

Abstract
The recent application of electron transfer dissociation (ETD) to measure the hydrogen exchange of proteins in solution at single-residue resolution (HX-ETD) paves the way for mass spectrometry-based analyses of biomolecular structure at an unprecedented level of detail. The approach requires that activation of polypeptide ions prior to ETD is minimal so as to prevent undesirable gas-phase randomization of the deuterium label from solution (i.e., hydrogen scrambling). Here we explore the use of ETD in a traveling wave ion guide of a quadrupole-time-of-flight (Q-TOF) mass spectrometer with a “Z-spray” type ion source, to measure the deuterium content of individual residues in peptides. We systematically identify key parameters of the Z-spray ion source that contribute to collisional activation and define conditions that allow ETD experiments to be performed in the traveling wave ion guide without gas-phase hydrogen scrambling. We show that ETD and supplemental collisional activation in a subsequent traveling wave ion guide allows for improved extraction of residue-specific deuterium contents in peptides with low charge. Our results demonstrate the feasibility, and illustrate the advantages of performing HX-ETD experiments on a high-resolution Q-TOF instrument equipped with traveling wave ion guides. Determination of parameters of the Z-spray ion source that contribute to ion heating are similarly pertinent to a growing number of MS applications that also rely on an energetically gentle transfer of ions into the gas-phase, such as the analysis of biomolecular structure by native mass spectrometry in combination with gas-phase ion-ion/ion-neutral reactions or ion mobility spectrometry.
Keywords: Hydrogen/deuterium exchange, Protein conformation, Electron transfer dissociation, Supplemental activation, Traveling wave ion guide
Introduction
Knowledge of protein structure can provide in-depth understanding of biology at the molecular level and lead to insights into the origins of human disease and the action of pharmaceutical drugs. The use of mass spectrometry to measure the hydrogen/deuterium exchange (HX-MS) of proteins in solution has become a recognized and widely applicable method to investigate the conformation and dynamics of proteins during function and molecular interactions [1–3]. The classic HX-MS approach is based on the time-resolved measurement of deuterium incorporated into backbone amide groups of either the intact protein (‘global exchange experiment’) or individual peptide segments generated by pepsin digestion (‘local exchange experiment’). Although information from a local exchange experiment can be mapped to regions of the intact protein, the obtainable resolution is limited by the size and diversity of the peptides generated during enzymatic proteolysis. With few exceptions [4], this prevents the extraction of hydrogen exchange profiles for individual amino acid residues.
Measurement of protein HX at single residue resolution was previously only attainable on a routine basis by NMR spectroscopy [5]. More recently, it has been shown that prompt gas-phase fragmentation techniques such as electron capture/transfer dissociation (ECD/ETD) [6, 7] can be incorporated into a HX-MS workflow to measure the HX of individual residues in a protein. By ECD/ETD of either peptic peptides [8–11] or the intact protein [12–15], the masses of fragment ions can be used to determine the solution deuterium uptake of individual backbone amide groups in a protein.
For such HX-ECD/ETD type experiments, it is imperative that the gas-phase fragmentation is performed on labeled peptide or proteins ions that have not experienced excessive heating or excitation. Despite the gentle or “soft” nature of the electrospray process, significant internal energy build-up can occur in peptide ions through energetic collisions with gas molecules in the ion source or other high-pressure regions of a mass spectrometer. It has been shown that vibrational excitation of peptide ions prior to electron-induced fragmentation can cause labile hydrogen atoms to become mobile and interchange their positions internally within the vibrationally excited gaseous polypeptide [8, 11]; a process referred to as hydrogen scrambling [16, 17]. If allowed to occur prior to gas-phase dissociation, hydrogen scrambling will erase the original deuterium labeling pattern from subsequent fragment ions and thus render the MS experiment invalid.
Collisional heating of analyte ions during HX-ECD/ETD experiments must be minimized by carefully controlling the relevant instrument parameters [8, 11]. Electrostatic potentials that accelerate ions in the ion source and the ion transfer optics are typically responsible for inducing undesirable collisional activation after ion formation in the ion source, but the degree to which this occurs is highly dependent on instrumental design. It is therefore important to systematically investigate the internal energy build-up in analyte ions during ionization and transport through the ion optics on different instrumental platforms. Hydrogen scrambling (and indirectly collisional activation) can be detected at high sensitivity in model peptides with a well-defined solution deuterium label as the exact contribution of hydrogen scrambling to the measured deuteration pattern can be accurately quantified [18, 19]. Furthermore, it was recently shown that by a straightforward inspection of the common loss of ammonia during ETD of peptic peptides actually analyzed in an HX-ETD experiment, one can ensure that the occurrence of gas-phase scrambling is minimized for routine applications without the use of model peptides [9].
Here we investigate the occurrence of hydrogen scrambling during ETD experiments in the traveling wave (T-Wave) ion guide of a quadrupole time of flight (Q-TOF) instrument (Waters SYNAPT G2). A T-Wave ion guide provides the ability to perform ion mobility separation (IMS) of analyte ions but more recently has also been used to harbor ion-neutral reactions [20–22] or ion-ion reactions for ETD [23]. Prior work using model peptides has defined the collisional activation threshold for hydrogen scrambling to occur during ECD/ETD in an ion trap and a FT-ICR mass spectrometer [8, 11]. Both of these instruments employ a heated transfer capillary (ion tube) ion source. Like many Q-TOF instruments, the SYNAPT G2 employs a “Z-spray” type ion source. The geometry and design of this ion source is considerably different than ion sources employing a heated transfer capillary. For instance, the process of electrospray is pneumatically assisted in the Z-spray ion source, desolvation is aided by curtain gasses and ion trajectories are forced along a Z-shaped path by the use of electrostatic potentials applied to two skimmer orifices. The contribution of this ion source design to inadvertent collisional activation of peptide ions during ionization has not been investigated in detail to our knowledge. Such information is critical for the use of ETD on this instrumental platform to measure the HX of proteins in solution. In addition, insight into the collisional activation experienced by ions in “Z-spray” type ion sources is also relevant to the growing number of applications of Q-TOF instruments in which proteins are ionized from native solution conditions and analyzed by gas-phase techniques (ion mobility separation [24–26] or gas-phase hydrogen/deuterium exchange [20–22]) to extract information about their structures in solution.
Presently, we have measured the contribution of individual Z-spray ion source parameters to the overall hydrogen scrambling experienced by a model peptide when traversing the high-pressure regions of the Q-TOF instrument. We define conditions for minimal gas-phase scrambling and inadvertent ion heating and demonstrate the feasibility of site-specific HX measurements by ETD in a T-Wave ion guide of a Q-TOF mass spectrometer.
Experimental
Materials and Reagents
The model peptide P1 with the sequence HHHHHHIIKIIK was obtained from Genscript Corp. (Piscataway, NJ, USA). D2O (99.9 atom % D) was purchased from Sigma-Aldrich (St. Louis, MO, USA). All other chemicals were of the highest grade commercially available.
Preparation of Selectively Labeled Peptide
The procedure for selective labeling of peptide P1 has been described previously [19]. Briefly, 500 nmol peptide P1 was dissolved in 50 μL 99.9% D2O (10 mM) and 5 μL of this stock was diluted into 99 μL 99.9% D2O (0.5 mM) and the solution was incubated for 12–18 h at 4 °C. Five μL of this stock solution of equilibrium labeled peptide P1 was then mixed with 245 μL pre-chilled ESI buffer (0.5 M acetic acid in 50% methanol, pH 2.5) and either loaded directly into a pre-chilled sample syringe or snap-frozen on dry ice for later use. Control samples of peptide P1 (referred to in the text as “unlabeled”) were prepared in a similar manner but were allowed to fully equilibrate (12 h at 25 °C) in the ESI buffer. The deuterium content per labile site of peptide P1 in control samples was measured to be 0.03 which is in good agreement with a theoretical value of 0.0287 [11].
ETD Tandem Mass Spectrometry of Peptide P1
Samples of labeled peptide were immediately transferred to a pre-chilled Hamilton syringe (825RN; Hamilton Company, Reno, NV, USA). The syringe was mounted on a syringe pump (Harvard Apparatus, Holliston, MA, USA) and cooled by placing a small re-sealable plastic bag containing dry-ice directly on top of the glass syringe barrel. Labeled peptide was infused into a SYNAPT G2 mass spectrometer equipped with ETD at 5–10 μL/min via a short PEEK tube (20 cm in length, 0.004 in. i.d.). A limited number of experiments were conducted using the nanoflow version of the Z-spray ion source where samples were infused from the syringe at a flow rate of 0.5 μL/min through a fused-silica capillary (20 cm in length). 4-Nitrotoluene was used as the ETD reagent and reagent vapor was introduced into the glow discharge anion source by a flow of nitrogen gas (Makeup Flow; 4.7–8.4 mL/min) across 4-nitrotoluene crystals contained in a sealed vial. Radical anions of the ETD reagent were generated as described previously [23] via a glow discharge anion source operated at 18–25 μA. During ETD experiments, peptide cations and radical anions were generated sequentially in the electrospray ion source. By automated switching of ion source polarity and quadrupole settings, multiply-charged peptide cations (+2/+3/+4) of model peptide P1 and singly-charged 4-nitrotoluene radical anions were delivered to the trap T-Wave ion guide for ETD reactions. MS/MS scans were acquired for 1 s and reagent anion-trap T-Wave refill times were 100 ms during the interscan period. The trap T-Wave ion guide was operated with a wave height of 0.2 V and a wave velocity of 250 m/s (conditions that were optimal for ETD efficiency). MS scans were recorded by increasing trap wave height to 1.5 V and removing the current on the glow-discharge needle. Mass spectra were acquired in a range of 50 to 2000m/z in both MS and MS/MS mode. Peptide ions were selected for ETD in the trap T-wave ion guide by the quadrupole using a selection window of ± 2.0 m/z units.
The occurrence of hydrogen scrambling in peptide P1 was monitored while varying ion source settings within the following ranges (the default value is indicated in brackets): capillary voltage, 2–4kV(3kV);desolvation gas flow, 0–900 L/h (600 L/h); cone gas flow, 0–600 L/h (0 L/h); source block temperature, 50–150 °C (75 °C); desolvation gas temperature 150–450 °C (150 °C); source pressure (Backing), 2.2–3.5 mBar (3.2 mBar); sample cone voltage, 5–65 V (45 V); extraction cone voltage, 0.5–10 V (4.1 V); source T-wave velocity, 150–1200 m/s (300 m/s); source T-wave height, 0.2–0.6 V (0.2 V). For the limited number of experiments performed using the nanoflow version of the Z-spray ion source, the nanoflow source was operated in similar ranges as indicated above but in the absence of a desolvation gas flow.
Data Analysis
Average masses of ETD product ions were determined from the intensity-weighted centroid mass of the individual isotopic envelopes in ETD mass spectra. The deuterium content of product ions was calculated by subtracting centroid masses of an unlabeled sample from the corresponding centroid masses of a labeled sample. Calculations of the degree of scrambling and the theoretical models for calculating scrambling from fragment ions of model peptide P1 were performed and validated as described previously [8, 11]. Levels of hydrogen scrambling calculated from c-type fragment ions (c2–c11) of peptide P1 showed an excellent correlation to corresponding values calculated from z-type fragment ions (z3–z5, z7–z11). The deuterium content of the z6 fragment ion could not be determined due to spectral overlap with an interfering ion.
Results and Discussion
Minimizing Gas-Phase Hydrogen Scrambling in a Z-Spray Ion Source
Sensitive and quantitative measurements of hydrogen scrambling were performed by the use of model peptide P1 (sequence: HHHHHHIIKIIK). Differences in intrinsic exchange rates in this amino acid sequence allows deuterium to be selectively retained only at the backbone amide hydrogen positions of the C-terminal residues of the peptide (i.e., -IIKIIK) [19]. In a systematic manner, the selectively labeled peptide P1 was infused into the Synapt G2 mass spectrometer and charge states +2, +3 and +4 of the peptide were subjected to ETD at different experimental settings. Reaction of 4-nitrotoluene radical anions with triply protonated ions of peptide P1 in the trap T-wave ion guide of the instrument gave rise to an extensive series of c-type (c2–c11) and z-type (z3–z11) fragment ions (Figure 1a). These ions originate from gas-phase cleavage of N–Cα bonds of the peptide backbone. ETD spectra (Figure 1b) of selectively labeled peptide P1 acquired at “default” settings of the ion source (see Experimental section) revealed a distinct increase in the deuterium content of fragment ions of the N-terminal half of the peptide compared with the equilibrium labeled control sample (an identical sample which had been equilibrated in the ESI buffer at room temperature for 12 h) (Figure 1d). As the selective deuterium label was exclusively confined to the C-terminal half of the peptide in solution prior to ionization, the mass spectrometry experiment conducted at default conditions of the Z-spray ion source had caused sufficient collisional activation of the peptide ion to induce the migration of deuterium from the C-terminal half of the gaseous peptide to the N-terminal half. An identical experiment was also recorded after reducing the electrostatic potential applied to the first skimmer orifice (sample cone) of the Z-spray ion source from 45 V to 25 V (Figure 1c). This moderate reduction in voltage caused the deuterium contents of fragment ions of the N-terminal half of the triply charged peptide (c2–c6) to drop to those of the equilibrium labeled control. This indicated that hydrogen scrambling could readily be eliminated by quite subtle tuning of the Z-spray ion source parameters. The reduced sample cone voltage setting also gave rise to decreased ion transmission, corresponding to a 4-fold reduction of the ion current of the intact reduced species in the corresponding ETD spectra.
Figure 1.
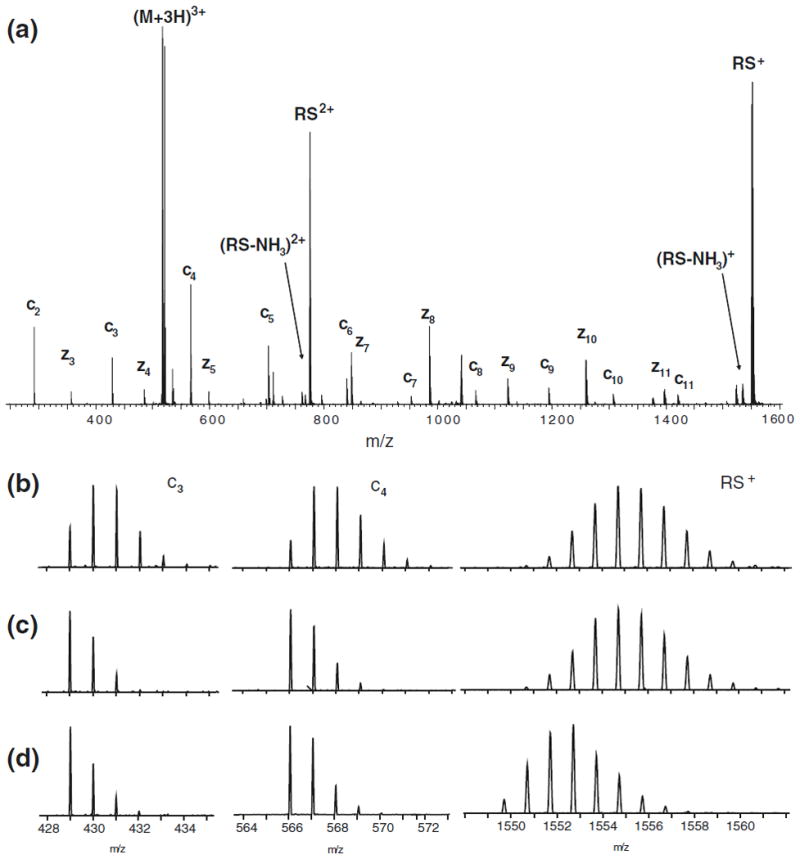
Elevated electrostatic potentials of the Z-spray ion source can induce gas-phase migration of deuterium in a selectively labeled peptide during ETD. (a) Full ETD spectrum of triply protonated peptide P1. Close-up spectra of the c3 fragment ion (left), the c4 fragment ion (middle), and the intact reduced species RS+ (right) of labeled (b)–(c) and unlabeled (d) peptide P1, recorded with a sample cone voltage of 45 V (b) and 25 V (c)–(d). The occurrence of gas-phase scrambling is directly detected by the mass shift in c3 and c4 fragment ions in ETD spectra recorded using a cone voltage of 45 V. The shift in mass observed in (b) corresponds to complete (100%) scrambling of all labile sites in peptide P1 prior to ETD.
Based on these initial findings, we investigated the influence, if any, on hydrogen scrambling of an extensive range of adjustable parameters of both the ion source and ion transfer regions of the instrument. These studies were performed by systematically changing one parameter while keeping all other parameters fixed at “default” settings. Of the numerous instrumental settings tested (see Experimental section), only three had a significant effect on hydrogen scrambling as observed in peptide P1: the sample cone voltage, the extraction cone voltage, and the velocity of the source T-wave ion guide (Figure 2a). The electrostatic potential applied to the first skimmer was found to have the greatest ability to induce gas-phase migration of deuterium in peptide P1. For instance, by changing the sample cone voltages from 5 to 55 V, the average hydrogen scrambling measured in ETD fragment ions (c2–c6)of peptide P1 was increased from 9% to 88% (Figure 2b). In comparison, increasing the potential applied to the second skimmer (extraction cone) from 0.5 to 10 V caused a more modest increase in scrambling from 9% to 30%, respectively (Figure 2c). This difference could be due to the lower electrostatic potential at which the second skimmer is operated relative to the first skimmer but also the lower pressure experienced by ions when they are accelerated past the second skimmer relative to the first. A pressure gradient exists between the main ion source chamber (approximate atmospheric pressure), across the two skimmer orifices to the source T-Wave ion guide (~1×10−3 mBar). The collisional activation of an ion accelerated at low pressure is smaller than if accelerated to the same extent at higher pressure as the probability of collisions with background gas increase with pressure [27]. It is therefore interesting to note that the third parameter observed to affect hydrogen scrambling was the wave velocity in the source T-Wave ion guide. This ion guide is placed in a lower pressure region adjacent to the ion source and the ion guide serves to transport ions efficiently from the high-pressure front end of the instrument to the quadrupole operated at much lower pressure (typically 10−5 mBar). A modest, yet significant, increase in the average hydrogen scrambling (6% to 29%) was observed in ETD fragment ions (c2 – c6) of peptide P1 upon increasing the velocity of the traveling wave that carries analyte ions through the ion guide from 150 m/s to 1200 m/s (Figure 2d). Evidently, a detectable level of hydrogen scrambling can be induced in peptide ions when traversing this ion guide at wave velocity settings above 300 m/s, despite the lower background pressure relative to the ion source region. Earlier studies at higher pressures have shown that a T-Wave ion guide operated at high speeds can cause dissociation of peptides [28]. The present findings indicate that a mild degree of activation can occur while operating the T-Wave in the current and significantly lower pressure regime. Notably, however, at wave velocities below 300 m/s, hydrogen scrambling was undetectable, indicating conditions for very gentle ion transport through the ion guide.
Figure 2.

Controlling hydrogen scrambling by adjusting key parameters of the Z-spray ion source. (a) Schematic of the Z-spray ion source of a Q-TOF (SYNAPT G2) mass spectrometer. (b)–(d) Plots of hydrogen scrambling (red curves) in ETD fragment ions of triply protonated peptide P1 as a function of the following ion source parameters: (b) sample cone voltage, (c) extraction cone voltage, and (d) velocity of the source T-wave. Hydrogen scrambling is plotted as an average of scrambling values determined in individual fragment ions (c2 – c6) from at least two replicate ETD experiments. The experimental error (one standard deviation) is indicated by error bars. The intensity of the reduced species (dotted green line) is shown to indicate the effect on ion transmission of varying the corresponding ion source parameter.
The experiments described so far were conducted using the standard ion source of the instrument. While HX-MS experiments are typically performed using flow rates (10–50 μL/min) that require this type of ion source other applications of this instrument such as ion mobility spectrometry often make use of the nano-flow version of the Z-spray ion source [25]. We therefore performed a limited set of experiments using the nano-flow ion source by infusing labeled peptide P1 into the mass spectrometer for ETD at a flow rate of 0.5 μL/min. Interestingly, the same parameters (i.e., the two skimmer orifice voltages and the source T-wave velocity) were observed to induce scrambling to a similar extent using the nano-flow ion source (data not shown). This suggests that hydrogen scrambling in the Z-spray ion source is induced primarily during acceleration of desolvated ions and is largely unaffected by both the size of the initial droplets in the electrospray plume and the efficiency of the desolvation process leading up to ion formation.
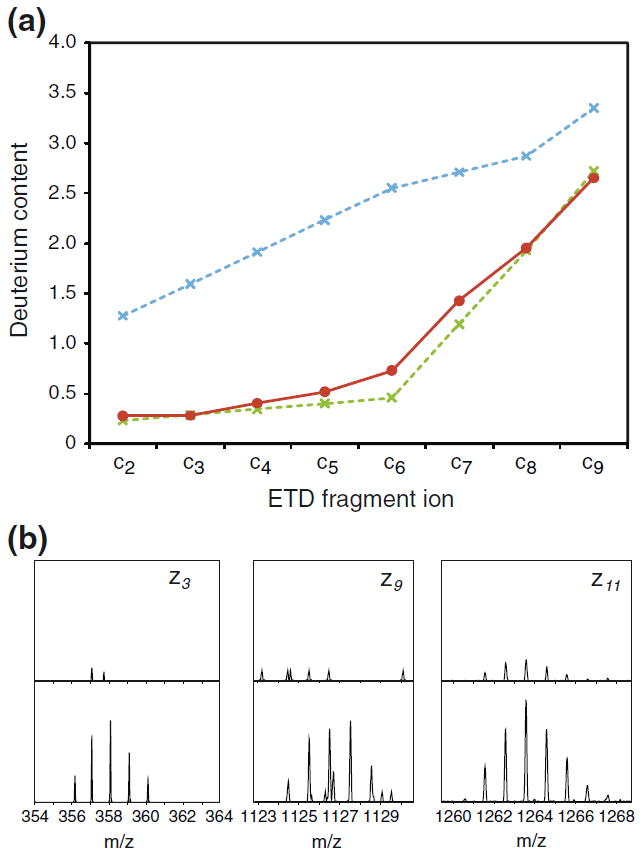
By tracking the extent of scrambling and the efficiency of ion transmission as a function of the three parameters found to induce scrambling, we were able to define a set of experimental settings that allowed maximal ion transmission while maintaining non-detectable scrambling levels: sample cone voltage 20 V, extraction cone voltage 2 V, and source T-wave velocity 300 m/s. At these “mild” conditions, the measured deuterium contents of either c-or z-type fragment ions of peptide P1 were in good agreement (i.e., ~0% scrambling) with the expected selective labeling pattern from solution (Figure 3a). In contrast, complete randomization (~100% scrambling) of the well-defined deuterium labeling of peptide P1 from solution could be induced in ETD fragment ions by adopting a set of conditions referred to as “harsh” conditions (sample cone voltage 65 V, extraction cone voltage 4.1, source T-wave velocity 300 m/s). Inspection of the deuterium level of peptides during loss of ammonia has recently been demonstrated to be a universal probe of scrambling in peptides during an HX-ETD experiment [9]. In our current experiments, the deuterium content of the deammoniated reduced species and the intact reduced species of peptide P1 was identical at mild ion source conditions, but a loss of 0.5D was observed for the deammoniated peptide at harsh conditions. By a calculation described earlier [9], this deuterium loss corresponds to 100% hydrogen scrambling in peptide P1, which is in good agreement with levels of scrambling calculated from deuterium contents of c-and z-type fragment ions observed in the same ETD spectrum.
Figure 3.

Minimizing hydrogen scrambling to negligible levels during ETD in the T-wave of a Q-TOF mass spectrometer. Plots of deuterium contents (red curves) of c-type fragment ions (left) and z-type fragment ions (right) of triply protonated peptide P1 during ETD experiments performed at (a) “mild” and (b) “harsh” ion source conditions (see text). The theoretical values in case of 0% (dotted green line) and 100% (dotted blue line) hydrogen scrambling are indicated. The deuterium content of the z6 fragment ion could not be determined due to spectral overlap with an interfering ion.
Peptides produced by pepsin proteolysis during a typical HX-MS experiment are mainly observed in the gas-phase as multiply (+2, +3, +4) protonated ions. While the occurrence of scrambling in doubly charged precursors during ETD has not been investigated in detail, it has previously been shown that quadruply protonated peptide ions are more prone to undergo gas-phase hydrogen scrambling than their triply protonated counterparts [8, 11]. Ions of high charge will attain greater kinetic energies than their lower charged counterparts when accelerated through the ion source by the same electrostatic potential and will, thus, undergo increased activation (i.e., vibrational excitation) upon collisions with residual gas molecules in the ion source. To maximize the number and diversity of fragment ions generated in an HX-ETD experiment, it would be highly desirable to be able to fragment peptide ions of all three charge states while maintaining minimal hydrogen scrambling. The ESI buffer and experimental conditions used in the current study allowed peptide P1 to populate multiple protonated forms (+2, +3, and +4) and it was therefore possible to investigate the occurrence of hydrogen scrambling during ETD for all three charge states of the peptide. ETD of the quadruply protonated peptide P1 produced abundant c-and z-type ions similar to the triply charged state. Complete scrambling in the gas-phase of the solution deuterium label of the quadruply protonated peptide occurred at a lower sample cone voltage (45 V) than observed in experiments with the triply protonated peptide (65 V) (Figure 4). This is in good accordance with previous ETD experiments performed with an ion trap mass spectrometer [11]. An identical ETD experiment at the “mild” Z-spray ion source settings established earlier for the triply protonated peptide showed that these settings were sufficiently gentle to also prevent gas-phase hydrogen scrambling in ETD fragment ions originating from the quadruply protonated peptide (Figure 4). Thus, despite the increased likelihood of mobilizing hydrogen atoms in quadruply protonated peptide ions, HX-ETD experiments could still be performed accurately on highly charged ions of model peptide P1 using the tuned conditions of the Z-spray ion source. This is an important finding as the use of fragment ions from quadruply protonated peptides in HX-ETD experiments using the current instrumental setup will allow the determination of deuterium levels for a greater number of individual residues in a protein compared with an earlier study in which data analysis was solely based on ETD of doubly and triply protonated peptides [10].
Figure 4.
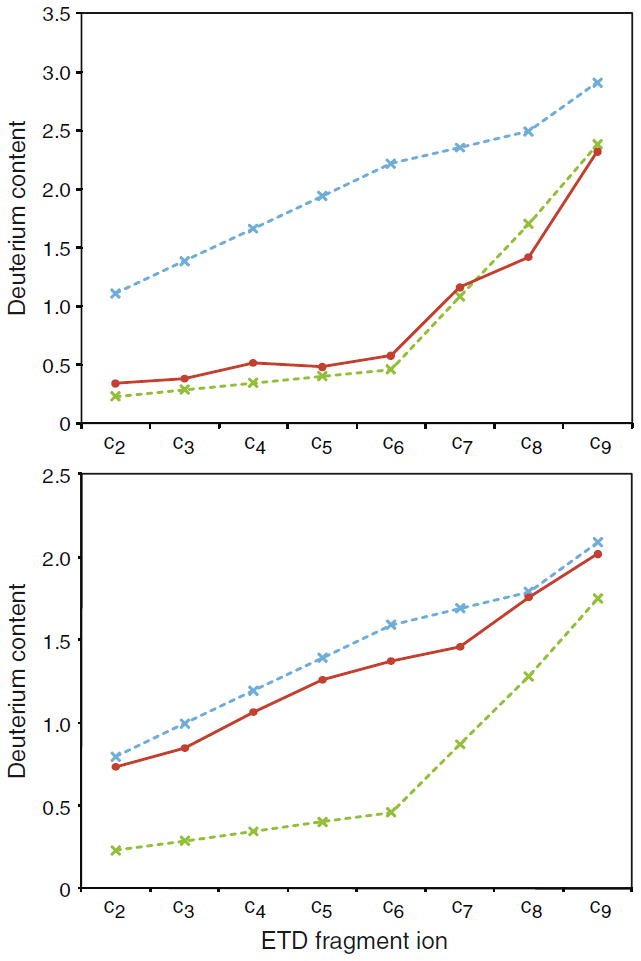
Hydrogen scrambling during ETD can be minimized to negligible levels in quadruply protonated peptides by tuning the Z-spray ion source. Deuterium contents (red curves) of c-type fragment ions of labeled quadruply protonated peptide P1 upon ETD at defined “mild” ion source conditions (upper panel) and after a moderate increase of the sample cone (45 V) and extraction cone (4.1 V) voltages (lower panel). The theoretical values in case of 0% (dotted green line) and 100% (dotted blue line) hydrogen scrambling are indicated.
ETD of the doubly protonated peptide P1 recorded at the “default” ion source conditions of the instrument (i.e., sample cone 45 V, extraction cone 4.1 V, see Experimental section) yielded only very few fragment ions (z11 and c10)of a signal intensity suitable for determination of average mass (data not shown). Low ETD efficiency is commonly observed for doubly charged peptide ions [29, 30], and experiments conducted at the “mild” ion source conditions failed altogether to yield any fragment ions of suitable quality. Interestingly, the deuterium contents of the two ETD fragment ions (z11 and c10) of the doubly charged peptide recorded at “default” ion source conditions showed an absence of scrambling even at these elevated voltage settings (data not shown). While doubly protonated peptide P1 only yielded few informative fragment ions, the fragmentation and number of fragment ions observed during ETD of doubly protonated peptides, in general, is highly dependent on both the charge density and the sites of protonation (i.e., the peptide sequence) [29]. In a prior HX-ETD study, the site-specific deuterium contents of 31% of the residues of a small protein could be extracted solely from ETD fragment ions originating from doubly protonated peptic peptides [10]. The present findings, therefore, indicate a significant opportunity for optimizing HX-ETD experiments by increasing the ionization and fragmentation efficiency of doubly protonated peptides without risking inadvertent hydrogen scrambling.
Prior work has shown that collisional activation either during or after an ETD reaction can increase c-and z-type fragment ion yields by further activation of charge reduced electron transfer (ET) products that did not completely dissociate (ETnoD) [29–31]. We sought to explore this in the context of an HX-ETD experiment and investigated the utility of collisional activation after ETD of deuterium labeled peptides. As the reaction between radical 4-nitrotoluene anions and protonated peptide ions was performed in transmission mode fashion in the trap T-Wave, we explored the possibility of subjecting ET product ions to supplemental activation as they traversed the adjacent transfer T-Wave ion guide before entering the time-of-flight tube for mass analysis. ETD experiments on labeled triply protonated peptide P1 were performed at defined “mild” ion source conditions while applying an increased ion injection “CE” voltage setting (from 4 V to 10 V) in the transfer T-Wave ion guide. Interestingly, the deuterium contents of the resulting c-and z-type fragment ions clearly retained the selective labeling pattern from solution, despite such post-ETD collisional activation (Figure 5a). The increase in c-and z-type fragment ion yields due to collisional activation during or after electron-transfer has been shown to be most pronounced during ETD of doubly charged peptides or peptides of low charge density [29–31]. While ETD and supplemental activation of triply protonated peptide P1 did not result in significant increases in fragment ion yields (data not shown), the same experiment was quite effective in countering the inherently poor ETD fragmentation efficiency of the doubly protonated peptide ion. A modest supplemental activation (10 V) during ETD of labeled doubly charged peptide P1 resulted in a significant increase in both the number (from 2 to 6) and abundance (2-to 5-fold) of ETD fragment ions (Figure 5b). In the absence of supplemental activation only two fragment ions (c10 and z11)were observed at suitable signal intensity, whereas conversion of the ETnoD product by supplemental activation in the transfer T-Wave gave rise to four additional informative fragment ions (z3, z8, z9, z10) of the doubly protonated peptide. Importantly, the measured deuterium contents of these fragment ions, as well as that of the ammonia-deficient reduced species of the doubly protonated peptide, were in good agreement with the expected values in case of ~0% scrambling (data not shown). This result correlated nicely with identical ETD experiments with supplemental activation described earlier for the triply protonated peptide. From a theoretical perspective, gas-phase mobilization of deuterium at amide positions could occur in long-lived peptide species that remain attached through noncovalent forces even after electron-induced cleavage [11]. To our knowledge, our current findings provide the first experimental indication that supplemental activation of ETnoD products can be performed without inducing hydrogen scrambling during HX-ETD type experiments. We note that the absence of detectable levels of hydrogen scrambling during our transmission mode ETD/CID-type experiments could be a result of the time-scale and the nature of the activation employed. Nevertheless, our results indicate that ETD and supplemental activation in neighboring T-Wave ion guides can provide for improved extraction of residue-specific deuterium levels from doubly protonated peptides in an HX-ETD experiment. Likewise, the benefits of supplemental activation should also extend to the significant population of peptic peptides analyzed in an HX-ETD experiment that have a higher charge (i.e., > +2) but a similarly low charge density (m/z >750) due to large size.
Figure 5.
Supplemental activation of product ions of the ETD reaction in the T-wave occurs without detectable hydrogen scrambling. (a) The deuterium content of c-type fragment ions of the labeled triply protonated peptide P1 generated by ETD in the trap T-wave ion guide and subsequent supplemental activation (“CE”=10V) in the transfer T-wave ion guide. (b) Mass spectra of z-type fragment ions produced by ETD of labeled doubly protonated peptide P1 without supplemental activation (upper spectra) and with supplemental activation (“CE”=10V) (lower spectra). Increased dissociation of the “ETnoD” product by supplemental activation is apparent by an increase in the abundance (2- to 5-fold) of z-type fragment ions of the doubly protonated peptide (the intensity axis in upper mass spectra is scaled to fit the intensity axis of the lower mass spectra).
Conclusions
Collisional activation during ionization and ion transport is detrimental to the use of ECD/ETD to measure the solution HX of individual residues in a protein. The present study provides detailed insight into factors contributing to ion activation in the front-end of a Q-TOF type instrument by performing a systematic investigation of hydrogen scrambling during ETD experiments on a model peptide. Our results indicate that a Q-TOF instrument with a Z-spray ion source and a traveling wave ion guide capable of ETD reactions in transmission mode provides a highly useful platform for integrating ETD into the classical HX-MS workflow. We demonstrate that hydrogen scrambling can be reduced to undetectable levels during HX-ETD experiments by simple tuning of the Z-spray ion source. Prior work with several different model peptides [11] has shown that mild ion source settings that ensured the absence of H/D scrambling in model peptides also prevented the occurrence of H/D scrambling in actual peptic peptides of a deuterium labeled protein analyzed in a HX-ETD experiment [10]. Settings of the Z-spray ion source shown presently to prevent H/D scrambling in model peptide P1 are therefore very likely to also do so for the diversity of peptides produced by pepsin proteolysis of a given protein. However, as described previously [9], this can be verified on a routine basis by monitoring the deuterium content of the ammonia-deficient product ion commonly observed in ETD spectra of deuterium labeled peptic peptides in a given HX-ETD experiment. Compared with an earlier setup that employed a 3D ion trap[10], we anticipate that the current TOF-based instrument will provide important advantages for HX-ETD analyses, including a high mass resolving power for handling complex peptide mixtures from one or more large proteins and the capacity for supplemental activation steps to improve residue-specific analysis of protein HX. The ability to limit undesirable activation of molecular species during ESI is also important for the growing applicability of mass spectrometry at native solution conditions to study protein structure and the stoichiometry of protein–ligand/protein–protein complexes. Excessive collisional activation can cause untimely gas-phase dissociation of noncovalent bonds resulting in disassembly of bound species or the unfolding of higher-order structure. If not limited and properly controlled, such effects could compromise MS analysis. This is particularly relevant for Q-TOF type instruments, which are increasingly being used to perform gas-phase ion mobility or gas-phase HX to probe native biomolecular structures. We conceive that information on the inadvertent activation of polypeptide ions in different ion sources, like the Z-spray ion source, can be used to develop new ion source designs capable of efficient ionization with lower concomitant ion heating and thereby improve future applications of mass spectrometry to investigate the higher-order structure of biomolecules.
Acknowledgments
The authors thank Professor Thomas J. D. Jørgensen for supplying batches of model peptide P1. They also thank Jim Langridge, Ian Campuzano, Tim Riley, Geoff Gerhardt, and Dan McCormick for their support during this project. The authors gratefully acknowledge financial support from the National Institutes of Health (R01-GM086507, J.R.E.), The Barnett Institute (this work is contribution no. 984, J.R.E), the Swiss Institute of Bioinformatics (K.D.R) and The International Human Frontier Science Program Organization (K.D.R). They finally gratefully acknowledge Professor Olivier Michielin for encouraging this work.
References
- 1.Konermann L, Pan J, Liu YH. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem Soc Rev. 2010;40:1224–1234. doi: 10.1039/c0cs00113a. [DOI] [PubMed] [Google Scholar]
- 2.Rand KD, Jorgensen TJ, Olsen OH, Persson E, Jensen ON, Stennicke HR, Andersen MD. Allosteric activation of coagulation factor VIIa visualized by hydrogen exchange. J Biol Chem. 2006;281:23018–23024. doi: 10.1074/jbc.M602968200. [DOI] [PubMed] [Google Scholar]
- 3.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 4.Del Mar C, Greenbaum EA, Mayne L, Englander SW, Woods VL., Jr Structure and properties of alpha-synuclein and other amyloids determined. Proc Natl Acad Sci USA. 2005;102:15477–15482. doi: 10.1073/pnas.0507405102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milne JS, Mayne L, Roder H, Wand AJ, Englander SW. Determinants of protein hydrogen exchange studied in equine cytochrome c. Protein Sci. 1998;7:739–745. doi: 10.1002/pro.5560070323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci USA. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zubarev RA, Kelleher NL, McLafferty FW. Electron capture dissociation of multiply charged protein cations. A nonergodic process. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
- 8.Rand KD, Adams CM, Zubarev RA, Jorgensen TJD. Electron capture dissociation proceeds with a low degree of intramolecular migration of peptide amide hydrogens. J Am Chem Soc. 2008;130:1341–1349. doi: 10.1021/ja076448i. [DOI] [PubMed] [Google Scholar]
- 9.Rand KD, Zehl M, Jensen ON, Jorgensen TJ. Loss of ammonia during electron-transfer dissociation of deuterated peptides as an inherent gauge of gas-phase hydrogen scrambling. Anal Chem. 2010;82:9755–9762. doi: 10.1021/ac101889b. [DOI] [PubMed] [Google Scholar]
- 10.Rand KD, Zehl M, Jensen ON, Jørgensen TJD. Protein hydrogen exchange measured at single-residue resolution by electron transfer dissociation mass spectrometry. Anal Chem. 2009;81:5577–5584. doi: 10.1021/ac9008447. [DOI] [PubMed] [Google Scholar]
- 11.Zehl M, Rand KD, Jensen ON, Jørgensen TJD. Electron transfer dissociation facilitates the measurement of deuterium incorporation into selectively labeled peptides with single residue resolution. J Am Chem Soc. 2008;130:17453–17459. doi: 10.1021/ja805573h. [DOI] [PubMed] [Google Scholar]
- 12.Abzalimov RR, Kaplan DA, Easterling ML, Kaltashov IA. Protein conformations can be probed in top-down HDX MS experiments utilizing electron transfer dissociation of protein ions without hydrogen scrambling. J Am Soc Mass Spectrom. 2009;20:1514–1517. doi: 10.1016/j.jasms.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan J, Han J, Borchers CH, Konermann L. Electron capture dissociation of electrosprayed protein ions for spatially resolved hydrogen exchange measurements. J Am Chem Soc. 2008;130:11574–11575. doi: 10.1021/ja802871c. [DOI] [PubMed] [Google Scholar]
- 14.Pan J, Han J, Borchers CH, Konermann L. Hydrogen/deuterium exchange mass spectrometry with top-down electron capture dissociation for characterizing structural transitions of a 17 kDa protein. J Am Chem Soc. 2009;131:12801–12808. doi: 10.1021/ja904379w. [DOI] [PubMed] [Google Scholar]
- 15.Kaltashov IA, Bobst CE, Abzalimov RR. H/D exchange and mass spectrometry in the studies of protein conformation and dynamics: Is there a need for atop-down approach? Anal Chem. 2009;81:7892–7899. doi: 10.1021/ac901366n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson RS, Krylov D, Walsh KA. Proton mobility within electrosprayed peptide ions. J Mass Spectrom. 1995;30:386–387. [Google Scholar]
- 17.Mueller DR, Eckersley M, Richter WJ. Hydrogen transfer-reactions in the formation of Y + 2 sequence ions from protonated peptides. Org Mass Spectrom. 1988;23:217–222. [Google Scholar]
- 18.Jorgensen TJD, Gardsvoll H, Ploug M, Roepstorff P. Intramolecular migration of amide hydrogens in protonated peptides upon collisional activation. J Am Chem Soc. 2005;127:2785–2793. doi: 10.1021/ja043789c. [DOI] [PubMed] [Google Scholar]
- 19.Rand KD, Jorgensen TJ. Development of a peptide probe for the occurrence of hydrogen ((1)H/(2)H) scrambling upon gas-phase fragmentation. Anal Chem. 2007;79:8686–8693. doi: 10.1021/ac0710782. [DOI] [PubMed] [Google Scholar]
- 20.Rand KD, Pringle SD, Murphy JP, Fadgen KE, Brown J, Engen JR. Gas-phase hydrogen/deuterium exchange in a traveling wave ion guide for the examination of protein conformations. Anal Chem. 2009;81:10019–10028. doi: 10.1021/ac901897x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fang J, Rand KD, Silva MC, Wales TE, Engen JR, Beuning PJ. Conformational dynamics of the Escherichia coli DNA polymerase manager proteins UmuD and UmuD’. J Mol Biol. 2010;398:40–53. doi: 10.1016/j.jmb.2010.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagy K, Redeuil K, Rezzi S. Online hydrogen/deuterium exchange performed in the ion mobility cell of a hybrid mass spectrometer. Anal Chem. 2009;81:9365–9371. doi: 10.1021/ac901736j. [DOI] [PubMed] [Google Scholar]
- 23.Williams JP, Brown JM, Campuzano I, Sadler PJ. Identifying drug metallation sites on peptides using Electron Transfer Dissociation (ETD), Collision Induced Dissociation (CID), and Ion Mobility-Mass Spectrometry (IM-MS) Chem Commun. 2010;46:5458–5460. doi: 10.1039/c0cc00358a. [DOI] [PubMed] [Google Scholar]
- 24.Heck AJR. Native mass spectrometry. A bridge between interactomics and structural biology. Nat Methods. 2008;5:927–933. doi: 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- 25.Ruotolo BT, Benesch JLP, Sandercock AM, Hyung SJ, Robinson CV. Ion mobility-mass spectrometry analysis of large protein complexes. Nat Protoc. 2008;3:1139–1152. doi: 10.1038/nprot.2008.78. [DOI] [PubMed] [Google Scholar]
- 26.Wang SC, Politis A, Di Bartolo N, Bavro VN, Tucker SJ, Booth PJ, Barrera NP, Robinson CV. Ion mobility mass spectrometry of two tetrameric membrane protein complexes reveals compact structures and differences in stability and packing. J Am Chem Soc. 2010;132:15468–15470. doi: 10.1021/ja104312e. [DOI] [PubMed] [Google Scholar]
- 27.Benesch JLP, Ruotolo BT, Simmons DA, Robinson CV. Protein complexes in the gas phase: Technology for structural genomics and proteomics. Chem Rev. 2007;107:3544. doi: 10.1021/cr068289b. [DOI] [PubMed] [Google Scholar]
- 28.Giles K, Pringle SD, Worthington KR, Little D, Wildgoose JL, Bateman RH. Applications of a traveling wave-based radio-frequency-only stacked ring ion guide. Rapid Commun Mass Spectrom. 2004;18:2401–2414. doi: 10.1002/rcm.1641. [DOI] [PubMed] [Google Scholar]
- 29.Pitteri SJ, Chrisman PA, McLuckey SA. Electron-transfer ion/ion reactions of doubly protonated peptides: Effect of elevated bath gas temperature. Anal Chem. 2005;77:5662–5669. doi: 10.1021/ac050666h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swaney DL, McAlister GC, Wirtala M, Schwartz JC, Syka JE, Coon JJ. Supplemental activation method for high-efficiency electron-transfer dissociation of doubly protonated peptide precursors. Anal Chem. 2007;79:477–485. doi: 10.1021/ac061457f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ledvina AR, McAlister GC, Gardner MW, Smith SI, Madsen JA, Schwartz JC, Stafford GC, Jr, Syka JE, Brodbelt JS, Coon JJ. Infrared photoactivation reduces peptide folding and hydrogen-atom migration following ETD tandem mass spectrometry. Angew Chem Int Ed. 2009;48:8526–8528. doi: 10.1002/anie.200903557. [DOI] [PMC free article] [PubMed] [Google Scholar]