

Abstract
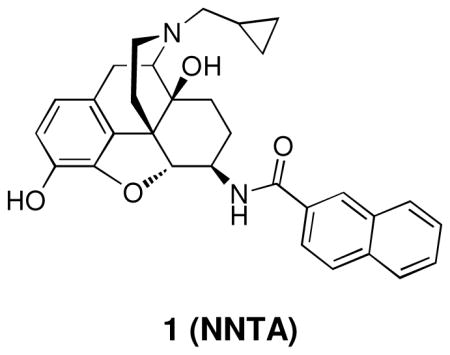
Using the selective mu-kappa agonist, NNTA 1, as the prototype ligand, a series of closely related naphthalene analogues were synthesized to study the chemical space around the naphthalene moiety in an effort to evaluate how receptor selectivity is affected by chemical modification. Nine analogues (2–10) of compound 1 were synthesized and tested on HEK-293 cells expressing homomeric and heteromeric opioid receptors, and in the mouse tail-flick assay. It was found that a small change in structure produces profound changes in selectivity in this series. This is exemplified by the discovery that introduction of a 6-fluoro group transforms 1 from a selective mu-kappa heteromeric receptor agonist to a delta-preferring agonist 7. The in vivo studies reveal that many of the ligands are more potent spinally than supraspinally and devoid of tolerance.
Introduction
Morphine and other opiates derived from opium have been employed as analgesics for well over a century. A feature common to these analgesics is that they produce side effects that include tolerance, physical dependence, constipation and respiratory depression. These ligands produce their effects through interaction with opioid receptors that are class A members of the G protein-coupled receptor (GPCR) superfamily1, 2.
The three major classes of opioid receptors known to mediate antinociception upon activation are designated mu (MOP), delta (DOP), and kappa (KOP)2. Since the establishment of these receptor types though binding, pharmacology, and cloning, selective opioid ligands have played an important role in deconvoluting the effects of opioid ligands acting at multiple opioid receptors3. This classification was based primarily assumption that opioid receptors exist as monomers4.
The concept of mu, delta, and kappa receptors as monomers was first challenged nearly three decades ago through studies with bivalent ligands that could bridge putative homomeric opioid receptors5, 6. Later studies in cultured cells revealed that class A–C GPCRs can exist as homodimers or heteromers7–9. This was followed by the discovery of mu-delta, kappa-delta, and mu-kappa heteromers10–13.
The existence of opioid receptor heteromers suggest a more complex level of action and regulation than the traditional view of monomeric receptor pharmacology. For example, morphine, fentanyl, and methadone, traditionally viewed as mu-selective analgesics, have been reported to be more potent in activating mu-delta heteromers in cultured cells14. Moreover, mu-delta heteromers also appear to be the principal target for producing antinociception in monkeys15.
Molecular tools that target opioid heteromers have been developed in an effort to sort out the effects mediated by such receptors. These have included ligands that selectively target mu-delta, mu-kappa, and delta-kappa heteromers. The recently reported, spinally selective mu-kappa agonist, N-naphthoyl-β-naltrexamine 1 (NNTA)16, selectively activates mu-kappa heteromers in HEK-293 cells and it produces exceptionally potent antinociception upon intrathecal (i.t.) administration in mice. Given the ~100-fold greater spinal potency of 1 than by the intracerebroventricular (i.c.v.) route, it is believed that the mu-kappa heteromers that mediate antinociception are localized in the spinal cord rather than supraspinally. Significantly, no tolerance was produced i.t., and only marginal tolerance was observed i.c.v. This profile prompted us to synthesize and evaluate derivatives related to ligand 1 in order to explore the structural requirements for selectivity.
Chemistry
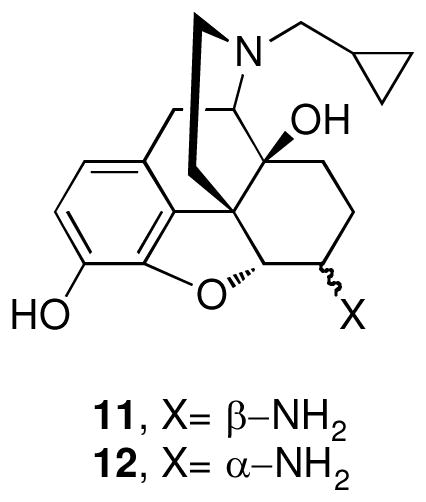
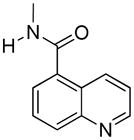
The series was designed based on conservative structural changes in the amide moiety. In this regard, the key question that we wished to address was the structural requirements for the mu-kappa selectivity of 1. Consequently, all members (2–10) of the series contain a naltrexamine opioid pharmacophore linked through an amide moiety to a substituted naphthalene or a heterocycle isosteric with naphthalene.
β-Naltrexamine 11 was employed as the opioid pharmacophore for all members of the series except 2, which was derived from α-naltrexamine 12, in order to evaluate the stereochemical role of the amide moiety in conferring selectivity. Both α- and β-naltrexamine were derived from naltrexone17.
The general procedure for synthesis of analogues 2–10 involved coupling of naltrexamines 11 or 12 in presence of benzotriazol-1-yl-oxy-tris-(dimethyamino)phosphonium hexafluorophosphate (BOP) and diisopropylethylamine (DIPEA) in dichloromethane (DCM)18. The target compounds 2–10 were thus obtained in moderate to high yields (58–84%) after chromatographic purification.
Biological Results
Intracellular Ca release studies
The target compounds were tested for agonist activity using an intracellular calcium release assay. Briefly, HEK-293 cells were transfected with a chimeric Δ6-Gαqi4-myr protein15 employed to measure intracellular Ca+2 ion release upon receptor activation.1 Cells stably expressing the chimeric protein were selected from transiently transfected cells in zeocin containing media (DMEM+10% fetal bovine serum+1% penicillin/streptomycin+0.1μg/ml zeocin). Opioid receptors were transiently transfected using different combinations of DNA for heteromers (mu-delta, mu-kappa, kappa-delta) or for singly expressing homomers (mu, delta, kappa). Intracellular calcium release was measured using a FLIPR calcium kit (Molecular Devices) in a FlexStation3 apparatus. For each compound, concentration-response profiles were established by measuring the fluorescence for 90 seconds after addition of the compound and determining the peak effect (maximum-minimum). A dose-response curve was plotted with the change in Relative Fluorescence Units (ΔRFU) vs. concentration (Table 1). Because the efficacy of 1 was substantially higher than other members of the series, the RFU scale for activity of 2–10 is expanded to 2.5x (0–1000) that of the ligand of reference 1(0–2500).
Table 1.
Intracellular Ca++ release profiles at multiple opioid receptors.
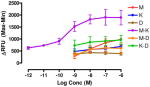
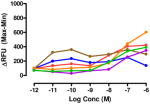
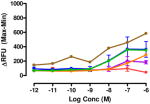
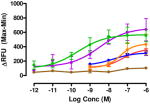
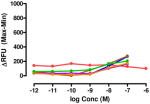
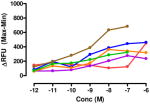
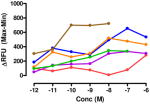
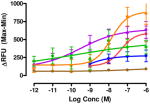
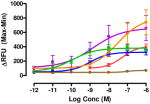
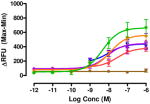
Data are mean ± SEM (n= 3–8). RFU, Relative Fluorescence Unit.
(1) data from ref 16.
The selectivity profile of 1 is displayed as the structurally related reference compound for the closely related series of compounds. Its 6α-epimer 2 was synthesized in order to evaluate the role of the C-6 stereocenter in the activation of μ-κ heteromers. It is noteworthy that its selectivity profile differs substantially from that of 1, in that mu-kappa receptor activation has been greatly reduced. Other opioid receptors also showed reduced activation. These data led us to explore only close structural modifications in the 6β-series.
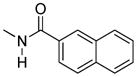
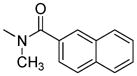
The activity of the N-methyl analogue 320 differed significantly from that of compound 1 in that activation of mu-kappa receptors was lost. There was an apparent small increase in activation at delta receptors in the higher concentration range. Conformational differences of the naphthyl moiety induced by the N-methyl group is one of several possibilities that could contribute to loss of mu-kappa selectivity.
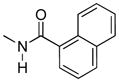
Regioisomer 4 activated both mu-kappa and delta-kappa heteromers to the same degree, but the mu-kappa component was ~3-fold less effective than 1. The curves for mu-kappa, mu-delta, and mu receptors were grouped together and have lower RFU values than the kappa-containing heteromers. No activation of delta receptors is apparent.
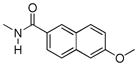
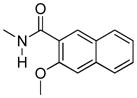
Various substituents were introduced at the 3′- and 6′-position of the naphthoyl group. Introduction of a 6′-methoxy (5) resulted in a loss of efficacy, suggesting that an electron-donating group in that position is not well tolerated. However, the same substituent in the 3′-position (6) afforded moderate delta selectivity. As the 3′-methoxy group could engage in intramolecular hydrogen bonding with the carboxamide NH, the delta selectivity might arise by restricting the naphthyl moiety to a conformation that differs from that of 5.
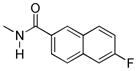
Interestingly, the congener with a 6′-fluoro group (7) was delta-selective and exhibited considerably greater agonist potency and efficacy for delta receptors than 6. That the activity of 7 is substantially more potent than 5 is consistent with the idea that an electron withdrawing group favors delta receptor activation in view of the inactivity of the 6′-methoxy analogue.
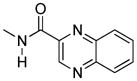
We also explored heterocyclic analogues (8–10) of 1 whose aromatic rings are isosteric with naphthalene. They included quinazoline 8 and isomeric quinolines 9 and 10. All of these analogues were inactive at delta receptors, but possessed nonselective opioid activity at heteromeric and homomeric receptors in the nanomolar range. In this regard, 8 and 9 had similar profiles with higher efficacy at mu-delta and mu-kappa receptors.
Pharmacological evaluation in mice
The in vivo profiles of compounds 2–10 were evaluated using the mouse tail-flick assay after the ligands were administered by the i.c.v and/or i.t routes21–23 (Table 2). Tolerance was measured only for selected compounds displaying a full agonist profile i.c.v and/or i.t by comparing the ED80–90 dose measured on day one to the same dose measured 24 hours later on the same mice.
Table 2.
Antinociceptive activity and tolerance of 2–10 after i.c.v. or i.t. administration in mice.
| Compounda | Injection site | ED50 pmol/mouse (95% C.I.) or | 24 hour toleranceb |
|---|---|---|---|
|
| |||
| 1 (NNTA) | i.t. | 18.7 (10.3 – 32.8) | No |
| i.c.v. | 2060 (1090 – 3270) | Yes1 | |
|
| |||
| 2 | i.t. | partial agonistc | - |
| i.c.v. | partial agonist | - | |
|
| |||
| 3 | i.t. | partial agonist | - |
| i.c.v. | partial agonist | - | |
|
| |||
| 4 | i.t. | 20.9 (13.5 – 32.7) | Yes |
| i.c.v. | partial agonist | - | |
|
| |||
| 5 | i.t. | partial agonist | - |
| i.c.v. | partial agonist | - | |
|
| |||
| 6 | i.t. | 115.1 (95.5 – 138.7) | No |
| i.c.v. | 312.4 (168.6 –578.7) | Yes | |
|
| |||
| 7 | i.t. | 81.1 (40.7 – 161.7) | No |
| i.c.v. | partial agonist | - | |
|
| |||
| 8 | i.t. | 50.8 (30.0 – 95.6) | No |
| i.c.v. | 757.2 (561.7 – 1021) | No | |
|
| |||
| 9 | i.t. | 1408 (931 – 2128) | No |
| i.c.v. | 2070 (1490 – 2876) | No | |
|
| |||
| 10 | i.t. | partial agonist | - |
| i.c.v. | partial agonist | - | |
Peak times for the dose response curves were as follows for i.t. 1, 5, 6, 8 all 5 min; 7, 9 both 20 min and for i.c.v. 5, 9 both 10 min; NNTA, 20 min.
24 hour tolerance was calculated using the highest dose of the dose-response curve on day 1 and repeated on day 2. If there was no significant difference between the two days the animals were said to be not tolerant.
Partial agonist is defined as when the maximum %MPE was ≤ 60%
The 6α-stereoisomer 2 and N-Me analogue 3 possessed partial agonist antinociceptive activity when administered by both routes. These data were supported by their low efficacy in the calcium mobilization assay as shown previously (Table 1).
Compound 4 displayed an i.t potency similar to the parent compound (20.98 pmol/mouse versus 18.7 pmol for 1 but it did not exhibit a full agonist profile when given supraspinally. The exceptional spinal activity was accompanied by tolerance 24 hours after i.t. administration.
The negative effect of a 6′-methoxy group (5) on efficacy, as revealed in the calcium mobilization assay, was supported by its i.t and i.c.v partial agonist profile. The activity of the 3′-isomer 6 was greater than that of 5 when injected spinally (ED50=115 pmol/mouse) or supraspinally (ED50= 312 pmol) and consistent with the cell-based results. No i.t tolerance was apparent, while some tolerance was observed after i.c.v injection.
The 6′-fluoro analogue 7 was found to be a fairly potent agonist by the i.t route, with an ED50 of 81.1 pmol. No tolerance was observed. Because 7 was found to be delta-selective in the calcium release assay, it was further evaluated using the selective antagonists, nor-BNI24 (kappa) and NTI25 (delta), and in mu receptor knock-out mice (Figure 1). After i.t administration of 7, NTI produced a 26-fold shift consistent with the delta selectivity observed in the cell-based assay. The finding that its activity was also antagonized by nor-BNI may be due to antagonism of the combined agonist effect mediated by kappa, kappa-delta, and kappa-mu receptors, given that 7 activates these receptor systems in the nM range as shown in the calcium release assay. Such antagonism by nor-BNI may be mediated through a negative allosteric effect mediated via the kappa receptor, as suggested previously26. Thus, nor-BNI may antagonize a mu or delta receptor in a heteromer through via an associated kappa protomer in vivo. This is consistent with the decreased antinociception of 7 in the mu-opioid receptors knock-out mice due to absence of mu-delta heteromers,
Figure 1.

Antinociception of compound 7 after treatment with opioid antagonists naltrindole (NTI) and norbinatorphimine (norBNI) after i.t administration in wild-type mice and in mu opioid receptor knockout (MOR-KO) mice.
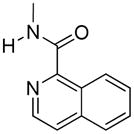
The quinoxaline analogue 8 was found to be a potent agonist by both i.c.v and i.t routes (ED50 50.76 pmol and 757.2 pmol, respectively), with 15-fold greater spinal potency. It is noteworthy that 8 exhibited no apparent tolerance by either route of administration. Quinoline analogue 9 also exhibited full i.t. and i.c.v. agonist activity without tolerance, but it was substantially less potent than that of 8. Interestingly, compound 4, the isosteric naphthalene analogue of 9, was exceptionally potent i.t., but displayed tolerance. The isomeric quinoline analogue 10 behaved as a partial agonist when administered spinally or supraspinally.
Discussion
Using the selective mu-kappa agonist 1, as the prototype ligand, a series of closely related naphthalene analogues were synthesized to study the chemical space around the naphthalene moiety in an effort to evaluate how receptor selectivity is affected by chemical modification. Nine analogues (2–10) were synthesized and tested on HEK-293 cells expressing homomeric and heteromeric opioid receptors, and in the mouse tail-flick assay.
Since 1 is derived from 6β-naltrexamine, we first explored the effect of configuration at the C6 chiral center by the synthesis of 2. As it appeared from the cell-based data that 2 gave a profound reduction in the activation of opioid receptors, particularly mu-kappa, relative to that of 1, we decided to focus on ligands in the 6β-series.
In this regard we first evaluated the effect of substitution on the 6β nitrogen atom with compound 3, as we suspected a N-methyl group would alter the rotational conformational preference of the naphthalene moiety. Once again, the activation of heteromeric and homomeric receptors was greatly reduced, suggesting that NH hydrogen bonding with the receptor(s) and/or conformational factors might be involved.
Interestingly, the 1-naphthyl analogue 4 exhibited improved ΔRFU values for activation of heteromeric mu-kappa and delta-kappa receptors relative to compound 3. As both of these heteromers have been reported to be potently activated in the spinal cord of rodents, it is not surprising that 4 produces high i.t. antinociception. Other receptors were activated to a lesser degree, and delta showed no activation.
The effect of substitution on the naphthyl group had a profound effect on selectivity and potency. Thus, a 6′-OMe substituent (5) greatly reduced activation of all homomers and heteromers, whereas its 3′-regioisomer (6) selectively activated delta receptors. These results are consistent with several possible explanations that include an unfavorable electronic or steric effect by the 6′-OMe in 5 and a favorable effect on the conformation of the naphthyl group of 6 due to possible intramolecular H-bonding between 6β-NH and the 3′-OMe.
The 6′-fluoro-substituted analogue 7 appeared to be more delta-selective than 6. One possible explanation could be due to the electron withdrawing effect of the 6′-fluoro group, in view of the electron donating 6′-OMe of 5 having the opposite effect on activity. It is noteworthy that 7 did not produce tolerance and was highly potent in mice by the i.t. route.
The quinoxaline analogue 8 and quinoline analogues (9, 10) were essentially nonselective agonists in HEK-293 cells. The trend seems to support greater activation for mu-delta and mu-kappa. Interestingly, these ligands failed to activate delta receptors.
Of the nine analogues tested in mice, five (4, 6–9) were highly potent full agonists after i.t. administration. Among these, only compound 4 exhibited tolerance. Analogues 2, 3, 5, and 10 produced partial agonism by this route. Partial agonism among members of the series was observed more frequently upon i.c.v. administration, and in this regard 2–5, 7, 10 produced this effect. The only members of the series that were full agonists both i.c.v. and i.t. were compounds 6, 8, 9. It is noteworthy that i.t. administration resulted in greater potency than by the i.c.v. route when the ED50 values could be determined after both i.t. and i.c.v. administration.
Conclusions
Judging from the calcium release studies for compounds 2–10 in HEK-293 cells, the high selectivity of 1 for activation of heteromeric mu-kappa receptors appears to be unique in this series. Intriguingly, the 6-fluoro analogue 7 appeared to selectively activate homomeric delta receptors in vitro and it induced potent i.t. antinociception without tolerance in mice. The finding that a small change in structure produces a profound change in selectivity exemplifies the complexity of the structure activity relationships in this series. The complex profiles of these compounds in activating the six combinations of opioid receptors makes it difficult to come to any firm conclusions regarding the relationship of receptor activation to potency and tolerance in vivo. Nevertheless, based on the i.t. and i.c.v. data, most of the analogues possess potent antinociceptive activity that arises from the selective activation of spinal opioid receptors.
Experimental section
All commercial reagents and anhydrous solvents were purchased from vendors and were used without further purification or distillation. Analytical thin-layer chromatography (TLC) was performed on EM Science silica gel 60 F254 (0.25 mm). Compounds were visualized by UV light. Flash column chromatography was performed on Fisher Scientific silica gel (230 – 400 mesh).
Melting points were determined on a Thomas-Hoover melting point apparatus and are uncorrected. 1H NMR spectra and 13C NMR spectra were taken on a Bruker Avance 400 MHz instruments and calibrated using an internal reference. Chemical shifts are expressed in ppm and coupling constants (J) are in hertz (Hz). Peak multiplicities are abbreviated: broad, br; singlet, s; doublet, d; triplet, t; and multiplet, m. ESI mode mass spectra were recorded on a BrukerBioTOF II mass spectrometer. Elemental analyses were performed by M-H-W Laboratories, Phoenix, AZ. Analytical data confirmed the purity of the products was ≥ 95%.
General procedure for the amidation of β-naltrexamine with a carboxylic acid
β-naltrexamine (100 mg, 0.29 mmol), heterocyclic carboxylic acid (0.58 mmol) and BOP (258 mg, 0.58 mmol) were dissolved in DCM (5 mL). To this solution, DIPEA (150 mL, 0.81 mmol) was added and the mixture was stirred at room temperature for 3 to 16 hours. The solution was concentrated under reduced pressure and the residue was taken up in MeOH (5 mL), and K2CO3 was added (300 mg). After 1 hour at room temperature, the mixture was concentrated to dryness. The final crude was purified by SiO2 chromatography to afford the target compound.
The title compound was then subsequently converted into the HCl salt for biological testing.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(2′-naphthyl)acetamido]morphinan (2)
Compound 2 was prepared according to the general procedure described above; combining α-naltrexamine (100 mg, 0.29 mmol), 2-naphthoic acid (117 mg, 0.58 mmol); BOP (258 mg, 0.58 mmol), and DIPEA (150 μL, 0.81 mmol) followed by basic solvolysis with K2CO3 gave the target compound which was purified by flash chromatography [EA/hexanes; 70/30], and then recrystallized from an acetone/hexanes mixture to provide 2 as a white solid (116 mg, 81%).
1H NMR (DMSO-d6) δ: 0.13 (m, 2H); 0.49 (m, 2H); 0.88 (m, 1H); 1.37–1.69 (m, 4H); 2.14–2.37 (m, 4H); 2.96–3.08 (m, 2H); 4.54 (m, 1H); 4.69 (d, 1H, JH5-H6= 3.8 Hz); 4.92 (bs, 1H, OH-14); 6.48 (d, 1H, JH1-H2= 8.1 Hz); 6.59 (d, 1H, JH2-H1= 8.1 Hz); 7.59–7.63 (m, 2H); 7.93–8.04 (m, 4H); 8.10 (d, 1H amide, J = 7.7 Hz); 8.47 (s, 1H); 8.89 (bs, 1H, OH-3); 13C NMR (DMSO-d6) δ: 3.40, 3.81, 9.13, 19.98, 22.31, 29.44, 33.53, 42.78, 46.52, 46.65, 58.75, 61.36, 69.23, 88.27, 117.22, 118.49, 124.40, 124.85, 126.67, 127.51, 127.58 (x2), 127.74, 128.76, 130.68, 131.88, 132.05, 134.06, 137.97, 145.82, 165.91. mp= 232–234 °C. Anal. Calcd. for C31H33ClN2O4: C, 69.85; H, 6.24; N, 5.26. Found: C, 69.98; H, 6.13; N, 5.32. ESI-TOF MS calculated for C31H32N2O4, m/z 496.236, found 519.303 (M+Na)+
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β- (1′-naphthyl)acetamido]morphinan (4)
Compound 4 was prepared according to the general procedure described above; combining β-naltrexamine (100 mg, 0.29 mmol), 1-naphthoic acid (117 mg, 0.58 mmol); BOP (258 mg, 0.58 mmol), and DIPEA (150 μL, 0.81 mmol) followed by basic solvolysis with K2CO3 gave the target compound which was purified by flash chromatography [EA/hexanes (75/25)], and then recrystallized from an acetone/hexanes mixture to provide 4 as a white solid (120 mg, 84%).
1H NMR (CDCl3) δ: 0.11 (m, 2H); 0.52 (m, 2H); 0.81 (m, 1H); 1.47 (m, 2H); 1.62–1.75 (m, 2H); 1.91 (m, 1H); 2.17 (m, 2H); 2.35 (m, 2H); 2.61 (m, 2H); 3.01 (d, 1H, J= Hz); 3.08 (m, 1H); 4.18 (m, 1H); 4.50 (d, 1H, JH5-H6= 7.8 Hz); 6.55 (d, 1H, JH1-H2= 8.1 Hz); 6.72 (d, 1H, JH2-H1= 8.1 Hz); 6.87 (d, 1H, J= 8.9 Hz); 7.38 (m, 1H); 7.48–7.53 (m, 2H); 7.58 (Dd, 1H, J= 7Hz, J= 1.2 Hz); 7.79–7.92 (m, 2H), 8.32 (m, 1H); 13C NMR (CDCl3) δ: 3.77, 3.97, 9.38, 22.64, 23.91, 31.23, 31.45, 43.96, 47.54, 51.00, 59.26, 62.26, 70.14, 93.40, 117.83, 119.19, 124.54, 124.72, 125.04, 125.42, 126.36, 127.12, 128.27, 130.10, 130.58, 130.86, 133.65, 134.44, 139.82, 142.97, 169.46; mp= 224–226 °C. Anal. Calcd. for C31H33ClN2O4: C, 74.98; H, 6.50; N, 5.64. Found: C, 75.27; H, 6.57; N, 5.74 C ESI-TOF MS calculated for C31H32N2O4, m/z 496.236, found 497.139 (M+H)+, 993.260 (2xM+H)+
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β- (6′-methoxy-2′-naphthyl)acetamido]morphinan (5)
Compound 5 was prepared according to the general procedure described above; combining β-naltrexamine (100 mg, 0.29 mmol), 6-methoxy-2-naphthoic acid (117 mg, 0.58 mmol); BOP (258 mg, 0.58 mmol), and DIPEA (150 μL, 0.81 mmol) followed by basic solvolysis with K2CO3 gave the target compound which was purified by flash chromatography [EA/hexanes; 75/25], and then recrystallized from an acetone/hexanes mixture to provide 5 as a white solid (114 mg, 75%).
1H NMR (DMSO-d6) δ: 0.15 (m, 2H); 0.50 (m, 2H); 0.88 (m, 1H); 1.23–1.62 (m, 4H); 1.82–1.91 (m, 2H); 2.08 (m, 1H); 2.27 (m, 2H); 2.36 (m, 2H); 3.03 (m, 2H); 3.75 (m, 1H); 3.90 (s, 3H); 4.77 (d, 1H, JH5-H6= 7.1 Hz); 4.91 (bs, 1H, OH-14); 6.57–6.60 (m, 2H); 7.23 (Dd, 1H, J= 9 Hz, J= 2.4 Hz); 7.38 (d, 1H, J= 1.9 Hz); 7.86–7.94 (m, 3H), 8.42 (s, 1H); 8.72 (D, 1H amide, JNH-H6= 8.1 Hz); 9.04 (bs, 1H, OH-3); 13C NMR (DMSO-d6) δ: 3.27, 3.45, 9.03, 22.32, 24.46, 29.89, 31.14, 38.81, 41.66, 45.71, 51.47, 55.27, 61.17, 69.63, 90.48, 105.82, 117.20, 118.76, 119.33, 123.45, 124.61, 126.57, 127.30, 127.42, 127.55, 129.28, 130.37, 135.70, 140.60, 142.11, 158.50, 165.67. mp= 199–201 °C. Anal. Calcd. for C32H35ClN2O5: C, 68.25; H, 6.27; N, 4.97. Found: C, 68.38; H, 6.13; N, 5.04. ESI-TOF MS calculated for C32H34N2O5:, m/z 526.247, found 527.298 (M+H)+
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β- (3′-methoxy-2′-naphthyl)acetamido]morphinan (6)
Compound 6 was prepared according to the general procedure described above; combining β-naltrexamine (100 mg, 0.29 mmol), 6-methoxy-2-naphthoic acid acid (117 mg, 0.58 mmol); BOP (258 mg, 0.58 mmol), and DIPEA (150 μL, 0.81 mmol) followed by basic solvolysis with K2CO3 gave the target compound which was purified by flash chromatography [EA/hexanes; 75/25], and then recrystallized from an acetone/hexanes mixture to provide 6 as a white solid (119 mg, 78%).
1H NMR (DMSO-d6) δ: 0.12 (m, 2H); 0.47 (m, 2H); 0.85 (m, 1H); 1.26–1.66 (m, 4H); 1.81–1.90 (m, 2H); 2.08 (m, 1H); 2.27 (m, 2H); 2.36 (m, 2H); 3.03 (m, 2H); 3.75 (m, 1H); 3.90 (s, 3H); 4.77 (d, 1H, JH5-H6= 7.1 Hz); 4.91 (bs, 1H, OH-14); 6.57–6.60 (m, 2H); 7.23 (Dd, 1H, J= 9 Hz, J= 2.4 Hz); 7.38 (d, 1H, J= 1.9 Hz); 7.86–7.94 (m, 3H), 8.23 (s, 1H); 8.50 (D, 1H amide, JNH-H6= 8.1 Hz); 9.04 (bs, 1H, OH-3); 13C NMR (DMSO-d6) δ: 3.45, 3.71, 9.17, 22.17, 24.30, 29.97, 30.36, 41.23, 45.67, 47.13, 51.56, 55.83, 61.72, 69.63, 90.63, 106.46, 115.91, 117.96, 124.20, 125.30, 126.33, 127.41, 127.66, 127.77, 128.17, 128.34, 130.66, 134.94, 140.43, 142.17, 154.41, 164.73. mp= 215–26 °C. Anal. Calcd. for C32H35ClN2O5: C, 68.25; H, 6.27; N, 4.97. Found: C, 68.47; H, 6.21; N, 5.02. ESI-TOF MS calculated for C32H34N2O5, m/z 526.247, found 527.301(M+H)+
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β- (6′-fluoro-2′-naphthyl)acetamido]morphinan (7)
Compound 7 was prepared according to the general procedure described above; combining β-naltrexamine (100 mg, 0.29 mmol), 6-fluoro-2-naphthoic acid (110 mg, 0.58 mmol); BOP (258 mg, 0.58 mmol), and DIPEA (150 μL, 0.81 mmol) followed by basic solvolysis with K2CO3 gave the target compound which was purified by flash chromatography [EA/hexanes; 80/20], and then recrystallized from an acetone/hexanes mixture to provide 7 as a white solid (118 mg, 79%).
1H NMR (CD3OD) δ: 0.23 (m, 2H); 0.59 (m, 2H); 0.94 (m, 1H); 1.47–1.79 (m, 4H); 2.03 (m, 1H); 2.17–2.35 (m, 2H); 2.44–2.52 (m, 2H); 2.70–2.76 (m, 2H); 3.13–3.23 (m, 2H); 4.01 (m, 1H); 4.72 (d, 1H, J = 7.7 Hz); 6.63 (d, 1H, J = 8.1 Hz); 6.68 (d, 1H, J = 8.1 Hz); 7.42 (td, 1H, J = 2.5 Hz, J = 9.8 Hz); 7.62 (dd, 1H, J = 2.4 Hz, J = 9.9 Hz); 7.93–7.99 (m, 2H); 8.08 (m, 1H), 8.45 (s, 1H); 13C NMR (CD3OD) δ: 4.14, 4.58, 10.23, 23.61, 25.59, 31.34, 31.42, 43.86, 49.01, 53.49, 60.19, 63.80, 71.82, 93.04, 111.69, 111.90, 117.93, 118.18, 118.67, 120.13, 126.10 (x2), 128.70, 128.91, 131.12, 132.55, 132.88, 134.05, 140.47, 143.74, 169.76; 19F NMR (CD3OD) δ: −114.03; mp= 189–190 °C. Anal. Calcd. for C31H32ClFN2O5: C, 67.57; H, 5.85; N, 5.08. Found: C, 67.91; H, 5.85; N, 5.17. ESI-TOF MS calculated for C31H31FN2O5, m/z 514.227, found 515.300 (M+H)+
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β- (2′-quinoxalyl)acetamido]morphinan (8)
Compound 8 was prepared according to the general procedure described above; combining β-naltrexamine (100 mg, 0.29 mmol), 2-quinoxalinecarboxylic acid (101 mg, 0.58 mmol); BOP (258 mg, 0.58 mmol), and DIPEA (150 μL, 0.81 mmol) followed by basic solvolysis with K2CO3 gave the target compound which was purified by flash chromatography [EA/hexanes (75/25)], and then recrystallized from an acetone/hexanes mixture to provide 8 as a white solid (110 mg, 77%).
1H NMR (DMSO-d6) δ: 0.01 (m, 2H); 0.37 (m, 2H); 0.72 (m, 1H); 1.28 (m, 1H); 1.49 (m, 2H); 1.85 (m, 2H); 2.05 (m, 2H); 2.25 (m, 2H); 2.55 (m, 2H); 2.93–3.02 (m, 2H); 4.00 (m, 1H); 4.37 (d, 1H, JH5-H6= 7.8 Hz); 6.46 (d, 1H, JH1-H2= 8.1 Hz); 6.55 (d, 1H, JH2-H1= 8.1 Hz); 7.31–7.36 (m, 3H); 7.44 (m, 1H); 7.81 (d, 1H, J= 8.3 Hz); 7.89 (d, 2H, J= 8.2 Hz); 8.15 (d, 1H, J= 8.7 Hz); 8.39 (s, 1H,); 13C NMR (DMSO-d6) δ: 3.52, 3.64, 9.36, 24.40, 25.26, 30.06, 30.27, 43.6, 47.43, 52.67, 58.37, 61.75, 69.56, 90.22, 110.87, 115.64, 118.31, 119.08, 126.54, 129.07, 129.38, 131.26, 139.76, 142.08, 142.84, 143.66, 144.36, 156.54, 162.88. mp= 177–180°C. Anal. Calcd. for C29H31ClN4O4: C. 65.10; H. 5.84; N; 10.47. Found: C. 64.81; H. 5.81; N. 10.55. ESI-TOF MS calculated for C29H30N4O4, m/z 498.227, found 499.121 (M+H)+, 1019.221 (2xM+Na)+
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β- (1′-isoquinolyl)acetamido]morphinan (9)
Compound 9 was prepared according to the general procedure described above; combining β-naltrexamine (100 mg, 0.29 mmol), isoquinoline-1-carboxylic acid (100.4 mg, 0.58 mmol); BOP (258 mg, 0.58 mmol), and DIPEA (150 μL, 0.81 mmol) followed by basic solvolysis with K2CO3 gave the target compound which was purified by flash chromatography [EA/hexanes (90/10)], and then recrystallized from an acetone/hexanes mixture to provide 9 as a white solid (90 mg, 63%).
1H NMR (CDCl3) δ: 0.13 (m, 2H); 0.53 (m, 2H); 0.85 (m, 1H); 1.17–1.68 (m, 4H); 1.83–2.04 (m, 3H); 2.18 (m, 2H); 2.25 (m, 2H); 2.38 (m, 2H); 2.61–2.67 (m, 2H); 4.06 (m, 1H); 4.60 (d, 1H, JH5-H6= 7.8 Hz); 6.58 (d, 1H, JH1-H2= 8.1 Hz); 6.73 (d, 1H, JH2-H1= 8.1 Hz); 7.58–7.78 (m, 4H); 8.39 (d, 1H, J= 5.5 Hz); 8.58 (d, 1H, J= 9.1 Hz), 9.49 (d, 1H amide, JNH= 8.2 Hz); 13C NMR (CDCl3) δ: 3.79, 3.97, 9.44, 22.70, 24.54, 30.27, 30.57, 44.11, 47.84, 51.34, 59.23, 62.37, 70.21, 93.82, 117.73, 119.17, 124.36, 124.44, 126.69, 126.92, 127.76, 128.56, 130.38, 131.15, 137.32, 140.06, 140.12, 142.56, 147.93, 165.89; mp= 152–154 °C. Anal. Calcd. for C30H32ClN3O4: C. 67.47; H. 6.04; N; 7.87. Found: C. 67.11; H. 6.08; N. 7.93. ESI-TOF MS calculated for C30H31N3O4, m/z 497.231, found 498.226 (M+H)+, 1017.425 (2xM+Na)+
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β- (5′-quinolyl)acetamido]morphinan (10)
Compound 10 was prepared according to the general procedure described above; combining β-naltrexamine (100 mg, 0.29 mmol), 5-quinoline-1-carboxylic acid (100.4 mg, 0.58 mmol); BOP (258 mg, 0.58 mmol), and DIPEA (150 μL, 0.81 mmol) followed by basic solvolysis with K2CO3 gave the target compound which was purified by flash chromatography [4%MeOH/DCM], and then recrystallized from an acetone/hexanes mixture to provide 10 as a white solid (83 mg, 58%).
1H NMR (CD3OD) δ: 0.18 (m, 2H); 0.56 (m, 2H); 0.91 (m, 1H); 1.45 (m, 1H); 1.58–1.65 (m, 2H); 1.84 (m, 1H); 2.00 (m, 1H); 2.13–2.36 (m, 2H); 2.43–2.51 (m, 2H); 2.69–2.73 (m, 2H); 3.10–3.20 (m, 2H); 3.99 (m, 1H); 4.59 (d, 1H, JH5-H6= 7.8 Hz); 6.62 (d, 1H, JH1-H2= 8.1 Hz); 6.69 (d, 1H, JH2-H1= 8.1 Hz); 7.62 (m, 1H); 7.82–7.85 (m, 2H); 8.14 (t, 1H, J = 5.3 Hz); 8.78 (d, 1H, J = 9.5 Hz); 8.90 (dd, 1H, J = 1.6 Hz, J = 4.2 Hz); 13C NMR (CD3OD) δ: 4.15, 4.54, 10.13, 23.59, 25.66, 31.44, 31.71, 45.44, 48.78, 53.54, 60.20, 63.75, 71.80, 92.95, 118.61, 120.18, 123.25, 125.36, 127.02, 127.24, 130.18, 131.60, 132.54, 136.16, 136.27, 142.08; 143.73, 148.69, 151.74, 170.64. mp= 165–167 °C. Anal. Calcd. for C30H32ClN3O4: C. 67.47; H. 6.04; N; 7.87. Found: C. 67.82; H. 6.09; N. 7.79. ESI-TOF MS calculated for C30H31N3O4, m/z 497.231, found 498.133 (M+H)+
Materials and Methods
Intracellular calcium release
To determine the functional selectivity of the above ligands, a modified intracellular calcium release assay was employed. As activation of opioid receptors does not regulate signaling mechanisms that cause calcium release because they are coupled to Gi/o, a chimeric G-protein (D6-Gqi4-myr), was employed. The construction of this chimera has been reported19. Human embryonic kidney cells (HEK-293) with stably expressing human Giq protein were grown at 37 °C and 10% CO2 in Dulbelcco’s modified medium (GIBCO) using zeocin as the antibiotic for selecting cells expressing the Gi G-protein.
HEK-293 cells containing the various opioid receptors were created by transiently transfecting opioid recptor DNA in OptiMEM medium (Invitrogen) at a concentration of 200 ng/20,000 cells using Lipofectamine 2000 (Invitrogen, Carlsbad CA). Twenty-four hours later, 20,000 cells/well were seeded into 96-well black plates (Corning Inc.). The FLIPR Calcium Explorer kit (Molecular devices) was used for the assay. On the third day, cells were incubated with a Ca++ chelating dye from the kit and incubated for 60 minutes at 37°C. The plates were then assayed in a FlexStation 3 apparatus (Molecular Devices) using a range of concentrations of the ligand tested. The response was measured as the change in Relative Fluorescence Units (ΔRFU = RFUmax-RFUmin) and the time of the response was measured in seconds. Following a 30 second equilibration period, the ligand was added to each well of the plate. RFUs were measured continuously for 60 seconds following the addition of the ligand (the response before calcium ion reuptake mechanisms bring the Ca2+ ion concentration back to basal levels). The ΔRFU was computed for each concentration, which was plotted as a concentration response curve and analyzed to obtain the IC50 value using nonlinear regression. The peak effect was also recorded. At least three independent replications with four internal replicates were used to evaluate each ligand in each of the cell types.
Animals
Male ICR-CD1 mice (17 – 25g; Harlan, Madison, WI) employed in the testing were housed in groups of 8 in a temperature- and humidity-controlled environment with unlimited access to food and water. They were maintained on a 12 h light/dark cycle. All experiments were approved by the Institutional Animal Care and Use Committee of the University of Minnesota (Minneapolis, MN).
Antinociceptive Testing
The tail flick assay for antinociception described by D’Amour21 and Smith and modified by Dewey et Al. was employed22. For the measurement, of the tail-flick latency, mice were held gently with the tail positioned in the apparatus (Tail Flick Analgesia Meter, Columbus Instruments, Columbus, Ohio) for radiant heat stimulus. The tail-flick response was elicited by applying radiant heat to the dorsal side of the tail. The intensity of the heat is set so that the mouse flicks its tail within 2 to 3 s. The test latency is measured before drug treatment (control) and again after the drug treatment (test) at the peak time of the compound, a 10 s maximum cut-off time is used to prevent damage to the tail. Antinociception is quantified according to the method of Harris and Pierson23 as the percent maximal possible effect (%MPE): %MPE = (Test - Control/10 – Control) × 100.
At least three groups of eight to ten mice are used for each dose response curve, and each mouse is used only once. ED50 values with 95% confidence intervals (C.I.) are computed with GraphPad Prism 4 by using nonlinear regression methods.
Agonism
All the compounds were dissolved in 10% DMSO and then diluted to less than 1% DMSO in the test solutions. Controls when given either i.c.v. or i.t. with 1% or less DMSO do not show any antinociception. All compounds were administered in a 5-μl volume in conscious mice according to the method of Haley and McCormick27 for i.c.v and Hylden and Wilcox28 for i.t. injections. A time course study (10, 20, 30 and 60 min) were used to determine the peak antinociception. Administration of ligands and antagonist were timed so that they peaked at the same time (nor-BNI (2.5 nmol/mouse – 20 minute peak time), NTI (5 nmol/mouse – 20 minute peak time)). Potency ratios (test ED50/control ED50) were deemed significant if their 95% confidence intervals do not overlap.
Acute tolerance was measured on select compounds by comparing the initial ED80 – 90 dose to the same dose measured 24 hours later on the same mouse.
Knock-out Studies
MOR-KO mice (male and female, 13–15 weeks old) were grouped with equal numbers of male and females so that there were eight mice per group. Each mouse was only used once. The MOR-KO mice were injected i.t. with the ED80 – 90 of the given agonist to see if there was a change in the %MPE. The point was considered significant if the 95% confidence intervals did not overlap.
Acknowledgments
We thank Dr. Evi Kostenis for providing us with the cDNA for Δ6-Gαqi4-myr chimeric Gα subunit, and Dr. Sabita Roy for the MOR-KO mice. This research was supported by National Institute of Health research grant DA01533.
References
- 1.IUPHAR receptor database, opioid receptors. http://uphar-db.org/ PRODGPCR.
- 2.Gutstein HB, Akil H. Goodman and Gillman’s the Pharmacological Basis of Therapeutics. 11. Chapter 21 McGraw-Hill; New York: 2006. Opioid Analgesics. [Google Scholar]
- 3.(a) Takemori AE, Portoghese PS. Selective naltrexone-derived opioid receptor antagonists. Annu Rev Pharmacol Toxicol. 1992;32:239–269. doi: 10.1146/annurev.pa.32.040192.001323. [DOI] [PubMed] [Google Scholar]; (b) Eguchi M. Recent advances in selective opioid receptor agonists and antagonists. Med Res Rev. 2004;24(2):182–213. doi: 10.1002/med.10059. [DOI] [PubMed] [Google Scholar]
- 4.Gershengorn MC, Osman R. Minireview: Insights into G protein-coupled receptor function using molecular models. Endocrinology. 2001;142:2–10. doi: 10.1210/endo.142.1.7919. [DOI] [PubMed] [Google Scholar]
- 5.Erez M, Takemori AE, Portoghese PS. Narcotic antagonistic potency of bivalent ligands which contain beta-naltrexamine. Evidence for bridging between proximal recognition sites. J Med Chem. 1982;25:847–849. doi: 10.1021/jm00349a016. [DOI] [PubMed] [Google Scholar]
- 6.Portoghese PS, Ronsisvalle G, Larson D, Takemori AE. Synthesis and opioid antagonist bivalent ligands with conformationally restricted spacers. J Med Chem. 1986;28:1650–1653. doi: 10.1021/jm00159a014. [DOI] [PubMed] [Google Scholar]
- 7.Angers S, Salahpour A, Bouvier M. Dimerization: An emerging concept for G Protein-Coupled Receptor ontogeny and function. Annu Rev Pharmacol Toxicol. 2002;42:409–435. doi: 10.1146/annurev.pharmtox.42.091701.082314. [DOI] [PubMed] [Google Scholar]
- 8.Pin J-P, Neubig R, Bouvier M, Devi L, Filizola M, Javitch JA, Lohse MJ, Milligan G, Palczewski K, Parmentier M, spedding M. International union of basic and clinical pharmacology. LXVII. Recommendations for the recognition and nomenclature of G Protein-coupled Receptor heteromultimers. Pharmacol Rev. 2007;59(5):5–13. doi: 10.1124/pr.59.1.5. [DOI] [PubMed] [Google Scholar]
- 9.Devi LA. Heterodimerization of G-protein-coupled receptors: pharmacology, signaling and trafficking. Trends Pharmacol Sci. 2001;22:532–537. doi: 10.1016/s0165-6147(00)01799-5. [DOI] [PubMed] [Google Scholar]
- 10.Jordan BA, Devi LA. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;5:697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gupta A, Decaillot FM, Devi LA. Targeting opioid receptor heterodimers: strategies for screening and drug development. AAPS J. 2006;10;8(1):E153–159. doi: 10.1208/aapsj080118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aldrich JV, Vigil-Cruz SC. Narcotic analgesics. In: Abraham DJ, editor. Burger’s Medicinal Chemistry and Drug Discovery. Vol. 6. New York, NY: John Wiley & Sons; 2003. pp. 329–481. [Google Scholar]
- 13.Friderichs E. Opioids. In: Buschmann H, Christoph T, Friderichs E, Maul C, Sundermann B, editors. Analgesics, From Chemistry and Pharmacology to Clinical Application. Weinheim, Germany: Wiley-VCH; 2002. pp. 127–150. [Google Scholar]
- 14.Yekkirala AS, Kalyuzhny AE, Portoghese PS. Standard opioid agonists activate heteromeric opioid receptors: Evidence for morphine and [D-Ala2-MePhe4-Glyol5]enkephalin as selective μ–δ agonists. ACS Chem Neurosci. 2010;1:146–154. doi: 10.1021/cn9000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yekkirala AS, Banks ML, Lunzer MM, Negus SS, Rice KC, Portoghese PS. Unpublished Results. [Google Scholar]
- 16.Yekkirala AS, Lunzer MM, McCurdy CR, Powers MD, Kalyuzhny AE, Roerig SC, Portoghese PS. N-Naphtoyl-b-Naltrexamine (NNTA), a highly selective and potent activator of heteromeric m/k opioid receptors. Proc Natl Acad Sci USA. 2011;108(12):5098–5103. doi: 10.1073/pnas.1016277108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sayre LM, Portoghese PS. Stereospecific synthesis of the 6.alpha.-and 6.beta.-amino derivatives of naltrexone and oxymorphone. J Org Chem. 1980;16:3366–3368. [Google Scholar]
- 18.(a) Castro B, Dormoy JR, Evin G, Selve CBOP. a new peptide coupling reagent exemplified in the synthesis of somatostatin. Pept, Proc Eur Pept Symp. 1976;14:79–84. [Google Scholar]; (b) Ghirmai S, Azar MR, Polgar WE, Berzetzei-Gurske I, Cashman JR. Synthesis and biological evaluation of alpha- and beta-6-amido derivatives of 17-cyclopropylmethyl-3,14beta-dihydroxy-4,5alpha-epoxymorphinan: potential alcohol-cessation agents. J Med Chem. 2008;51:1913–1924. doi: 10.1021/jm701060e. [DOI] [PubMed] [Google Scholar]
- 19.Kostenis E. Is G-alpha16 the optimal tool for fishing ligands of orphan G-protein-coupled receptors? Trends Pharmacol Sci. 2001;22:560–564. doi: 10.1016/s0165-6147(00)01810-1. [DOI] [PubMed] [Google Scholar]
- 20.(a) Kawai K, Hayakawa J, Miyamoto T, Imamura Y, Yamane S, Wakita H, Fujii H, Kawamura K, Matsuura H, Izumimoto N, Kobayashi R, Endoh Y, Nagase H. Design, synthesis and structure-activity relationship of novel opioid k-agonists. Bioorg Med Chem. 2008;16(20):9188–9201. doi: 10.1016/j.bmc.2008.09.011. [DOI] [PubMed] [Google Scholar]; (b) Nagase H, Hayakawa J, Kawamura H, Kawai K, Endoh T. Preparation of morphinan derivatives as brain cell protecting agents. CODEN: PIXXD2 WO 9503308. PCT Int Appl. :A1 19950202.
- 21.D’Amour FE, Smith DL. A method for determining loss of pain sensation. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]
- 22.Dewey WL, Harris LS, Howes JF, Nuite JA. The effect of various neurochemical modulators on the activity of morphine and the narcotic antagonists in the tail-flick and phenylquinone tests. J Pharmacol Exp Ther. 1970;175:435–442. [PubMed] [Google Scholar]
- 23.Harris LS, Pierson AK. Some narcotic antagonists in the benzomorphan series. J Pharmacol Exp Ther. 1964;143:141–148. [PubMed] [Google Scholar]
- 24.Portoghese PS, Lipkowski AW, Takemori AE. Binaltorphimine and nor binaltorphimine, potent and selective kappa opioid receptor antagonists. Life Sci. 1987;40:1287–1292. doi: 10.1016/0024-3205(87)90585-6. [DOI] [PubMed] [Google Scholar]
- 25.Portoghese PS, Sultana M, Takemori AE. Design of peptidomimetic δ opioid receptor antagonists using the message-address concept. J Med Chem. 1990;33:1714–1720. doi: 10.1021/jm00168a028. [DOI] [PubMed] [Google Scholar]
- 26.Lunzer MM, Portoghese PS. Selectivity of d- and k-opioid ligands depends on the route of central administration in mice. J Pharmacol Exp Ther. 2007;322:166–171. doi: 10.1124/jpet.107.120279. [DOI] [PubMed] [Google Scholar]
- 27.Haley TJ, McCormick WG. Pharmacological effects produced by intracerebral injection of drugs in the conscious mouse. Br J Pharmacol Chemother. 1957;12:12–15. doi: 10.1111/j.1476-5381.1957.tb01354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hylden JL, Wilcox GL. Intrathecal morphine in mice: A new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]