

Abstract
IQ motif-containing GTPase-activating proteins IQGAP1 and IQGAP2 are highly homologous multidomain scaffolding proteins. Their major function consists of integration of Rho GTPase and Ca2+/calmodulin signals with cell adhesive and cytoskeletal reorganizational events. Recent studies showed that they play an important role in carcinogenesis. There is growing evidence that IQGAP2 is a novel tumor suppressor counteracting the effects of IQGAP1, an oncogene, in several cancers, especially in hepatocellular carcinoma (HCC). While HCC is highly prevalent and one of the deadliest cancers worldwide, the signaling pathways involved are not fully understood and treatment of advanced disease still represents an area of high unmet medical need. This paper compiles various findings from studies in mouse models, cell lines, and patient samples that support future development of IQGAPs into new therapeutic targets. It also discusses distinct features of IQGAP2 in an attempt to provide insight into the mechanism of the seemingly paradoxical opposing roles of the two very similar IQGAP proteins in carcinogenesis.
1. Introduction
This paper focuses on the recently recognized role of IQ motif-containing GTPase-activating proteins (IQGAPs) in hepatic carcinogenesis. Hepatocellular carcinoma (HCC), the most common form of primary liver cancer [1], is the second and the sixth leading cause of cancer-related death worldwide in men and women, respectively [2]. HCC is responsible for between 500,000 and 1 million worldwide deaths annually [3]. HCC etiologies are diverse and include chronic hepatitis B (HBV) and C (HCV), chronic excessive alcohol consumption, steatosis, diabetes, and exposure to toxic agents such as aflatoxin B1, or any hepatic disease associated with cirrhosis. While the highest HCC rates are found in East and South East Asia, HCC incidence is increasing in many parts of the world, including the United States, most probably due to the rising incidence of HCV infection [4]. In the US, HCC incidence rates tripled between 1975 and 2005 [1]. In 2005, HCC incidence was estimated at 667,000 cases globally and 17,550 cases in the United States [5]. Overall survival of patients with HCC has not improved in the last two decades. Since chronic liver disease can often be asymptomatic in early stages, the majority of HCC patients are diagnosed late in the course of their disease and the five-year survival rate in such patients is only 5% [6]. While the only significant improvement in overall survival for patients with advanced HCC was reported for the multikinase inhibitor sorafenib [7], the clinical benefit derived with sorafenib occurs mainly through disease stabilization and curative agents for advanced HCC are yet to be developed.
The diverse etiology of HCC infers a considerable genomic heterogeneity and involvement of multiple signaling pathways in the disease pathogenesis. The heterogeneity and instability of human tumors pose a serious impediment to identification of target genes for cancer therapy, making genetically well-defined mouse models increasingly important in cancer research. A multitude of molecules has been identified as potential therapeutic targets for HCC and described in many excellent reviews [8–10]. Here, we survey involvement of a significantly understudied protein, IQGAP2, and its homolog, IQGAP1, in cancer with more specific emphasis on the development of HCC. Compelling recent data have introduced IQGAP2 as the newest addition to the long list of HCC-related tumor suppressors and potential molecular targets for much needed curative therapy.
2. Multifunctional IQGAP Protein Family
IQGAPs represent a small subgroup of an evolutionally conserved superfamily of GTPase-activating proteins (GAPs) [11]. In humans and mice, the IQGAP protein family consists of three members-IQGAP1, IQGAP2 and IQGAP3 [12–15]. IQGAPs are highly homologous multidomain proteins that integrate Rho GTPase and Ca2+/calmodulin signals with cell adhesive and cytoskeletal reorganizational events [16]. All three are large cytoplasmic scaffolding proteins (MW 180–190 kDa). Their domain structure includes an actin-binding calponin homology (CH) domain, a single WW domain capable of binding various proline-rich proteins, four IQ motifs binding calmodulin, a large GTPase binding domain (GBD) known to bind Rho GTPases Rac1 and cdc42, and a RasGAP C-terminus domain (RGCT) (Figure 1) [16]. The GTPase binding domain in IQGAPs lacks an arginine residue, essential for GTP-hydrolysis, which may explain why none of the IQGAPs demonstrated in vitro GAP activity toward GTPases [17]. Rather, IQGAPs are believed to stabilize the active GTP-bound form of both Rac1 and cdc42 [18, 19]. Among the three homologs, IQGAP1 is the most extensively studied to date. IQGAP1 has been shown to play a role in multiple cellular processes requiring cytoskeletal rearrangement, including cell motility, polarity, proliferation, and differentiation [16]. Significant amount of evidence also implicates IQGAP1 in promoting tumorigenesis in various cancers [20, 21], characterizing it as a bona fide oncogene. Unlike its ubiquitously expressed homolog, IQGAP2 is predominantly expressed in liver, kidney and platelets [13, 22], and its physiological role remains largely understudied. Both IQGAP1 and IQGAP2 appear to have functional significance in platelets. IQGAP1 may modulate platelet procoagulant function by regulating the secretory pathway of α-granule exocytosis [23], while IQGAP2 functions as a unique scaffolding protein linking thrombin activation to platelet cytoskeletal actin assembly and reorganization, a finding consistent with the IQGAP2 domain structure [22]. Peculiar chromosomal localization of the IQGAP2 gene within the proteinase activated receptor (PAR) gene cluster in both the human and mouse genome [22, 24] also supports the notion that IQGAP2 and PARs may be components of a functional genomic unit uniquely evolved to facilitate thrombin-mediated signaling. The most recently discovered member of the IQGAP family, IQGAP3, is expressed predominantly in the brain and regulates neurite outgrowth [15]. There also have been two reports on potential function of IQGAP3 in liver. A genome-wide microarray expression profiling of 24 HCC tumors revealed upregulation of the IQGAP3 gene, along with several other genes located at 1q22, a region commonly amplified in HCC [25]. IQGAP3 expression was also shown to be increased in proliferating hepatocytes in a mouse liver regeneration model [26].
Figure 1.
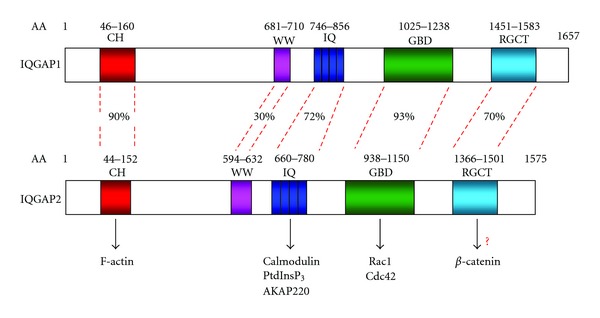
The domain structure of human IQGAP1 and IQGAP2. CH (calponin homology) domain, WW, polyproline binding domain, IQ calmodulin-binding motif, GBD-GTPase binding domain, and RGCT-RasGAP C-terminus domain. Domain percent homology is shown. Adapted from [15, 16]. Also shown binding partners of IQGAP2 identified to date. While it has been confirmed that IQGAP2 co-immunoprecipitates with β-catenin [27], RGCT is marked as a domain responsible for β-catenin binding based on analogy with IQGAP1. A list of IQGAP1 numerous binding partners can be found in [28].
3. IQGAP1 and IQGAP2 Play Opposing Roles in Hepatic Carcinogenesis in Mice
While the molecular mechanisms leading to HCC may differ by etiology, it generally evolves through a multistep process involving hepatocyte destruction, proliferation and regeneration. At the molecular level, both genetic and epigenetic alterations have been observed in HCC tumors that result in abnormal expression of genes involved in cell cycle control, cell growth and proliferation, apoptosis, and cell-cell interactions [29]. Recurrent allelic losses or gains have been detected on 14 chromosome arms in more than 30% of all HCC analyzed [29]. Notably, 5q13, the region of localization of IQGAP2 gene, is not found among them. Similarly, IQGAPs were not among proteins implicated in the development of HCV-related HCC based on an expansive gene profiling study [30]. Still, given the heterogeneity of this disease, it is highly unlikely that all genes involved have been identified to date. Unlike IQGAP1, IQGAP2 has never been implicated in carcinogenesis until now.
Generation of a conventional knockout mouse lacking the Iqgap2 gene [27] provided a first insight into the physiological functions of IQGAP2. Iqgap2 −/− mice demonstrated an age-dependent increase in apoptosis and a structural mitochondrial defect in hepatocytes [27]. Furthermore, 86% of Iqgap2 −/− mice (N = 18/21) developed HCC by the age of 18–24 months [27]. Both sexes were affected equally and no other malignancies were evident. At the molecular level, Iqgap2 −/− HCC tumors were characterized by an 8-fold increase in cyclin D1 levels (a β-catenin nuclear target), a 9-fold upregulation of cytoplasmic IQGAP1 expression and loss of membrane E-cadherin expression. HCC development in Iqgap2 −/− mice was linked to the Wnt-β-catenin pathway activation, supported by, in addition to cyclin D1 upregulation, β-catenin translocation from the cellular membrane and accumulation of its dephosphorylated (active) form. Interestingly, more moderate (∼2.5-fold) increase in cyclin D1 and IQGAP1 expression in the livers of younger Iqgap2 −/− mice without HCC was also evident, establishing that IQGAP1 upregulation and cyclin D1 activation predated histological evidence for HCC development in these mice [27]. An elevated serum level of aspartate aminotransferase (AST) was detected in Iqgap2 −/− mice as early as 4 months of age. Late onset of HCC development in Iqgap2 −/− mice was consistent with the timeframe of human disease, which undergoes multiple phases and often takes decades to progress to carcinoma. While IQGAP2 was found in abundance in platelets [22], Iqgap2 −/− mice displayed platelet function indistinguishable from the wild-type littermates, probably due to a certain functional redundancy of IQGAPs in platelet activation.
Even more surprisingly, when Iqgap2 −/− mice were crossed with Iqgap1 −/− mice [31], the resultant Iqgap1 −/− /Iqgap2 −/− mice showed a lower incidence and smaller size of HCC tumors, and improvement of overall survival compared to Iqgap2 −/− mice [27]. These data suggest that inactivation of IQGAP1 in mouse liver impairs tumorigenesis caused by IQGAP2 deficiency. According to a proposed model, IQGAP2 plays the role of a tumor suppressor by being a part of the APC/AXIN/GSK3β complex, binding cytoplasmic β-catenin and preventing it from dephosphorylation and, subsequently, from activating β-catenin's nuclear target genes (Figure 2). Data on HCC development in Iqgap1 −/− / Iqgap2 −/− mice suggest that its mechanism is also IQGAP1-dependent, and, at least in the mouse model, IQGAP1 antagonizes IQGAP2 in liver carcinogenesis. The fact that Iqgap1 −/−/Iqgap2 −/− mice develop HCC, albeit at a lower rate, shows that in the absence of IQGAP2, destabilization of the E-cadherin/β-catenin axis is a primary cause of HCC. Noteworthy, IQGAP2 appears to be critical for maintaining cell adhesion during embryonic development of Xenopus, where silencing of IQGAP2 resulted in a loss of β-catenin and IQGAP1 from cell borders in the ectoderm [32]. Unattended downstream effects of upregulated IQGAP1 in liver may have the role of a “second hit”, exacerbating the HCC development. IQGAP1 has been shown to translocate into the nucleus in late G1/early S phase of the cell cycle and is believed to stimulate DNA replication and progression of the cell cycle [33]. Therefore, it is possible that the observed overexpression of IQGAP1 in Iqgap2 −/− livers may result in activation of currently unidentified nuclear oncogenic targets of IQGAP1. Yet, it remains unestablished whether isolated IQGAP1 overexpression is sufficient for HCC development. A study in a rat model of oxidative stress-induced hepatotumorigenesis showed a significant stepwise IQGAP1 upregulation at both the transcript and protein levels throughout tumor progression [34]. Interestingly, in this model IQGAP1 upregulation was tightly linked to increased levels of vimentin, a cytoplasmic intermediate filament protein synthesized in cells of mesenchymal origin, and these findings were confirmed by microarray data mining in human HCC tumors [34].
Figure 2.
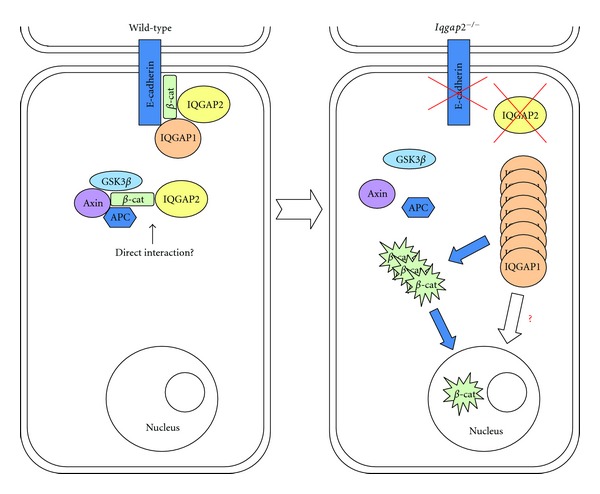
Schema summarizing the proposed hypothesis for IQGAP1 and IQGAP2 involvement in HCC development. In wild-type hepatocytes, IQGAP2 exists in two pools-bound to β-catenin and anchored at the submembrane region along with E-cadherin and IQGAP1, and as a part of the β-catenin destruction complex consisting of GSK3β kinase, Axin and Adenomatous polyposis coli (APC). The β-catenin destruction complex prevents β-catenin activation and translocation to the nucleus. In Iqgap2 −/− hepatocytes, E-cadherin disappears from the membrane, while β-catenin escapes the destruction complex, accumulates in the cytoplasm, and enters the nucleus, where it initiates transcription of various target genes. Simultaneously, IQGAP1, released from the submembrane region, is upregulated and perhaps acts in the similar to active β-catenin manner. Overexpressed IQGAP1 may also stimulate activity of destabilized β-catenin.
Alternatively, overexpressed IQGAP1 may realize its oncogenic effect in HCC liver by stimulating β-catenin transcriptional activity in the nucleus, which has been described earlier in colon cancer cells [35]. The observation that IQGAP1 has the ability to bind RNA directly [36] supports this hypothesis. It is also known that at the membrane, IQGAP1 competes with α-catenin for binding to E-cadherin [37], and destabilization of the β-catenin-E-cadherin membrane complex may be another consequence of the IQGAP1 overexpression in IQGAP2-deficiency (Figure 2). Of note, targeted disruption of the murine Iqgap1 gene caused no defect except for a late onset of gastric mucosal hyperplasia [31]. Given the IQGAP2 multidomain structure, it is feasible to postulate that IQGAP2 may contribute to hepatic carcinogenesis via several mutually nonexclusive and possibly converging mechanisms, which are likely to be identified in the future.
4. IQGAP2 Is Silenced in Human Hepatic, Gastric, and Prostate Cancers
As of now, genetic alterations in the IQGAP2 gene have not been identified in patients, and no human condition has been definitively linked to the gene. Still, single nucleotide polymorphism (SNP) research has recently started to yield evidence for IQGAP2 involvement in certain diseases. A recent genome-wide SNP study identified several SNPs in the human IQGAP2 gene associated with insulin-dependent and noninsulin-dependent type 2 diabetes mellitus [38], emphasizing the significance of exploring the IQGAP2-insulin resistance link. A study in Iqgap2 −/− mice supports this notion by showing that these mice are hypoglycemic and protected from diet-induced hepatic steatosis and insulin resistance [39]. Also, the presence of the rs153317CT/TT IQGAP2 genetic variant was linked to a 2-fold increased risk of shorter survival in patients with pancreatic cancer receiving radiation treatment [40]. IQGAP2 SNP rs457717C/T was correlated with age-dependent hearing impairment in yet another genome-wide association study [41]. Finally, IQGAP2 SNP rs3797418 was shown to influence sensitivity of 174 patient lymphoblastoid cell lines to the cytidine analogues gemcitabine and arabinosylcytosin [42]. This is an important finding since gemcitabine is used to treat many solid tumors, including pancreatic, and this SNP might be developed into a predictive marker for drug response.
Immunohistochemical analysis of a cohort of HCC patient liver specimens showed that IQGAP2 protein expression was reduced in 78% of the samples (64/82) and, consistent with the Iqgap2 −/− mouse model, the IQGAP1 protein was overexpressed in 84% of tumors studied [43], thus confirming the relevance of the Iqgap2 −/− mouse model to human disease. Both liver samples from normal donors and from patients with cirrhosis showed the reverse trend, suggesting that a hepatic IQGAP1 to IQGAP2 ratio may be developed into a promising biomarker for accurate HCC diagnosis. IQGAP1 protein overexpression in 58% of HCC patient tumors (32/55) compared to normal adjacent tissue was confirmed in another study [44]. A microarray analysis also showed upregulation of IQGAP1 and downregulation of IQGAP2 in late-stage HCC tumors [45]. Lastly, subcellular tissue proteomics analysis of HCC patient tumors revealed a 7.2-fold decrease in IQGAP2 expression and validated IQGAP2 as one among 21 novel candidate molecular targets of HCC [46]. Further studies into a possible epigenetic cause of IQGAP2 silencing in human HCC ruled out promoter hypermethylation as a potential regulatory mechanism of IQGAP2 expression [43]. This is somewhat surprising, since IQGAP2 promoter hypermethylation was identified as a cause of IQGAP2 silencing in gastrointestinal cancers [47]. Additionally, IQGAP2 was shown to be downregulated in gastric cancer by DNA copy number loss [48].
A vast number of recent publications showed that microRNAs (miRNAs), a class of small noncoding RNAs, are involved in posttranscriptional negative regulation of both oncogenes and tumor suppressors in multiple cancers, including HCC [49–51]. Various miRNAs have been implicated in different types of liver disease [52]. In the past several years, miRNAs have been found to be frequently deregulated in HCC, capable of being both oncogenes and tumor suppressors during tumor development and progression [53, 54]. A link between miRNAs and the clinicopathological features of HCC tumors has also been recently established. The most frequently deregulated miRNAs in HCC include the let-7 family (downregulated), miR-122 (downregulated), and miR-221/222 (upregulated) [52, 53]. Of note, miR-122 represents 70% of the total hepatic miRNA population. miRNA expression profiling has also been shown to be useful for classification of HCC molecular subclasses [55]. While the knowledge of how, in turn, miRNA expression is controlled in cancer remains limited, many HCC-related miRNAs have been shown to be silenced as a result of CpG hypermethylation, whereas others, such as miR-151, are overexpressed in HCC because of a chromosomal region gain (8q24 in case of miR-151) [53]. It has been reported recently that miR-124, and miR-203 may regulate iqgap1 expression in HCC [56]. This is the first report describing regulation of an IQGAP family member by miRNA. MiR-124 and miR-203 were identified as epigenetically silenced in HCC as a result of assessment of the methylation status of 43 loci containing CpG islands around 39 mature miRNA genes in a panel of HCC cell lines and noncancerous liver tissues as controls. Overexpression of miR-124 and miR-203 suppressed HCC cell growth in vitro. The 3′-untranslated region (3′-UTR) of IQGAP1 gene was identified as a direct target for both miRNAs, along with cycline-dependent kinase 6, vimentin, and several other proteins. This study characterized miR-124 and miR-203 as novel tumor-suppressive miRNAs in HCC and may provide an explanation for the observed upregulation of IQGAP1 in human HCC tumors [43]. Since IQGAP genes are highly homologous, screening for IQGAP2-specific HCC-relevant miRNAs is well justified and may provide new therapeutic targets. In 2009, an AAV-mediated hepatic delivery of a tumor-suppressing miR-26a proved to be effective in treatment of HCC in a mouse model [57].
Recently, the tumor suppressing repertoire of IQGAP2 has been expanded to include prostate cancer [58]. IQGAP2 was shown to be expressed at significantly reduced levels in tumor specimens from patients with both advanced and androgen-independent prostate cancers. Ectopic overexpression of IQGAP2 reduced proliferation of both DU145 and PC3 prostate cancer cell lines, as well as invasiveness of DU145 cells. This was linked to inhibition of Akt kinase activation [58].
5. Diverse Binding Partners May Explain How IQGAP2 Counteracts the Effects of IQGAP1
While IQGAP1 and IQGAP2 proteins share a domain structure and possess significant sequence homology, they appear to have opposing functions in vivo, at the very least in the pathogenesis of cancer. Such an apparent paradoxical phenomenon can be attributed to their distinct tissue expression, subcellular localization and protein binding partners [20], and dissecting the mechanisms underlying the divergent functions of IQGAPs could lead to developing these scaffolding proteins into novel molecular targets for HCC. Other precedents of homologous proteins counteracting each other have been described. For instance, structurally similar heparin-degrading endosulfatases sulfatase 1 (SULF1) and sulfatase 2 (SULF2) have been reported recently as having a role of a tumor suppressor and oncogene, respectively, in HCC by regulating different signaling pathways [59]. Moreover, IQGAP1 levels of expression positively correlated with SULF2 levels in HCC specimens, while IQGAP2 showed negative correlation with SULF2 [59]. SULFs might be regulating the Wnt/β-catenin pathway and epithelial-mesenchymal transition (EMT), and functional relationships between IQGAPs and SULFs should be further investigated.
An impressive list of confirmed IQGAP1 binding partners has been growing continuously in the past decade. The most recent reviews on the topic [28, 60] list over 40 proteins associated with IQGAP1. Less is known for IQGAP2. Among the proteins associated with IQGAP2 are actin, calmodulin, Rac1, and cdc42 GTPases [20], and also the arp2/3 complex in platelets [22], β-catenin in liver [27], protein kinase A-anchoring protein AKAP220 [61] and, most recently identified, phospholipid PtdInsP3 [62] (Figure 1). While both IQGAP1 and IQGAP2 bind Rac1 and cdc42, it appears that IQGAP1 selectively binds an inactive, GDP-bound form of these GTPases [18, 63], while IQGAP2 does not discriminate between their GTP- and GDP-bound forms [13, 64]. This is of particular relevance to HCC, because targeted ablation of cdc42 in mouse hepatocytes and bile ducts resulted in the development of HCC, closely resembling the cancer in Iqgap2 −/− mice in terms of late onset and molecular signature of the tumors [65]. It was also reported recently that different IQ-motifs in different IQGAPs display selectivity for calmodulin and related proteins, such as myosin essential light chain and S100B [66].
Another point of IQGAPs divergence that may translate into functional differences is IQGAPs phosphorylation by distinct kinases. It has been reported that phosphorylation of IQGAP1 at Ser1443 by protein kinase Cε (PKCε) increases IQGAP1 binding to nucleotide-free Cdc42, leading to the loss of cell-cell contacts [67]. Later, it was shown that phosphorylation controls IQGAP1 switching between pro-growth and pro-cell division/migration conformations, and that failure to switch between them leads to uncontrolled cell proliferation and transformation [68]. While three-dimensional structure is not currently available for either full-length IQGAP, it is anticipated that these proteins possess considerable capacity for conformational change, enabling integration and processing of diverse signals [69]. Most recently, phosphorylation of IQGAP2 has been confirmed [61]. It occurs at Thr716 by cAMP-dependent protein kinase (PKA) and was shown to enhance IQGAP2 binding to the active, GTP-bound form of Rac1 [61]. PKCε and PKA kinases regulate distinct signaling pathways, and while both have a role in cancer progression, their dissimilar activators and targets may also hold a key to deciphering the mechanisms behind the opposing functions of IQGAP1 and IQGAP2 in HCC. PKCε kinase belongs to the so-called novel, calcium-independent, PKCs as opposed to the classical calcium-dependent PKC group, which includes PKCα, β, and γ. All PKCs are major targets of tumor promoting phorbol esters and diacylglycerol (DAG) in response to the activation of growth factor receptors [70]. PKCε has been described as an oncogene and a tumor biomarker in many cancers, and it realizes its oncogenic effects mostly through the modulation of the Ras signaling cascade, phosphorylation of the Bcl-2 family proteins and activation of the Akt signaling pathway [71]. PKCε, in turn, is regulated by the phosphoinositide-dependent kinase 1 (PDK1) and also by autophosphorylation [72]. PKA kinase modulates cell adhesion-related events, including migration, along with glucose and lipid metabolism [73]. It is regulated by integrin-mediated cell adhesion to extracellular matrix (ECM). Molecular targets of PKA are numerous and include actin, α4 integrins, VASP, Rho GTPases, p21-activated kinase-1 (PAK1), and Src, to name only a few. A-kinase anchoring proteins (AKAPs) associate PKA with the actin cytoskeleton, thereby enhancing its signaling [73].
It remains to be evaluated whether other kinases may phosphorylate IQGAP2 at different sites. Another recent work showed that Deleted in liver cancer 1 (DLC1), a Rho GTPase-activating protein, is a substrate for phosphorylation by Akt, and this phosphorylation negatively regulates the tumor suppressor function of DLC1 in liver [74]. Moreover, IQGAP1 was shown to coimmunoprecipitate with Akt in heart [75] and liver [44]. High homology and similar domain structure between IQGAP1 and IQGAP2 supports the notion that IQGAP2 might be an Akt substrate as well. Overactivation of the PI3K/Akt signaling pathway (evident by increased phosphorylation of both Akt and GSK3β) was observed in Iqgap2 −/− livers [39]. This suggests that in IQGAP2-deficiency, overactivation of the PI3K/Akt signaling pathway may contribute to the development of HCC, along with the aberrantly activated Wnt/β-catenin pathway [27]. More direct evidence of IQGAP2 involvement in the PI3K signaling network has been provided as a result of solving the crystal structure of the IQGAP2 C-terminal domain [62]. It was shown that the extreme C-terminus of IQGAPs is responsible for binding to phosphatidylinositol 3,4,5-trisphosphate (PtdInsP3). Given that both IQGAP1 and IQGAP2 were reported to be able to bind PtdInsP3 with the similar affinity, further studies will be needed to distinguish the roles of the two IQGAPs in PI3K signaling.
6. Concluding Remarks
Mounting compelling evidence in support of IQGAP2 acting as a tumor suppressor in HCC and other cancers calls for further thorough studies of this intriguing protein. The fact that it counteracts oncogenic effects of its very close homolog, IQGAP1, makes these studies even more urgent and opens additional avenues for development of principally new therapeutics for HCC and perhaps other malignancies. Further research employing targeted functional proteomics will hopefully identify the whole spectrum of IQGAP2 binding partners and domains responsible for these interactions. Identification of downstream molecular effectors unique for each IQGAP would provide additional candidate targets. Likewise, means for specific blocking of IQGAP1 domains need to be explored, although this should be approached with caution. For instance, it was demonstrated that IQGAP1 is essential for the integrity of actin structures around bile canaliculi and inhibition of IQGAP1 would impact maintenance of stable adherens junctions in liver [76]. Finally, a better understanding of tissue-specific regulation of the IQGAP1 and IQGAP2 genes would provide additional options for urgently needed therapeutic intervention. Clearly, there are many gaps yet to be filled in our knowledge of IQGAPs.
Acknowledgment
Work in the Schmidt laboratory is supported by the Research Scholar Grant (RSG-09-033-01-CSM) from the American Cancer Society.
References
- 1.Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. Journal of Clinical Oncology. 2009;27(9):1485–1491. doi: 10.1200/JCO.2008.20.7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: Cancer Journal for Clinicians. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.Lee HC, Kim M, Wands JR. Wnt/frizzled signaling in hepatocellular carcinoma. Frontiers in Bioscience. 2006;11(2):1901–1915. doi: 10.2741/1933. [DOI] [PubMed] [Google Scholar]
- 4.Alsowmely AM, Hodgson HJ. Non-surgical treatment of hepatocellular carcinoma. Alimentary Pharmacology and Therapeutics. 2002;16(1):1–15. doi: 10.1046/j.1365-2036.2002.01149.x. [DOI] [PubMed] [Google Scholar]
- 5.Jemal A, Murray T, Ward E, et al. Cancer statistics, 2005. CA: A Cancer Journal for Clinicians. 2005;55(1):10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 6.Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology. 2005;42(5):1208–1236. doi: 10.1002/hep.20933. [DOI] [PubMed] [Google Scholar]
- 7.Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. The New England Journal of Medicine. 2008;359(4):378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 8.Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29(36):4989–5005. doi: 10.1038/onc.2010.236. [DOI] [PubMed] [Google Scholar]
- 9.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nature Reviews Cancer. 2006;6(9):674–687. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 10.Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene. 2006;25(27):3787–3800. doi: 10.1038/sj.onc.1209556. [DOI] [PubMed] [Google Scholar]
- 11.Bernards A. GAPs galore! a survey of putative Ras superfamily GTPase activating proteins in man and Drosophila. Biochimica Et Biophysica Acta. 2003;1603(2):47–82. doi: 10.1016/s0304-419x(02)00082-3. [DOI] [PubMed] [Google Scholar]
- 12.Weissbach L, Settleman J, Kalady MF, et al. Identification of a human RasGAP-related protein containing calmodulin- binding motifs. The Journal of Biological Chemistry. 1994;269(32):20517–20521. [PubMed] [Google Scholar]
- 13.Brill S, Li S, Lyman CW, et al. The Ras GTPase-activating-protein-related human protein IQGAP2 harbors a potential actin binding domain and interacts with calmodulin and Rho family GTPases. Molecular and Cellular Biology. 1996;16(9):4869–4878. doi: 10.1128/mcb.16.9.4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Briggs MW, Sacks DB. IQGAP proteins are integral components of cytoskeletal regulation. EMBO Reports. 2003;4(6):571–574. doi: 10.1038/sj.embor.embor867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang S, Watanabe T, Noritake J, et al. IQGAP3, a novel effector of Rac1 and Cdc42, regulates neurite outgrowth. Journal of Cell Science. 2007;120(4):567–577. doi: 10.1242/jcs.03356. [DOI] [PubMed] [Google Scholar]
- 16.Brown MD, Sacks DB. IQGAP1 in cellular signaling: bridging the GAP. Trends in Cell Biology. 2006;16(5):242–249. doi: 10.1016/j.tcb.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 17.Scheffzek K, Ahmadian MR, Wittinghofer A. GTPase-activating proteins: helping hands to complement an active site. Trends in Biochemical Sciences. 1998;23(7):257–262. doi: 10.1016/s0968-0004(98)01224-9. [DOI] [PubMed] [Google Scholar]
- 18.Hart MJ, Callow MG, Souza B, Polakis P. IQGAP1, a calmodulin-binding protein with a rasGAP-related domain, is a potential effector for cdc42Hs. EMBO Journal. 1996;15(12):2997–3005. [PMC free article] [PubMed] [Google Scholar]
- 19.Swart-Mataraza JM, Li Z, Sacks DB. IQGAP1 is a component of Cdc42 signaling to the cytoskeleton. The Journal of Biological Chemistry. 2002;277(27):24753–24763. doi: 10.1074/jbc.M111165200. [DOI] [PubMed] [Google Scholar]
- 20.White CD, Brown MD, Sacks DB. IQGAPs in cancer: a family of scaffold proteins underlying tumorigenesis. FEBS Letters. 2009;583(12):1817–1824. doi: 10.1016/j.febslet.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson M, Sharma M, Henderson BR. IQGAP1 regulation and roles in cancer. Cellular Signalling. 2009;21(10):1471–1478. doi: 10.1016/j.cellsig.2009.02.023. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt VA, Scudder L, Devoe CE, Bernards A, Cupit LD, Bahou WF. IQGAP2 functions as a GTP-dependent effector protein in thrombin-induced platelet cytoskeletal reorganization. Blood. 2003;101(8):3021–3028. doi: 10.1182/blood-2002-09-2807. [DOI] [PubMed] [Google Scholar]
- 23.Bahou WF, Scudder L, Rubenstein D, Jesty J. A shear-restricted pathway of platelet procoagulant activity is regulate by IQGAP1. The Journal of Biological Chemistry. 2004;279(21):22571–22577. doi: 10.1074/jbc.M402561200. [DOI] [PubMed] [Google Scholar]
- 24.Cupit LD, Schmidt VA, Miller F, Bahou WF. Distinct PAR/IQGAP expression patterns durinq murine development: implications for thrombin-associated cytoskeletal reorganization. Mammalian Genome. 2004;15(8):618–629. doi: 10.1007/s00335-004-2370-8. [DOI] [PubMed] [Google Scholar]
- 25.Skawran B, Steinemann D, Weigmann A, et al. Gene expression profiling in hepatocellular carcinoma: upregulation of genes in amplified chromosome regions. Modern Pathology. 2008;21(5):505–516. doi: 10.1038/modpathol.3800998. [DOI] [PubMed] [Google Scholar]
- 26.Kunimoto K, Nojima H, Yamazaki Y, Yoshikawa T, Okanoue T, Tsukita S. Involvement of IQGAP3, a regulator of Ras/ERK-related cascade, in hepatocyte proliferation in mouse liver regeneration and development. Journal of Cellular Physiology. 2009;220(3):621–631. doi: 10.1002/jcp.21798. [DOI] [PubMed] [Google Scholar]
- 27.Schmidt VA, Chiariello CS, Capilla E, Miller F, Bahou WF. Development of hepatocellular carcinoma in Iqgap2-deficient mice is IQGAP1 dependent. Molecular and Cellular Biology. 2008;28(5):1489–1502. doi: 10.1128/MCB.01090-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White CD, Erdemir HH, Sacks DB. IQGAP1 and its binding proteins control diverse biological functions. Cell Signaling. 2012;24(4):826–834. doi: 10.1016/j.cellsig.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nature Genetics. 2002;31(4):339–346. doi: 10.1038/ng0802-339. [DOI] [PubMed] [Google Scholar]
- 30.Wurmbach E, Chen YB, Khitrov G, et al. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology. 2007;45(4):938–947. doi: 10.1002/hep.21622. [DOI] [PubMed] [Google Scholar]
- 31.Li S, Wang Q, Chakladar A, Bronson RT, Bernards A. Gastric hyperplasia in mice lacking the putative Cdc42 effector IQGAP1. Molecular and Cellular Biology. 2000;20(2):697–701. doi: 10.1128/mcb.20.2.697-701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamashiro S, Abe H, Mabuchi I. IQGAP2 is required for the cadherin-mediated cell-to-cell adhesion in Xenopus laevis embryos. Developmental Biology. 2007;308(2):485–493. doi: 10.1016/j.ydbio.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 33.Johnson M, Sharma M, Brocardo MG, Henderson BR. IQGAP1 translocates to the nucleus in early S-phase and contributes to cell cycle progression after DNA replication arrest. International Journal of Biochemistry and Cell Biology. 2011;43(1):65–73. doi: 10.1016/j.biocel.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 34.Tsubota A, Matsumoto K, Mogushi K, et al. IQGAP1 and vimentin are key regulator genes in naturally occurring hepatotumorigenesis induced by oxidative stress. Carcinogenesis. 2010;31(3):504–511. doi: 10.1093/carcin/bgp313. [DOI] [PubMed] [Google Scholar]
- 35.Briggs MW, Li Z, Sacks DB. IQGAP1-mediated stimulation of transcriptional co-activation by β-catenin is modulated by calmodulin. The Journal of Biological Chemistry. 2002;277(9):7453–7465. doi: 10.1074/jbc.M104315200. [DOI] [PubMed] [Google Scholar]
- 36.Willingham AT, Orth AP, Batalov S, et al. Molecular biology: a strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science. 2005;309(5740):1570–1573. doi: 10.1126/science.1115901. [DOI] [PubMed] [Google Scholar]
- 37.Kuroda S, Fukata M, Nakagawa M, et al. Role of IQGAP1, a target of the small GTPases Cdc42 and Rac1, in regulation of E-cadherin-mediated cell-cell adhesion. Science. 1998;281(5378):832–835. doi: 10.1126/science.281.5378.832. [DOI] [PubMed] [Google Scholar]
- 38.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chiariello CS, LaComb JF, Bahou WF, Schmidt VA. Ablation of Iqgap2 protects from diet-induced hepatic steatosis due to impaired fatty acid uptake. Regulatory Peptides. 2012;173(1–3):36–46. doi: 10.1016/j.regpep.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeng H, Yu H, Lu L, et al. Genetic effects and modifiers of radiotherapy and chemotherapy on survival in pancreatic cancer. Pancreas. 2011;40(5):657–663. doi: 10.1097/MPA.0b013e31821268d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Laer L, Huyghe JR, Hannula S, et al. A genome-wide association study for age-related hearing impairment in the Saami. European Journal of Human Genetics. 2010;18(6):685–693. doi: 10.1038/ejhg.2009.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li L, Fridley BL, Kalari K, et al. Gemcitabine and arabinosylcytosin pharmacogenomics: genome-wide association and drug response biomarkers. PLoS ONE. 2009;4(11) doi: 10.1371/journal.pone.0007765.e7765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.White CD, Khurana H, Gnatenko DV, et al. IQGAP1 and IQGAP2 are reciprocally altered in hepatocellular carcinoma. BMC Gastroenterology. 2010;10(1, article 125) doi: 10.1186/1471-230X-10-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen F, Zhu HH, Zhou LF, Wu SS, Wang J, Chen Z. IQGAP1 is overexpressed in hepatocellular carcinoma and promotes cell proliferation by Akt activation. Experimental and Molecular Medicine. 2010;42(7):477–483. doi: 10.3858/emm.2010.42.7.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liao YL, Sun YM, Chau GY, et al. Identification of SOX4 target genes using phylogenetic footprinting-based prediction from expression microarrays suggests that overexpression of SOX4 potentiates metastasis in hepatocellular carcinoma. Oncogene. 2008;27(42):5578–5589. doi: 10.1038/onc.2008.168. [DOI] [PubMed] [Google Scholar]
- 46.Lee YY, McKinney KQ, Ghosh S, et al. Subcellular tissue proteomics of hepatocellular carcinoma for molecular signature discovery. Journal of Proteome Research. 2011;10(11):5070–5083. doi: 10.1021/pr2005204. [DOI] [PubMed] [Google Scholar]
- 47.Jin SH, Akiyama Y, Fukamachi H, Yanagihara K, Akashi T, Yuasa Y. IQGAP2 inactivation through aberrant promoter methylation and promotion of invasion in gastric cancer cells. International Journal of Cancer. 2008;122(5):1040–1046. doi: 10.1002/ijc.23181. [DOI] [PubMed] [Google Scholar]
- 48.Fan B, Dachrut S, Coral H, et al. Integration of DNA copy number alterations and transcriptional expression analysis in human gastric cancer. PLoS ONE. 2012;7(4) doi: 10.1371/journal.pone.0029824.e29824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Le XF, Merchant O, Bast RC, Calin GA. The roles of microRNAs in the cancer invasion-metastasis cascade. Cancer Microenvironment. 2010;3(1):137–147. doi: 10.1007/s12307-010-0037-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mott JL. MicroRNAs involved in tumor suppressor and oncogene pathways: implications for hepatobiliary neoplasia. Hepatology. 2009;50(2):630–637. doi: 10.1002/hep.23010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lovat F, Valeri N, Croce CM. MicroRNAs in the pathogenesis of cancer. Seminars in Oncology. 2011;38(6):724–733. doi: 10.1053/j.seminoncol.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 52.Bala S, Marcos M, Szabo G. Emerging role of microRNAs in liver diseases. World Journal of Gastroenterology. 2009;15(45):5633–5640. doi: 10.3748/wjg.15.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang S, He X. The role of microRNAs in liver cancer progression. British Journal of Cancer. 2011;104(2):235–240. doi: 10.1038/sj.bjc.6606010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Braconi C, Henry JC, Kogure T, Schmittgen T, Patel T. The role of microRNAs in human liver cancers. Seminars in Oncology. 2011;38(6):752–763. doi: 10.1053/j.seminoncol.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Toffanin S, Hoshida Y, Lachenmayer A, et al. MicroRNA-based classification of hepatocellular carcinoma and oncogenic role of miR-517a. Gastroenterology. 2011;140(5):1618–1628.e16. doi: 10.1053/j.gastro.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Furuta M, Kozaki KI, Tanaka S, Arii S, Imoto I, Inazawa J. miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis. 2009;31(5):766–776. doi: 10.1093/carcin/bgp250. [DOI] [PubMed] [Google Scholar]
- 57.Kota J, Chivukula RR, O’Donnell KA, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137(6):1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xie Y, Yan J, Cutz JC, et al. IQGAP2, A candidate tumour suppressor of prostate tumorigenesis. Biochimica Et Biophysica Acta. 2012;1822(6):875–884. doi: 10.1016/j.bbadis.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 59.Yang JD, Sun Z, Hu C, et al. Sulfatase 1 and sulfatase 2 in hepatocellular carcinoma: associated signaling pathways, tumor phenotypes, and survival. Genes Chromosomes and Cancer. 2011;50(2):122–135. doi: 10.1002/gcc.20838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shannon KB. IQGAP family members in yeast, Dictyostelium, and mammalian cells. International Journal of Cell Biology. 2012;2012:14 pages. doi: 10.1155/2012/894817.894817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Logue JS, Whiting JL, Tunquist B, Langeberg LK, Scott JD. Anchored protein kinase A recruitment of active Rac GTPase. The Journal of Biological Chemistry. 2011;286(25):22113–22121. doi: 10.1074/jbc.M111.232660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dixon MJ, Gray A, Schenning M, et al. IQGAP proteins reveal an atypical phosphoinositide (aPI) binding domain with a pseudo C2 domain fold. The Journal of Biological Chemistry. 2012;287(27):22483–22496. doi: 10.1074/jbc.M112.352773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Joyal JL, Annan RS, Ho YD, et al. Calmodulin modulates the interaction between IQGAP1 and Cdc42: identification of IQGAP1 by nanoelectrospray tandem mass spectrometry. The Journal of Biological Chemistry. 1997;272(24):15419–15425. doi: 10.1074/jbc.272.24.15419. [DOI] [PubMed] [Google Scholar]
- 64.McCallum SJ, Wu WJ, Cerione RA. Identification of a putative effector for Cdc42Hs with high sequence similarity to the RasGAP-related protein IQGAP1 and a Cdc42Hs binding partner with similarity to IQGAP2. The Journal of Biological Chemistry. 1996;271(36):21732–21737. doi: 10.1074/jbc.271.36.21732. [DOI] [PubMed] [Google Scholar]
- 65.van Hengel J, D’Hooge P, Hooghe B, et al. Continuous cell Injury promotes hepatic tumorigenesis in Cdc42-deficient mouse liver. Gastroenterology. 2008;134(3):781–792. doi: 10.1053/j.gastro.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 66.Atcheson E, Hamilton E, Pathmanathan S, Greer B, Harriott P, Timson DJ. IQ-motif selectivity in human IQGAP2 and IQGAP3: binding of calmodulin and myosin essential light chain. Bioscience Reports. 2011;31(5):371–379. doi: 10.1042/BSR20100123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grohmanova K, Schlaepfer D, Hess D, Gutierrez P, Beck M, Kroschewski R. Phosphorylation of IQGAP1 modulates its binding to Cdc42, revealing a new type of Rho-GTPase regulator. The Journal of Biological Chemistry. 2004;279(47):48495–48504. doi: 10.1074/jbc.M408113200. [DOI] [PubMed] [Google Scholar]
- 68.Wang JB, Sonn R, Tekietsadik YK, Samorodnitsky D, Osman MA. IQGAP1 regulates cell proliferation through a novel CDC42-mTOR pathway. Journal of Cell Science. 2009;122(12):2024–2033. doi: 10.1242/jcs.044644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Elliott SF, Allen G, Timson DJ. Biochemical analysis of the interactions of IQGAP1 C-terminal domain with CDC42. World Journal of Biological Chemistry. 2012;3(3):53–60. doi: 10.4331/wjbc.v3.i3.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nature Reviews Cancer. 2007;7(4):281–294. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 71.Gorin MA, Pan Q. Protein kinase Cε: an oncogene and emerging tumor biomarker. Molecular Cancer. 2009;8, article 9 doi: 10.1186/1476-4598-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dutil EM, Toker A, Newton AC. Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK-1) Current Biology. 1998;8(25):1366–1375. doi: 10.1016/s0960-9822(98)00017-7. [DOI] [PubMed] [Google Scholar]
- 73.Howe AK. Regulation of actin-based cell migration by cAMP/PKA. Biochimica et Biophysica Acta. 2004;1692(2-3):159–174. doi: 10.1016/j.bbamcr.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 74.Ko FCF, Chan LK, Tung EKK, Lowe SW, Ng IOL, Yam JWP. Akt phosphorylation of deleted in liver cancer 1 abrogates its suppression of liver cancer tumorigenesis and metastasis. Gastroenterology. 2010;139(4):1397–1407.e6. doi: 10.1053/j.gastro.2010.06.051. [DOI] [PubMed] [Google Scholar]
- 75.Sbroggiò M, Carnevale D, Bertero A, et al. IQGAP1 regulates ERK1/2 and AKT signalling in the heart and sustains functional remodelling upon pressure overload. Cardiovascular Research. 2011;91(3):456–464. doi: 10.1093/cvr/cvr103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Emadali A, Muscatelli-Groux B, Delom F, et al. Proteomic analysis of ischemia-reperfusion injury upon human liver transplantation reveals the protective role of IQGAP1. Molecular and Cellular Proteomics. 2006;5(7):1300–1313. doi: 10.1074/mcp.M500393-MCP200. [DOI] [PubMed] [Google Scholar]