the crystal structure, 5PTI.34,35 Using these same five structures and the crystal structure as references, we calculated RMSD values (heavy backbone atoms) along our 500 ns aMD simulation, shown in Figure 1. We color each point according to which of the five structures it is closest to in terms of RMSD, allowing one to see the transitions from the different structural states. RMSD results show we sample structures from all five states but spend the majority of the time near the blue state and the least amount of time close to the green state. The RMSD with respect to the X-ray structure shows that the protein moves away from the crystal structure 49 ns into the simulation and achieves a maximum RMSD of 2.7 Å before coming back to within 0.89 Å, emphasizing that the system is sampling states both far from and near to the crystal structure, as was seen in the long 1 ms cMD simulation. The largest deviation from the crystal structure is 3.29 Å, which occurs at 195 ns.
Figure 1.
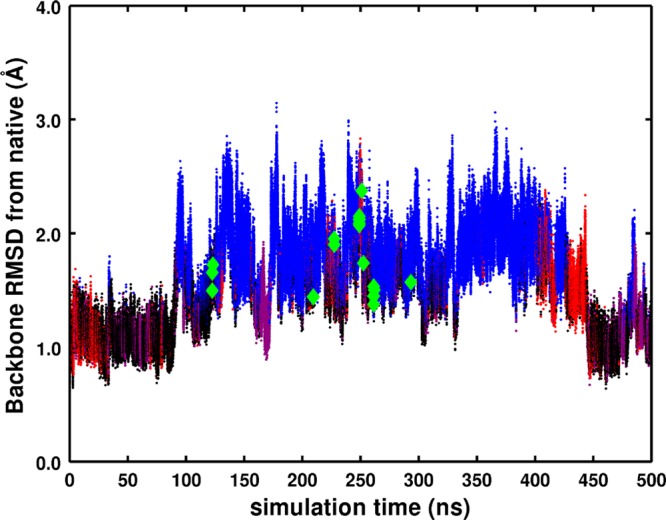
RMSD of the aMD trajectory from the crystal structure. Red, blue, green, purple, and black colors correspond to the kinetic clusters identified in the 1 ms cMD BPTI simulation.34 For each frame, we identify what cluster we are closest to and color it accordingly. Only a handful of transitions to the green state were observed, and we highlight them with diamond markers.
To characterize the conformational states explored in more detail, principal component analysis was carried out on the 1 ms simulation using Bio3D.36 Figure 2a displays the two-dimensional representation of the structural data set as a projection of the Boltzmann reweighted distribution onto the subspace defined by the first and second principal component vectors (PC1 and PC2) built from the C-α atoms spanning residues 4 to 54. In this analysis, PC1 and PC2 describe 35% and 21%, respectively, of the total variance of the motions in the simulation (Movies S1 and S2). The five long-lived structures and the crystal structure were then projected into this space. The 500 ns cMD control simulation and the 500 ns aMD simulation were also projected into the subspace defined by the 1 ms simulation (Figure 2b,c). It is clear from Figure 2b that the 500 ns cMD control simulation does not explore the amount of conformational space that the aMD simulation does and remains trapped in the basin localized around the crystal structure.
Figure 2.

The free energy principal component projection of (a) 1 ms simulation, (b) 500 ns cMD ff99SB-I simulation, and (c) 500 ns aMD ff99SB-I simulation onto (PC1, PC2) defined by the 1 ms simulation. The long-lived structures are projected onto the free energy surface and are labeled as red, blue, green, purple, and black triangles. The crystal structure, 5PTI, is demarked by the red diamond (see also Movies S1 and S2).
The aMD simulation (Figure 2c) exhibits a rather broad pathway from the crystallographic basin (0.0, −5.0) to the region (−10.0, −5.0) which is not present in the 1 ms simulation (Figure 2a). Flattening of the potential energy surface in the two basins could promote transitions over a large barrier separating them, but the 1 ms simulation may also not have sampled these regions frequently enough to explore this pathway. The 1 ms simulation spent the majority of the time in the basin around (2.5, 1.0) which is ultimately responsible for the observed populations in the 1 ms cMD simulation differing substantially from experiment. In the Shaw et al. paper, this is attributed to inaccuracies in the underlying force field, and indeed we also spend a considerable amount of our 500 ns simulation in the same basin, as one would expect using the same force field. In future work, to determine accurately the conformational changes mapped out by the PC space using an aMD simulation, one could use the technique employed by Wereszczynski and McCammon.37
NMR Observables
The recent analysis carried out by Xue et al. examined in detail the χ1, χ2, and χ3 dihedral angles associated with the disulfide bond formed between cysteine 14 and cysteine 38 during the course of the 1 ms cMD BPTI simulation.23 In this manuscript, a similar analysis was performed. Figure 3c shows the Boltzmann reweighted χ1(C14) vs χ1(C38) free energy surface explored throughout the 500 ns aMD simulation. In contrast, the 500 ns control cMD simulation never visits the minima explored by the aMD simulation and remains trapped in a state close to the crystal structure (Figure 3b), as was seen in the PC projections. Comparing the free energy surface explored by the 1 ms cMD simulation (Figure 3a) to that of our aMD simulation, it is clear the aMD simulation explores the same states as the unbiased simulation.
Figure 3.

The χ1–C14 vs χ1–C38 dihedral angle free energy surfaces of (a) 1 ms cMD simulation, (b) 500 ns cMD ff99SB-I simulation, and (c) 500 ns aMD ff99SB-I simulation. The long-lived stable states are plotted onto the free energy surface and are labeled as red, blue, green, purple, and black triangles. The crystal structure, 5PTI, is demarked by the red diamond. The major state M is labeled along with the minor states mC14 and mC38.
The χ2 and χ3 dihedral angles associated with Cys14 and Cys38 can be used to further describe isomerization configurations of the disulfide bond: the major state M, which consists of three substates (M1, M2, and M3), and the two minor or excited states mC38 and mC14.23 A detailed description of these states is included in the Supporting Information (Figure S1), which can be compared to those analyzed by Xue et al. for the 1 ms cMD simulation. In contrast with the 1 ms cMD simulation, which predicts the excited state mC14 to be the most populated, we find the major state M to be the most populated in the aMD simulations after Boltzmann reweighting our distribution (Table S1); however, to fully converge the population distribution would require a longer simulation than was used in this study.
Structures from states M1, mC38, and mC14 were extracted from the trajectory by using the lowest energy structure in the M1 state as a reference to select an ensemble of structures with similar energy from each of the three basins for performing the chemical shift analysis. Using the SHIFTX2 software package,38 chemical shift differences were computed between the ensembles representing the different substates (Table 1). A RMS deviation of 2.1 ppm was obtained from the ff99SB-I aMD simulation (computed for the shifts with known sign) compared to 2.7 ppm computed from the 1 ms cMD simulation. In general, good agreement is achieved from the aMD simulation compared to values calculated from the 1 ms cMD simulation and the experimental values.39,40 We would like to highlight the fact that one cannot compute these differences using only 500 ns of cMD since the simulation never explores the excited states during the course of such a short simulation (Figure 3b).
Table 1. 15N Chemical Shift Differences between the Conformational States M1(M), mC14, and mC38.
Water Occupancy
Examination of the water occupancy throughout the aMD simulation correctly identifies the four long-lived waters in agreement with experimental results.41 The longest-lived water, W122, is identified by the 1 ms cMD simulation as having a lifetime of 14 μs. During the course of the aMD simulation, several exchange events are captured at this location and an interesting “revolving door” mechanism is identified, whereby the disulfide bond rotates around and pushes the water out of the pocket (highlighted in Movie S3). The longest binding events of water molecules at this site occur when in the crystallographic basin of the aMD simulation, consistent with the 1 ms cMD simulation.
Conclusion
This work shows that, using conventional off the shelf GPU hardware combined with an enhanced sampling algorithm, events taking place on the millisecond time scale can be effectively sampled with dynamics simulations orders of magnitude shorter (2000×) than those time scales. The implementation is validated in this work by comparison with a long unbiased cMD simulation and experimental data. Structurally, we show the long-lived states identified by the 1 ms cMD simulation have been sampled both in terms of RMSD and coverage in PC space. We demonstrate that access to these states cannot be obtained with the same length of simulation carried out using conventional MD. Important structural waters were preserved and found to exhibit the same occupancies as those experimentally. Chemical shift differences computed from the aMD simulations were found to be in agreement with those calculated from the long 1 ms cMD simulation, and similar dihedral populations were also observed. However, we point out that, while aMD is great for exploration of conformational space, it does not reproduce the exact dynamics of the system. We conclude by emphasizing never before has the aMD been benchmarked against a long cMD simulation, and we commend D. E. Shaw Research for providing their data to the community.
Acknowledgments
This research used resources of the Keeneland Computing Facility at the Georgia Institute of Technology, which is supported by the National Science Foundation under contract OCI-0910735. This work was funded in part by the National Science Foundation through the Scientific Software Innovations Institutes program—NSF SI2-SSE (NSF1047875 & NSF1148276) grants to R.C.W, the NSF XSEDE program, and also by a University of California (UC Lab 09-LR-06-117792) grant to R.C.W. Computer time was provided by the San Diego Supercomputer Center through National Science Foundation award TGMCB090110 to R.C.W. The work was also supported by a CUDA fellowship to R.C.W. from NVIDIA. The J.A.M group is supported by NSF, NIH, HHMI, NBCR, and CTBP.
Supporting Information Available
Simulation setup, theory of aMD, details of how aMD parameters were selected, and reweighting protocol. Movies S1–S3, Figure S1, and Table S1. This information is available free of charge via the Internet at http://pubs.acs.org/.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kubelka J.; Chiu T. K.; Davies D. R.; Eaton W. A.; Hofrichter J. Sub-microsecond protein folding. J. Mol. Biol. 2006, 359, 546–53. [DOI] [PubMed] [Google Scholar]
- Schaeffer R. D.; Fersht A.; Daggett V. Combining experiment and simulation in protein folding: closing the gap for small model systems. Curr. Opin. Struct. Biol. 2008, 18, 4–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freddolino P. L.; Schulten K. Common structural transitions in explicit-solvent simulations of villin headpiece folding. Biophys. J. 2009, 97, 2338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson M. K.; Zhou H. X. Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 21–42. [DOI] [PubMed] [Google Scholar]
- Lindahl E.; Sansom M. S. Membrane proteins: molecular dynamics simulations. Curr. Opin. Struct. Biol. 2008, 18, 425–31. [DOI] [PubMed] [Google Scholar]
- Khalili-Araghi F.; Gumbart J.; Wen P. C.; Sotomayor M.; Tajkhorshid E.; Schulten K. Molecular dynamics simulations of membrane channels and transporters. Curr. Opin. Struct. Biol. 2009, 19, 128–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubmüller H. Predicting slow structural transitions in macromolecular systems: conformational flooding. Phys. Rev. E 1995, 52. [DOI] [PubMed] [Google Scholar]
- Lange O. F.; Schäfer L. V.; Grubmüller H. Flooding in GROMACS: Accelerated barrier crossings in molecular dynamics. J. Comput. Chem. 2006, 27, 1693–1702. [DOI] [PubMed] [Google Scholar]
- Voter A. F. Hyperdynamics: Accelerated Molecular Dynamics of Infrequent Events. Phy. Rev. Lett. 1997, 78, 3908–3911. [Google Scholar]
- Voter A. F. A method for accelerating the molecular dynamics simulation of infrequent events. J. Chem. Phys. 1997, 106, 4665–4677. [Google Scholar]
- Bussi G.; Laio A.; Parrinello M. Equilibrium Free Energies from Nonequilibrium Metadynamics. Phy. Rev. Lett. 2006, 96, 090601. [DOI] [PubMed] [Google Scholar]
- Leone V.; Marinelli F.; Carloni P.; Parrinello M. Targeting biomolecular flexibility with metadynamics. Curr. Opin. Struct. Biol. 2010, 20, 148–154. [DOI] [PubMed] [Google Scholar]
- Darve E.; Pohorille A. Calculating free energies using average force. J. Chem. Phys. 2001, 115, 9169–9183. [Google Scholar]
- Darve E.; Rodriguez-Gomez D.; Pohorille A. Adaptive biasing force method for scalar and vector free energy calculations. J. Chem. Phys. 2008, 128(14), 144120. [DOI] [PubMed] [Google Scholar]
- Henin J.; Fiorin G.; Chipot C.; Klein M. L. Exploring Multidimensional Free Energy Landscapes Using Time-Dependent Biases on Collective Variables. J. Chem. Theory Comput. 2009, 6, 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw D. E.; Deneroff M. M.; Dror R. O.; Kuskin J. S.; Larson R. H.; Salmon J. K.; Young C.; Batson B.; Bowers K. J.; Chao J. C.; Eastwood M. P.; Gagliardo J.; Grossman J. P.; Ho C. R.; Ierardi D. J.; Kolossvary I.; Klepeis J. L.; Layman T.; McLeavey C.; Moraes M. A.; Mueller R.; Priest E. C.; Shan Y.; Spengler J.; Theobald M.; Towles B.; Wang S. C. Anton, a special-purpose machine for molecular dynamics simulation. Commun. ACM 2008, 51, 91–97. [Google Scholar]
- Dror R. O.; Arlow D. H.; Maragakis P.; Mildorf T. J.; Pan A. C.; Xu H.; Borhani D. W.; Shaw D. E. Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 18684–18689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y.; Kim E. T.; Eastwood M. P.; Dror R. O.; Seeliger M. A.; Shaw D. E. How Does a Drug Molecule Find Its Target Binding Site?. J. Am. Chem. Soc. 2011, 133, 9181–9183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey M. J.; Giupponi G.; Fabritiis G. D. ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [DOI] [PubMed] [Google Scholar]
- Friedrichs M. S.; Eastman P.; Vaidyanathan V.; Houston M.; Legrand S.; Beberg A. L.; Ensign D. L.; Bruns C. M.; Pande V. S. Accelerating molecular dynamic simulation on graphics processing units. J. Comput. Chem. 2009, 30, 864–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz A. W.; Williamson M. J.; Xu D.; Poole D.; Le Grand S.; Walker R. C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande V. S.; Beauchamp K.; Bowman G. R. Everything you wanted to know about Markov State Models but were afraid to ask. Methods 2010, 52, 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y.; Ward J. M.; Yuwen T.; Podkorytov I. S.; Skrynnikov N. R. Microsecond time-scale conformational exchange in proteins: using long molecular dynamics trajectory to simulate NMR relaxation dispersion data. J. Am. Chem. Soc. 2012, 134, 2555–62. [DOI] [PubMed] [Google Scholar]
- Hamelberg D.; Mongan J.; McCammon J. A. Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–29. [DOI] [PubMed] [Google Scholar]
- Hamelberg D.; de Oliveira C. A.; McCammon J. A. Sampling of slow diffusive conformational transitions with accelerated molecular dynamics. J. Chem. Phys. 2007, 127, 155102. [DOI] [PubMed] [Google Scholar]
- Fajer M.; Hamelberg D.; McCammon J. A. Replica-Exchange Accelerated Molecular Dynamics (REXAMD) Applied to Thermodynamic Integration. J. Chem. Theory Comput. 2008, 4, 1565–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wereszczynski J.; McCammon J. A. Using Selectively Applied Accelerated Molecular Dynamics to Enhance Free Energy Calculations. J. Chem. Theory Comput. 2010, 6, 3285–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinko W.; de Oliveira C. A. F.; Pierce L. C. T.; McCammon J. A. Protecting High Energy Barriers: A New Equation to Regulate Boost Energy in Accelerated Molecular Dynamics Simulations. J. Chem. Theory Comput. 2012, 8, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Harrison C. B.; Schulten K.; McCammon J. A. Implementation of Accelerated Molecular Dynamics in NAMD. Comput. Sci. Discovery 2011, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Darden T. A.; Cheatham T. E. III Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Walker R. C.; Zhang W.; Merz K. M.; Roberts B.; Hayik S.; Roitberg A.; Seabra G.; Swails J.; Goetz A. W.; Kolossvai I.; Wong K. F.; Paesani F.; Vanicek J.; Wolf R. M.; Liu J.; Wu X.; Brozell S.R.; Steinbrecher T.; Gohlke H.; Cai Q.; Ye X.; Wang J.; Hsieh M.-J.; Cui G.; Roe D.R.; Mathews D.H.; Seetin M.G.; Salomon-Ferrer R.; Sagui C.; Babin V.; Luchko T.; Gusarov S.; Kovalenko A. Kollman P. A.. Amber 12; University of California: San Francisco, CA, 2012. [Google Scholar]
- Le Grand S.; Walker R. C.. SPFP: Speed without compromise - a mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 2012, not supplied.
- Otting G.; Liepinsh E.; Wuethrich K. Proton exchange with internal water molecules in the protein BPTI in aqueous solution. J. Am. Chem. Soc. 1991, 113, 4363–4364. [Google Scholar]
- McCammon J. A.; Gelin B. R.; Karplus M. Dynamics of folded proteins. Nature 1977, 267, 585–590. [DOI] [PubMed] [Google Scholar]
- Shaw D. E.; Maragakis P.; Lindorff-Larsen K.; Piana S.; Dror R. O.; Eastwood M. P.; Bank J. A.; Jumper J. M.; Salmon J. K.; Shan Y.; Wriggers W. Atomic-Level Characterization of the Structural Dynamics of Proteins. Science 2010, 330, 341–346. [DOI] [PubMed] [Google Scholar]
- Wlodawer A.; Walter J.; Huber R.; Sjolin L. Structure of bovine pancreatic trypsin inhibitor. Results of joint neutron and X-ray refinement of crystal form II. J. Mol. Biol. 1984, 180, 301–29. [DOI] [PubMed] [Google Scholar]
- Grant B. J.; Rodrigues A. P.; ElSawy K. M.; McCammon J. A.; Caves L. S. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–6. [DOI] [PubMed] [Google Scholar]
- Wereszczynski J.; McCammon J. A.. Nucleotide-dependent mechanism of Get3 as elucidated from free energy calculations. Proc. Natl. Acad. Sci. U. S. A. 2012, not supplied. [DOI] [PMC free article] [PubMed]
- Han B.; Liu Y.; Ginzinger S. W.; Wishart D. S. SHIFTX2: significantly improved protein chemical shift prediction. J. Biomol. NMR 2011, 50, 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey M. J.; Wang C.; Palmer A. G. 3rd. Disulfide bond isomerization in basic pancreatic trypsin inhibitor: multisite chemical exchange quantified by CPMG relaxation dispersion and chemical shift modeling. J. Am. Chem. Soc. 2003, 125, 14324–35. [DOI] [PubMed] [Google Scholar]
- Berndt K. D.; Beunink J.; Schroeder W.; Wuethrich K. Designed replacement of an internal hydration water molecule in BPTI: structural and functional implications of a Gly-to-Ser mutation. Biochemistry 1993, 32, 4564–4570. [DOI] [PubMed] [Google Scholar]
- Persson E.; Halle B. Nanosecond to microsecond protein dynamics probed by magnetic relaxation dispersion of buried water molecules. J. Am. Chem. Soc. 2008, 130, 1774–87. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.