

Abstract
Transcriptional regulation is a critical mediator of many normal cellular processes, as well as disease progression. Transcription factors (TFs) often co-localize at cis-regulatory elements on the DNA, form protein complexes, and collaboratively regulate gene expression. Machine learning and Bayesian approaches have been used to identify TF modules in a one-dimensional context. However, recent studies using high throughput technologies have shown that TF interactions should also be considered in three-dimensional nuclear space. Here, we describe methods for identifying TF modules and discuss how moving from a one-dimensional to a three-dimensional paradigm, along with integrated experimental and computational approaches, can lead to a better understanding of TF association networks.
Keywords: Chromatin Immunoprecipitation (ChIP), Chromatin Structure, Protein-DNA interaction, Transcription Factors, Transcriptional Regulation, High Throughput, Binding Sites, Modules, Networks
Introduction
Transcriptional regulation is a critical step in transmission of information from genotype (i.e. DNA) to phenotype (e.g. expression of proteins and noncoding RNAs or microRNAs). A large number of proteins, namely transcription factors (TFs),2 play an essential role in gene regulatory networks by binding to short DNA sequences called cis-regulatory elements (CREs), which include promoters, enhancers, repressors, and insulators (1). In many cases, multiple TFs function as a regulatory complex (hereafter referred to as a TF module) at a CRE. The concept of cooperative regulation by TFs bound near each other in the genome has been recognized for decades. For example, the enhancer located upstream of the interferon-β gene (IFNB1) has served as a wonderful example of cooperative regulation (2, 3). Another example of TF modules is the regulatory region upstream of the Drosophila gene eve. Expression of the eve gene is modulated by different combinations of multiple TFs that bind to the upstream CRE (4). Although the identification of modules composed of TFs bound to adjacent genomic sites is increasing, due in part to ChIP-seq analysis of a large set of TFs by the ENCODE Consortium,3 it has recently become clear that cooperative regulation can be achieved by means other than the interaction of TFs bound next to each other on the genome. TFs can also be brought into close spatial proximity with other TFs bound to a regulatory element located a great distance away via the three-dimensional conformation of a chromosome. In fact, three-dimensional genomic organization, which brings together two distant loci, has been shown to be involved in both gene regulation and nuclear compartmentalization (5–8). The regulatory effects of a TF bound to a CRE can be either active or repressive, often switching from one to the other depending on other interacting factors. Therefore, a detailed understanding of the association of TFs with other TFs bound at adjacent or distal sites is required to comprehend the complex molecular mechanisms involved in transcriptional regulation of the genome. Also, elucidating cell type-specific TF modules may help to understand the mechanisms driving cell differentiation and disease progression. New experimental techniques facilitated by high throughput sequencing allow investigators to more globally address questions concerning the relationship between three-dimensional chromatin organization and TF modules. However, it is a remarkably complex task to extend the analysis of TF modules from a one-dimensional to a three-dimensional scale, requiring tremendous efforts from both experimental and computational biologists, as well as effective communication and collaboration among these specialists. In this minireview, we focus on the experimental and computational methods involved in the identification of TF modules, concluding with a suggested pathway by which investigators can identify both one- and three-dimensional TF modules.
Experimental Methods
Profiling TF-binding Sites (TFBSs)
Although a variety of methods have been developed to investigate TF binding throughout the genome (9), the technique of chromatin immunoprecipitation (ChIP) is the most common. This technique, which was developed during the 1980s and 1990s, has been modified extensively for the analysis of site-specific factors and histones (10–17). The steps in a ChIP experiment include 1) cross-linking TFs to the genome, 2) shearing DNA (usually by sonication) to fragments ranging from 100 to 500 bp in length, 3) enriching for TF-DNA complexes using target TF-specific antibodies, 4) removing proteins by reversing the cross-links, and 5) purifying the enriched DNA fragments for further analyses (Fig. 1A). When ChIP was first developed, a polymerase chain reaction (PCR) assay would be performed to determine whether the TF bound to a specific genomic position. Although this assay is still used to study single loci, the sequencing of the human genome (18–20) and the development of high throughput technologies (21) have enabled genome-wide profiling of TFBSs. ChIP-chip, a high throughput technique that hybridizes the enriched DNA fragments to microarrays (22–25), was first used to survey TFBSs genome-wide. ChIP-seq (26–31), a new technology that combines ChIP and massively parallel sequencing (on platforms such as the Illumina Genome Analyzer and HiSeq machines, the ABI SOLiD system, and the Roche 454 system), was first developed in 2007 and has rapidly proved to have several advantages such as the complete coverage of the unique portions of the genome along with high resolution and sensitivity.
FIGURE 1.

Experimental techniques to investigate TFBSs and chromatin interactions. A, schematic representation of major steps in one-dimensional ChIP-based high throughput methods used to identify TFBSs. Briefly, cells are treated with formaldehyde to cross-link the TFs to genomic binding sites, the genomic DNA is sheared, and bound fragments are selected by immunoprecipitation using an antibody to a TF of interest. The cross-links are then reversed, and the fragments are purified and applied to microarrays (ChIP-chip) or sequenced (ChIP-seq). B, assays used to study three-dimensional chromatin structure. ChIA-PET is similar to ChIP in that fragments bound to a TF of interest are immunoprecipitated. However, unlike ChIP assays, fragments brought into close proximity by DNA looping are ligated prior to the immunoprecipitation step. Hi-C is similar to ChIA-PET in that fragments in close proximity are ligated. However, Hi-C does not rely on immunoprecipitation by an antibody to a TF but rather uses biotin labeling of the ligation sites, followed by avidin-based purification. The fragments are then subjected to paired-end sequencing. The 3C, 4C, and 5C assays also detect pairs of genomic loci that are in close proximity in the three-dimensional space of the nucleus. Formaldehyde is used to cross-link spatially close chromatin regions, the DNA is digested with a restriction enzyme, and fragments within the cross-linked complexes are joined by ligation. In 3C, the joined regions are analyzed using PCR. In 4C, a second enzyme restriction digestion step is performed to shorten the hybrid fragments, which are circularized and subjected to inverse PCR; the products of inverse PCR are hybridized to a custom microarray. In 5C, a LMA step allows the ligation junctions of all the hybrid fragments in the 3C library to be analyzed using microarrays or next-generation sequencing. Note that this figure shows greatly simplified versions of the different technologies; for detailed descriptions, please see the original papers.
Identifying Chromatin Interactions
Although the aforementioned techniques provide a detailed map of TFBSs, they are not sufficient to identify TF modules coordinated by higher order chromatin organization. In the past, co-localization methods such as fluorescent in situ hybridization (FISH) have been used to investigate three-dimensional chromatin structures (32). However, the higher resolution of the chromosome conformation capture (3C) technique (33) has greatly improved our ability to examine the effects of chromatin conformation on transcriptional regulation. The 3C assay can detect pairs of genomic loci that are in close proximity in the three-dimensional space of the nucleus. In a 3C experiment, formaldehyde is used to cross-link non-adjacent regions of chromatin that are spatially close. The DNA is then digested with a restriction enzyme, and the fragments within the cross-linked complexes are joined by ligation. This is followed by cross-link reversal and PCR using primers specific for two different genomic regions. A high signal for the hybrid DNA sequence indicates a high ligation rate between the two genomic loci, which is likely produced by their close proximity and high interaction frequency (Fig. 1B). Several high throughput variations on the 3C assay have been developed that allow a larger scale screening of chromatin interactions. For example, chromosome conformation capture-on-chip (4C) (34, 35) detects many genomic regions interacting with one particular locus using a microarray containing a set of specifically designed probes. After the cross-link reversal step of 3C, a second enzyme restriction digestion step is performed to shorten the hybrid fragments, and then the small hybrid fragments are circularized and subjected to inverse PCR. To identify the interacting regions for the locus of interest (which is called the bait), specific primers within the bait region of the circularized hybrid fragment are designed such that they face the portion of the circularized fragment that is derived from the interacting region. After amplification, the products of inverse PCR are hybridized to the custom microarray. The major obstacle for wide application of the 4C assay is that it can detect only regions interacting with one chosen genomic locus per experiment. Another 3C-based large-scale DNA interaction profiling method is chromosome conformation capture carbon copy (5C) (36). Similar to 4C, 5C allows detection of many potential chromatin interactions, but a multiplex ligation-mediated amplification (LMA) step distinguishes it from 4C. The universal primers of LMA are designed to fit near the restriction enzyme cutting sites and have a specific orientation so that the amplification products in the 5C library theoretically contain the ligation junctions of all the hybrid fragments in the 3C library. The 5C library is analyzed using microarrays or next-generation sequencing. Both 4C and 5C involve multiplex primers or probe designs, which substantially increase the cost and decrease the applicability of the assay. Two newly developed next-generation sequencing-based techniques, ChIA-PET (37, 38) and Hi-C (39), are more suitable for unbiased identification of chromatin interactions across the entire genome. ChIA-PET incorporates enrichment of cross-linked complexes that contain a target protein using antibody-based immunoprecipitation before cross-link reversal; thus, it has been used mainly to interrogate interactomes of a specific TF such as estrogen receptor-α (ER-α) (37) or CCCTC-binding factor (CTCF) (38). On the other hand, Hi-C uses biotin labeling of the ligation sites, which are then purified using avidin. In contrast to ChIA-PET, Hi-C can map the location of all possible interacting loci in the genome in an unbiased manner, but it does not provide information as to which TFs are involved in formation of the different interactions. However, by combining other experimental and computational assays with Hi-C (as described below), one can link TFs to sets of interacting loci, allowing the identification of three-dimensional TF modules.
Computational Methods
Detecting TFBSs
In a ChIP-seq experiment, millions of short DNA sequence tags are aligned with a reference genome, and binding sites of the target protein are identified as genomic regions that are enriched within the set of sequenced tags. However, not all enriched regions correspond to binding sites, and therefore, computational methods have been developed to identify true binding sites. Model-based analysis of ChIP-seq (MACS) (40), PeakSeq (41), QuEST (17), site identification from short sequence reads (SISSRs) (42), Sole-Search (43, 44), and other peak identification programs (45–48) are available for ChIP-seq data analysis, and each of these programs applies a different algorithm (see Ref. 49 for a review of these tools). For example, because only the end of a ChIP fragment is sequenced, the MACS algorithm begins by shifting the sequenced tags toward the binding site for a certain number of base pairs and then locates the binding site by calculating the summit within a peak region. Instead of tag shifting, PeakSeq utilizes a strategy in which tags are extended to better represent the precipitated fragments. QuEST identifies binding sites using a tag enrichment profile of a peak region. SISSRs screens binding sites in a certain window by a threshold of tag counts on both forward and reverse strands that is calculated based on a Poisson distribution. Sole-Search extends the tags to represent the length of the precipitated fragments and allows the identification of both narrow peaks and longer binding regions based on prior knowledge of the factor being studied. Another recent method applies a mixture model to provide higher resolution of TFBS localization and the ability to distinguish closely positioned TFBSs (50). wBELT uses a bin-based enrichment threshold to identify TFBSs and applies tag shifting and statistical methods to define a false discovery rate (FDR); this software has been integrated into a user-friendly web-based application called W-ChIPeaks (51).
Identifying TF Modules from One-dimensional Omics Data
Accurate identification of individual TFBSs can be achieved using ChIP-seq and ChIP-chip. However, as discussed above, TFs usually do not function alone. Cooperating factors can influence the specificity and affinity of TF binding and can significantly alter the function of a bound TF, greatly influencing gene regulation. For example, the serum response factor (SRF) activates distinct sets of genes via interaction with different cofactors upon serum stimulation (52, 53). By interacting with different cofactors, the Mcm1 protein can either promote or repress transcription of a group of genes (54). Thus, to completely understand the function of a TF, it is important to characterize the TF at the level of interacting modules. Many computational approaches have been developed to search for TF modules, most of which are based on the fact that TFs bind to a specific sequence of DNA called a motif. A motif can be represented by a position weight matrix (PWM), which is a probability matrix that indicates the chance of each position of the motif being a certain nucleotide; motifs for many TFs are collected in the TRANSFAC data base (56). A high concurrence of motifs for two different TFs within a relatively short region of DNA may indicate a potential TF module. One example of a computational approach to predict TF modules is CisModule (55), which applies Bayesian inference and uses a two-layer hierarchical mixture model, in which the first layer represents the mixture of modules and the second layer represents the mixture of motifs in the modules. Parameters, including the product multinomial parameters for each motif, the width of each motif, the probability of a module start, and the probability of a motif start, are updated with each iteration. Studies have shown that methods that take advantage of both the TRANSFAC TF motif data base (56) and sequence conservation information within multiple organisms (57–60) generally outperform other algorithms in terms of the accuracy of prediction of TF binding. More recently, integrated computational and experimental genomics approaches have been combined to identify one-dimensional TF modules from ChIP-chip and ChIP-seq data (61). In these approaches, the binding sites of a TF are first identified using peak detection software. Then, regions adjacent to a set of high confidence TFBSs are searched for putative motifs of other TFs whose PWMs have been characterized in the TRANSFAC data base (56). One such method called ChIPModules employs a classification and regression tree model to generate TF modules based on the co-occurrence rate of the PWMs. Using this method, E2F1 target genes were classified into distinct groups regulated by five different modules. ChIP-chip analysis demonstrated that one predicted cofactor, activating enhancer binding protein 2α, did in fact form binding modules with E2F1. Another example of this type of method is hypergeometric optimization of motif enrichment (HOMER) (62), which includes a set of programs for de novo motif discovery. A recent study using HOMER found that PU.1 co-localizes with distinct sets of TFs in macrophages versus B cells (62). These findings demonstrated that different TF modules are indeed lineage-specific and responsible for the development of characteristic features of macrophages and B cells. Cistrome is another tool suite that includes a de novo motif discovery algorithm and allows cofactor identification through co-localization analysis of TF motifs (63). A recent study utilized Cistrome and epigenetic information to define CREs and predict TFBSs with high accuracy (64). Although this method was applied primarily to predict TFBSs, it could also increase the accuracy of predicting cell type-specific TF modules by eliminating false positive regions within closed chromatin. Studies using programs such as ChIPModules, HOMER, and Cistrome clearly demonstrate that searching for motifs in regions located near TFBSs can identify putative collaborating TFs. However, to find TF associations that occur through higher order chromatin structure, one must also integrate chromatin interaction data.
Identifying Chromatin Interactions
Among the chromatin interaction profiling methods, the Hi-C technique has the potential to identify a whole genome interactome in an unbiased manner without relying on known protein interactions. In the original description of the Hi-C assay (39), a probability matrix at 1-Mb scale was used to model the data. However, this method identified only low resolution interacting loci and is thus not suitable for analysis of the fine structure of the chromatin. To increase the resolution and better utilize the great resources of Hi-C data, a more recent method utilized a mixture Poisson regression model (MPRM) to increase the resolution of the identified interacting loci to 20 kb (65). There are three major types of hybrid fragments in the Hi-C library: 1) proximate ligation, formed by the joining of closely positioned DNA fragments; 2) random ligation, formed by the random interaction of two “floating” fragments; and 3) self-ligation, formed when the two ends of a single fragment are joined. In the MPRM, the self-ligation fragments are easily discounted by their distinctive characteristics, but the proximate ligation events and random ligation events are considered as two independent Poisson distributions; thus, the overall ligation events can be represented by a latent class model with two hidden variables. An expectation maximization algorithm is used to estimate the hidden variables, and a FDR is calculated based on the cumulative distribution function of the Poisson distribution. The MPRM identified 96,137 interacting loci with a FDR of 5.76%. Consistent a the previous study (39), the majority of the identified interactions were intrachromosomal and within a distance of 1 million bp and occurred close to CREs marked by histone depletion and flanked by H3K4me1 (65). Because of sequencing depth limitations in the existing data, it was estimated that the identified interacting loci represent only ∼25% of the complete set of interacting loci in K562 cells. These analyses suggest that the future Hi-C analysis of human cells should be derived from sequence information of at least 100 million proximate ligation hybrid fragments. Another method developed to explore Hi-C data is based on a probabilistic model, which takes into account several systematic biases that reside in the Hi-C protocol (66). These biases include spurious ligation fragments, the size of the enzyme-digested DNA fragments, the ligation efficiency, the CpG content of the ligation sites, and the mappability of the sequenced tags. This method also showed a high interaction rate of nucleosome-depleted regions and active promoters. Furthermore, this study demonstrated that the genome can be divided into active and inactive domains based on the connection intensity between CREs in those domains. As experimental techniques such as Hi-C become more commonly used, there will be an urgent demand for optimized methodologies to analyze the derived data. Perhaps by combining the advantages of the above-mentioned approaches, new methods with high resolution that appropriately consider system biases can provide a robust reconstruction of the chromatin structure.
Identifying TF Modules from Three-dimensional Omics Data
As described above, 3C has been used to study looping between defined genomic regions such as the β-globin locus control region (LCR) and DNase I-hypersensitive sites upstream or downstream of the locus (67). In another study, expression of UBE2C was shown to be regulated by FoxA1- and MED1-mediated chromatin interactions between the UBE2C promoter and enhancer (68). In breast cancer cells, the 3C assay was used to demonstrate that ER-α drives chromatin looping at a cluster of genes on genomic region 16p11.2 (69). In these cases, specific TFs were analyzed at specific loci. Because very few genome-wide chromatin interactions studies have been published, there are only a few examples in which TF binding data have been integrated with large-scale genomic structure information. However, a recent study (70) integrated Hi-C information with CTCF ChIP-chip and ChIP-seq data and found that strong interchromosomal interactions are highly correlated with CTCF-binding sites, suggesting that CTCF plays an important role in the organization of the human genome. This study served as a proof of principle that it is possible to identify chromosomal hubs that are associated with a specific TF. The extensive ChIP-seq data that are now available from the ENCODE Consortium provide investigators with the opportunity to identify associations of interacting loci with TF binding, chromatin modifications, and open chromatin. A recent study used 5C to comprehensively interrogate long-range looping interactions between genes and distal elements in the 1% of the human genome representing the ENCODE Pilot regions.4 These 5C maps of ENCODE regions in GM12878, K562, HeLa, and human embryonic stem cells (hESCs) identify thousands of long-range interactions and provide new insights into the cell-type specificity of chromatin looping. We have used data from the ENCODE Consortium to develop a TF interaction network using integrated genome-wide Hi-C data, epigenomic profiles, open chromatin, and TF binding data (65). In this study, the Apriori algorithm (71) was used to search for the association of TFs bound at the two ends of sets of interacting loci, e.g. if TF1 was bound to a set of loci and if binding of TF2 was statistically enriched in the genomic regions interacting with those loci, this suggested a potential association of TF1 and TF2. By incorporating ChIP-seq for 45 different TFs, the analysis showed 1) a high concurrence of CTCF and RAD21 at the ends of interacting loci, which is consistent with their similar binding preference and with previous reports (72); 2) that, consistent with previous studies, E2F4 and RNA polymerase II are highly linked (73); and 3) that c-Jun, GATA1, GATA2, INI1, and BRG1 are closely linked, which suggests interactions between chromatin modifiers and cell type-specific TFs. We note that this type of detailed TF network analysis is possible only if the genome-wide experimental data sets for chromatin interactions and TF binding are available for the same cell line. To date, Hi-C data are available only for K562 and GM06990 cells (39). Although Hi-C data from additional cell lines will become more readily available due to reduced sequencing costs, it will be a longer period before hundreds of TFs are characterized by ChIP-seq in all cell types. The ENCODE Consortium is producing large ChIP-seq data sets for numerous commonly used cell lines. However, most of these cell lines are derived from cancer cells. The analysis of primary cells and/or specific subpopulations of cells from different tissues will be problematic. Although it is possible to obtain enough material from primary cells for Hi-C, current ChIP-seq technologies use large numbers of cells, and thus, it is difficult to obtain enough primary cells for hundreds of ChIP-seq experiments. In this case, searching for putative TFBSs using motif analysis and the TRANSFAC data base (56) can serve as an alternative method to integrate chromatin structure and TF information. This approach has been used to correlate CTCF motifs with Hi-C data (70). However, it is critical to keep in mind that binding of TFs to the genome is highly influenced by certain histone modifications; a consensus motif may not be bound by a TF if the chromatin is in a closed confirmation (i.e. it is marked by H3K27me3 or H3K9me3). For example, ER-α was shown to bind to consensus motifs that are in open chromatin and marked by the histone modification H3K4me1 (64). Thus, if motifs are used to predict TF binding, it is imperative that epigenomic information be included to identify regions that are biologically relevant. Fortunately, relatively small amounts of chromatin are needed for epigenetic analyses, which can be easily performed on primary cells (Roadmap Epigenomics Project). Putative motifs can then be identified within interacting loci that fall within regions of open chromatin and are marked by H3K4me1 or H3K27ac. A TF association network can be predicted using methods such as the Apriori algorithm, a Bayesian approach, neural networks, etc. We note that a derived network using only motif information will be more complex than a network produced using experimentally determined TFBSs. Therefore, it is essential that putative TF associations be validated by downstream experimental approaches.
Pathway for Identification of Three-dimensional TF Modules
High throughput techniques such as ChIP-seq, ChIP-chip, ChIP-PET, ChIA-PET, and Hi-C can provide detailed information of genome-wide binding of TFs, histone modifications, and higher order organization of the chromatin. Integration of these different types of data has greatly facilitated our understanding of TF associations and has brought our view of transcriptional regulation from a linear paradigm to a three-dimensional model. Although such studies are in their infancy, it is clear that elucidating the relationship between TFs and chromatin interactions is essential to understand the complexity of the underlying biology of transcriptional regulation. Fig. 2 illustrates a possible workflow for a genome-wide identification of three-dimensional TF modules. In this pathway, epigenetic profiling to identify sites of DNA methylation, modified histones, and open chromatin is performed to segment the genome into different epigenomic states (Step 1). Next, the Hi-C method is used to identify all the interacting loci in the genome (Step 2). Then, the clustering of interacting loci based on epigenetic status is performed to identify distinct sets of interacting chromatin loci (Step 3). TF binding data, either predicted (Step 4A) or experimentally determined (Step 4B), are integrated with the chromatin structure information (Step 5), and computational methods such as the Apriori algorithm, Bayesian approaches, or neural networks are used to develop TF modules and association networks (Step 6). Finally, experimental validation of the predicted TF associations can be performed (Step 7). We also note that although moving from a one-dimensional to a three-dimensional model of gene regulation has been a major advance in the field, it is critical to realize that a complete understanding of transcription requires that we take into account changes caused by drug treatments, environment challenges, and time (74–80). There is increasing evidence suggesting a dynamic model of nuclear organization and gene regulation. For example, studies have shown that the interaction of TFs with chromatin can change in a cyclical manner after treatment with hormone, and there is an intensive chromatin reorganization induced by estrogen treatment of Matrigel-derived endothelial cells (69). These observations support a new transcriptional regulatory paradigm in which sets of associated TFs serve as a driving force for highly dynamic formations and dissociations of chromatin interactions, resulting in a profound impact on transcription. Thus, we look forward to the future when a series of three-dimensional TF networks can be combined to provide a four-dimensional motion picture of gene regulation and chromatin organization.
FIGURE 2.
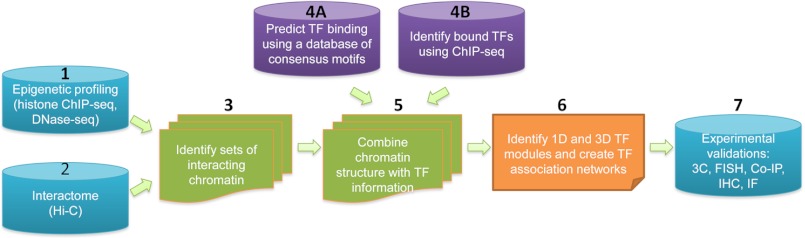
Pathway to identify one- and three-dimensional TF modules. The steps in identifying TF modules include the following: step 1, perform epigenomic profiling (histone ChIP-seq) and identify open chromatin (DNase-seq) in the cell type of interest; step 2, identify interacting chromosomal loci in the same cell type using the Hi-C method; step 3, use the epigenomic data to cluster the interacting chromosomal loci into distinct sets; step 4, either predict TF binding using a data base of TF consensus motifs (4A) or identify bound TFs using experimental ChIP-seq data (4B); step 5, integrate the chromatin structure information with the TF binding information; step 6, create one-dimensional (1D) and three-dimensional (3D) TF modules and TF association networks using computational methods such as the Apriori algorithm, a Bayesian approach, or a neural network; step 7, experimentally validate TF associations via methods such as 3C, fluorescent in situ hybridization (FISH), co-immunoprecipitation (Co-IP), immunohistochemistry (IHC), and immunofluorescence (IF).
This work was supported, in whole or in part, by National Institutes of Health Grants CA45250 and 1U54HG004558 from the United States Public Health Service (to P. J. F.). This work was also supported by the Department of Biomedical Informatics of The Ohio State University (to V. X. J.). This is the fourth article in the Thematic Minireview Series on Results from the ENCODE Project: Integrative Global Analyses of Regulatory Regions in the Human Genome.
The ENCODE Consortium (2012) Integrative analysis of the human genome. Nature, in press.
J. Dekker, personal communication.
- TF
- transcription factor
- CRE
- cis-regulatory element
- TFBS
- TF-binding site
- LMA
- multiplex ligation-mediated amplification
- ER-α
- estrogen receptor-α
- CTCF
- CCCTC-binding factor
- FDR
- false discovery rate
- PWM
- position weight matrix
- MPRM
- mixture Poisson regression model.
REFERENCES
- 1. Vaquerizas J. M., Kummerfeld S. K., Teichmann S. A., Luscombe N. M. (2009) A census of human transcription factors: function, expression, and evolution. Nat. Rev. Genet. 10, 252–263 [DOI] [PubMed] [Google Scholar]
- 2. Panne D. (2008) The enhanceosome. Curr. Opin. Struct. Biol. 18, 236–242 [DOI] [PubMed] [Google Scholar]
- 3. Maniatis T., Falvo J. V., Kim T. H., Kim T. K., Lin C. H., Parekh B. S., Wathelet M. G. (1998) Structure and function of the interferon-β enhanceosome. Cold Spring Harb. Symp. Quant. Biol. 63, 609–620 [DOI] [PubMed] [Google Scholar]
- 4. Howard M. L., Davidson E. H. (2004) cis-Regulatory control circuits in development. Dev. Biol. 271, 109–118 [DOI] [PubMed] [Google Scholar]
- 5. Göndör A., Ohlsson R. (2009) Chromosome cross-talk in three dimensions. Nature 461, 212–217 [DOI] [PubMed] [Google Scholar]
- 6. Ling J. Q., Li T., Hu J. F., Vu T. H., Chen H. L., Qiu X. W., Cherry A. M., Hoffman A. R. (2006) CTCF mediates interchromosomal co-localization between Igf2/H19 and Wsb1/Nf1. Science 312, 269–272 [DOI] [PubMed] [Google Scholar]
- 7. Osborne C. S. (2007) Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 5, e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sutherland H., Bickmore W. A. (2009) Transcription factories: gene expression in unions? Nat. Rev. Genet. 10, 457–466 [DOI] [PubMed] [Google Scholar]
- 9. Vogel M. J., Peric-Hupkes D., van Steensel B. (2007) Detection of in vivo protein-DNA interactions using DamID in mammalian cells. Nat. Protoc. 2, 1467–1478 [DOI] [PubMed] [Google Scholar]
- 10. O'Geen H., Echipare L., Farnham P. J. (2011) Using ChIP-seq technology to generate high-resolution profiles of histone modifications. Methods Mol. Biol. 791, 265–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. O'Geen H., Frietze S., Farnham P. J. (2010) Using ChIP-seq technology to identify targets of zinc finger transcription factors. Methods Mol. Biol. 649, 437–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Farnham P. J. (2009) Insights from genomic profiling of transcription factors. Nat. Rev. Genet. 10, 605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oberley M. J., Tsao J., Yau P., Farnham P. J. (2004) High throughput screening of chromatin immunoprecipitates using CpG island microarrays. Methods Enzymol. 376, 315–334 [DOI] [PubMed] [Google Scholar]
- 14. Weinmann A. S., Farnham P. J. (2002) Identification of unknown target genes of human transcription factors using chromatin immunoprecipitation. Methods 26, 37–47 [DOI] [PubMed] [Google Scholar]
- 15. Lefrançois P., Zheng W., Snyder M. (2010) ChIP-Seq using high throughput DNA sequencing for genome-wide identification of transcription factor-binding sites. Methods Enzymol. 470, 77–104 [DOI] [PubMed] [Google Scholar]
- 16. Hawkins R. D., Hon G. C., Ren B. (2010) Next-generation genomics: an integrative approach. Nat. Rev. Genet. 11, 476–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Valouev A., Johnson D. S., Sundquist A., Medina C., Anton E., Batzoglou S., Myers R. M., Sidow A. (2008) Genome-wide analysis of transcription factor-binding sites based on ChIP-seq data. Nat. Methods 5, 829–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lander E. S., Linton L. M., Birren B., Nusbaum C., Zody M. C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., Funke R., Gage D., Harris K., Heaford A., Howland J., Kann L., Lehoczky J., LeVine R., McEwan P., McKernan K., Meldrim J., Mesirov J. P., Miranda C., Morris W., Naylor J., Raymond C., Rosetti M., Santos R., Sheridan A., Sougnez C., Stange-Thomann N., Stojanovic N., Subramanian A., Wyman D., Rogers J., Sulston J., Ainscough R., Beck S., Bentley D., Burton J., Clee C., Carter N., Coulson A., Deadman R., Deloukas P., Dunham A., Dunham I., Durbin R., French L., Grafham D., Gregory S., Hubbard T., Humphray S., Hunt A., Jones M., Lloyd C., McMurray A., Matthews L., Mercer S., Milne S., Mullikin J. C., Mungall A., Plumb R., Ross M., Shownkeen R., Sims S., Waterston R. H., Wilson R. K., Hillier L. W., McPherson J. D., Marra M. A., Mardis E. R., Fulton L. A., Chinwalla A. T., Pepin K. H., Gish W. R., Chissoe S. L., Wendl M. C., Delehaunty K. D., Miner T. L., Delehaunty A., Kramer J. B., Cook L. L., Fulton R. S., Johnson D. L., Minx P. J., Clifton S. W., Hawkins T., Branscomb E., Predki P., Richardson P., Wenning S., Slezak T., Doggett N., Cheng J. F., Olsen A., Lucas S., Elkin C., Uberbacher E., Frazier M., Gibbs R. A., Muzny D. M., Scherer S. E., Bouck J. B., Sodergren E. J., Worley K. C., Rives C. M., Gorrell J. H., Metzker M. L., Naylor S. L., Kucherlapati R. S., Nelson D. L., Weinstock G. M., Sakaki Y., Fujiyama A., Hattori M., Yada T., Toyoda A., Itoh T., Kawagoe C., Watanabe H., Totoki Y., Taylor T., Weissenbach J., Heilig R., Saurin W., Artiguenave F., Brottier P., Bruls T., Pelletier E., Robert C., Wincker P., Smith D. R., Doucette-Stamm L., Rubenfield M., Weinstock K., Lee H. M., Dubois J., Rosenthal A., Platzer M., Nyakatura G., Taudien S., Rump A., Yang H., Yu J., Wang J., Huang G., Gu J., Hood L., Rowen L., Madan A., Qin S., Davis R. W., Federspiel N. A., Abola A. P., Proctor M. J., Myers R. M., Schmutz J., Dickson M., Grimwood J., Cox D. R., Olson M. V., Kaul R., Shimizu N., Kawasaki K., Minoshima S., Evans G. A., Athanasiou M., Schultz R., Roe B. A., Chen F., Pan H., Ramser J., Lehrach H., Reinhardt R., McCombie W. R., de la Bastide M., Dedhia N., Blocker H., Hornischer K., Nordsiek G., Agarwala R., Aravind L., Bailey J. A., Bateman A., Batzoglou S., Birney E., Bork P., Brown D. G., Burge C. B., Cerutti L., Chen H. C., Church D., Clamp M., Copley R. R., Doerks T., Eddy S. R., Eichler E. E., Furey T. S., Galagan J., Gilbert J. G., Harmon C., Hayashizaki Y., Haussler D., Hermjakob H., Hokamp K., Jang W., Johnson L. S., Jones T. A., Kasif S., Kaspryzk A., Kennedy S., Kent W. J., Kitts P., Koonin E. V., Korf I., Kulp D., Lancet D., Lowe T. M., McLysaght A., Mikkelsen T., Moran J. V., Mulder N., Pollara V. J., Ponting C. P., Schuler G., Schultz J., Slater G., Smit A. F., Stupka E., Szustakowski J., Thierry-Mieg D., Thierry-Mieg J., Wagner L., Wallis J., Wheeler R., Williams A., Wolf Y. I., Wolfe K. H., Yang S. P., Yeh R. F., Collins F., Guyer M. S., Peterson J., Felsenfeld A., Wetterstrand K. A., Patrinos A., Morgan M. J., de Jong P., Catanese J. J., Osoegawa K., Shizuya H., Choi S., Chen Y. J. (2001) Initial sequencing and analysis of the human genome. Nature 409, 860–921 [DOI] [PubMed] [Google Scholar]
- 19. Pollack J. R., Iyer V. R. (2002) Characterizing the physical genome. Nat. Genet. 32, 515–521 [DOI] [PubMed] [Google Scholar]
- 20. Venter J. C., Adams M. D., Myers E. W., Li P. W., Mural R. J., Sutton G. G., Smith H. O., Yandell M., Evans C. A., Holt R. A., Gocayne J. D., Amanatides P., Ballew R. M., Huson D. H., Wortman J. R., Zhang Q., Kodira C. D., Zheng X. H., Chen L., Skupski M., Subramanian G., Thomas P. D., Zhang J., Gabor Miklos G. L., Nelson C., Broder S., Clark A. G., Nadeau J., McKusick V. A., Zinder N., Levine A. J., Roberts R. J., Simon M., Slayman C., Hunkapiller M., Bolanos R., Delcher A., Dew I., Fasulo D., Flanigan M., Florea L., Halpern A., Hannenhalli S., Kravitz S., Levy S., Mobarry C., Reinert K., Remington K., Abu-Threideh J., Beasley E., Biddick K., Bonazzi V., Brandon R., Cargill M., Chandramouliswaran I., Charlab R., Chaturvedi K., Deng Z., Di Francesco V., Dunn P., Eilbeck K., Evangelista C., Gabrielian A. E., Gan W., Ge W., Gong F., Gu Z., Guan P., Heiman T. J., Higgins M. E., Ji R. R., Ke Z., Ketchum K. A., Lai Z., Lei Y., Li Z., Li J., Liang Y., Lin X., Lu F., Merkulov G. V., Milshina N., Moore H. M., Naik A. K., Narayan V. A., Neelam B., Nusskern D., Rusch D. B., Salzberg S., Shao W., Shue B., Sun J., Wang Z., Wang A., Wang X., Wang J., Wei M., Wides R., Xiao C., Yan C., Yao A., Ye J., Zhan M., Zhang W., Zhang H., Zhao Q., Zheng L., Zhong F., Zhong W., Zhu S., Zhao S., Gilbert D., Baumhueter S., Spier G., Carter C., Cravchik A., Woodage T., Ali F., An H., Awe A., Baldwin D., Baden H., Barnstead M., Barrow I., Beeson K., Busam D., Carver A., Center A., Cheng M. L., Curry L., Danaher S., Davenport L., Desilets R., Dietz S., Dodson K., Doup L., Ferriera S., Garg N., Gluecksmann A., Hart B., Haynes J., Haynes C., Heiner C., Hladun S., Hostin D., Houck J., Howland T., Ibegwam C., Johnson J., Kalush F., Kline L., Koduru S., Love A., Mann F., May D., McCawley S., McIntosh T., McMullen I., Moy M., Moy L., Murphy B., Nelson K., Pfannkoch C., Pratts E., Puri V., Qureshi H., Reardon M., Rodriguez R., Rogers Y. H., Romblad D., Ruhfel B., Scott R., Sitter C., Smallwood M., Stewart E., Strong R., Suh E., Thomas R., Tint N. N., Tse S., Vech C., Wang G., Wetter J., Williams S., Williams M., Windsor S., Winn-Deen E., Wolfe K., Zaveri J., Zaveri K., Abril J. F., Guigo R., Campbell M. J., Sjolander K. V., Karlak B., Kejariwal A., Mi H., Lazareva B., Hatton T., Narechania A., Diemer K., Muruganujan A., Guo N., Sato S., Bafna V., Istrail S., Lippert R., Schwartz R., Walenz B., Yooseph S., Allen D., Basu A., Baxendale J., Blick L., Caminha M., Carnes-Stine J., Caulk P., Chiang Y. H., Coyne M., Dahlke C., Mays A., Dombroski M., Donnelly M., Ely D., Esparham S., Fosler C., Gire H., Glanowski S., Glasser K., Glodek A., Gorokhov M., Graham K., Gropman B., Harris M., Heil J., Henderson S., Hoover J., Jennings D., Jordan C., Jordan J., Kasha J., Kagan L., Kraft C., Levitsky A., Lewis M., Liu X., Lopez J., Ma D., Majoros W., McDaniel J., Murphy S., Newman M., Nguyen T., Nguyen N., Nodell M., Pan S., Peck J., Peterson M., Rowe W., Sanders R., Scott J., Simpson M., Smith T., Sprague A., Stockwell T., Turner R., Venter E., Wang M., Wen M., Wu D., Wu M., Xia A., Zandieh A., Zhu X. (2001) The sequence of the human genome. Science 291, 1304–1351 [DOI] [PubMed] [Google Scholar]
- 21. Elnitski L., Jin V. X., Farnham P. J., Jones S. J. (2006) Locating mammalian transcription factor-binding sites: a survey of computational and experimental techniques. Genome Res. 16, 1455–1464 [DOI] [PubMed] [Google Scholar]
- 22. Iyer V. R., Horak C. E., Scafe C. S., Botstein D., Snyder M., Brown P. O. (2001) Genomic binding sites of the yeast cell cycle transcription factors SBF and MBF. Nature 409, 533–538 [DOI] [PubMed] [Google Scholar]
- 23. Kim T. H., Barrera L. O., Zheng M., Qu C., Singer M. A., Richmond T. A., Wu Y., Green R. D., Ren B. (2005) A high-resolution map of active promoters in the human genome. Nature 436, 876–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ren B., Robert F., Wyrick J. J., Aparicio O., Jennings E. G., Simon I., Zeitlinger J., Schreiber J., Hannett N., Kanin E., Volkert T. L., Wilson C. J., Bell S. P., Young R. A. (2000) Genome-wide location and function of DNA-binding proteins. Science 290, 2306–2309 [DOI] [PubMed] [Google Scholar]
- 25. Weinmann A. S., Yan P. S., Oberley M. J., Huang T. H., Farnham P. J. (2002) Isolating human transcription factor targets by coupling chromatin immunoprecipitation and CpG island microarray analysis. Genes Dev. 16, 235–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K. (2007) High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 [DOI] [PubMed] [Google Scholar]
- 27. Johnson D. S., Mortazavi A., Myers R. M., Wold B. (2007) Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497–1502 [DOI] [PubMed] [Google Scholar]
- 28. Mikkelsen T. S., Ku M., Jaffe D. B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T. K., Koche R. P., Lee W., Mendenhall E., O'Donovan A., Presser A., Russ C., Xie X., Meissner A., Wernig M., Jaenisch R., Nusbaum C., Lander E. S., Bernstein B. E. (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Robertson G., Hirst M., Bainbridge M., Bilenky M., Zhao Y., Zeng T., Euskirchen G., Bernier B., Varhol R., Delaney A., Thiessen N., Griffith O. L., He A., Marra M., Snyder M., Jones S. (2007) Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 4, 651–657 [DOI] [PubMed] [Google Scholar]
- 30. Wang Z., Zang C., Rosenfeld J. A., Schones D. E., Barski A., Cuddapah S., Cui K., Roh T. Y., Peng W., Zhang M. Q., Zhao K. (2008) Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 40, 897–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen X., Xu H., Yuan P., Fang F., Huss M., Vega V. B., Wong E., Orlov Y. L., Zhang W., Jiang J., Loh Y. H., Yeo H. C., Yeo Z. X., Narang V., Govindarajan K. R., Leong B., Shahab A., Ruan Y., Bourque G., Sung W. K., Clarke N. D., Wei C. L., Ng H. H. (2008) Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–1117 [DOI] [PubMed] [Google Scholar]
- 32. van Steensel B., Dekker J. (2010) Genomics tools for unraveling chromosome architecture. Nat. Biotechnol. 28, 1089–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dekker J., Rippe K., Dekker M., Kleckner N. (2002) Capturing chromosome conformation. Science 295, 1306–1311 [DOI] [PubMed] [Google Scholar]
- 34. Simonis M., Klous P., Splinter E., Moshkin Y., Willemsen R., de Wit E., van Steensel B., de Laat W. (2006) Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat. Genet. 38, 1348–1354 [DOI] [PubMed] [Google Scholar]
- 35. Zhao Z. (2006) Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat. Genet. 38, 1341–1347 [DOI] [PubMed] [Google Scholar]
- 36. Dostie J. (2006) Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 16, 1299–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fullwood M. J., Liu M. H., Pan Y. F., Liu J., Xu H., Mohamed Y. B., Orlov Y. L., Velkov S., Ho A., Mei P. H., Chew E. G., Huang P. Y., Welboren W. J., Han Y., Ooi H. S., Ariyaratne P. N., Vega V. B., Luo Y., Tan P. Y., Choy P. Y., Wansa K. D., Zhao B., Lim K. S., Leow S. C., Yow J. S., Joseph R., Li H., Desai K. V., Thomsen J. S., Lee Y. K., Karuturi R. K., Herve T., Bourque G., Stunnenberg H. G., Ruan X., Cacheux-Rataboul V., Sung W. K., Liu E. T., Wei C. L., Cheung E., Ruan Y. (2009) An estrogen receptor-α-bound human chromatin interactome. Nature 462, 58–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Handoko L., Xu H., Li G., Ngan C. Y., Chew E., Schnapp M., Lee C. W., Ye C., Ping J. L., Mulawadi F., Wong E., Sheng J., Zhang Y., Poh T., Chan C. S., Kunarso G., Shahab A., Bourque G., Cacheux-Rataboul V., Sung W. K., Ruan Y., Wei C. L. (2011) CTCF-mediated functional chromatin interactome in pluripotent cells. Nat. Genet. 43, 630–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lieberman-Aiden E., van Berkum N. L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B. R., Sabo P. J., Dorschner M. O., Sandstrom R., Bernstein B., Bender M. A., Groudine M., Gnirke A., Stamatoyannopoulos J., Mirny L. A., Lander E. S., Dekker J. (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S. (2008) Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rozowsky J., Euskirchen G., Auerbach R. K., Zhang Z. D., Gibson T., Bjornson R., Carriero N., Snyder M., Gerstein M. B. (2009) PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat. Biotechnol. 27, 66–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jothi R., Cuddapah S., Barski A., Cui K., Zhao K. (2008) Genome-wide identification of in vivo protein-DNA binding sites from ChIP-seq data. Nucleic Acids Res. 36, 5221–5231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Blahnik K. R., Dou L., O'Geen H., McPhillips T., Xu X., Cao A. R., Iyengar S., Nicolet C. M., Ludäscher B., Korf I., Farnham P. J. (2010) Sole-Search: an integrated analysis program for peak detection and functional annotation using ChIP-seq data. Nucleic Acids Res. 38, e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Frietze S., O'Geen H., Blahnik K. R., Jin V. X., Farnham P. J. (2010) ZNF274 recruits the histone methyltransferase SETDB1 to the 3′-ends of ZNF genes. PLoS One 5, e15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fejes A. P., Robertson G., Bilenky M., Varhol R., Bainbridge M., Jones S. J. (2008) FindPeaks 3.1: a tool for identifying areas of enrichment from massively parallel short-read sequencing technology. Bioinformatics 24, 1729–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ji H., Jiang H., Ma W., Johnson D. S., Myers R. M., Wong W. H. (2008) An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat. Biotechnol. 26, 1293–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zang C., Schones D. E., Zeng C., Cui K., Zhao K., Peng W. (2009) A clustering approach for identification of enriched domains from histone modification ChIP-seq data. Bioinformatics 25, 1952–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Z. D., Rozowsky J., Snyder M., Chang J., Gerstein M. (2008) Modeling ChIP sequencing in silico with applications. PLoS Comput. Biol. 4, e1000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Park P. J. (2009) ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 10, 669–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang X., Robertson G., Krzywinski M., Ning K., Droit A., Jones S., Gottardo R. (2011) PICS: probabilistic inference for ChIP-seq. Biometrics 67, 151–163 [DOI] [PubMed] [Google Scholar]
- 51. Lan X., Bonneville R., Apostolos J., Wu W., Jin V. X. (2011) W-ChIPeaks: a comprehensive web application tool for processing ChIP-chip and ChIP-seq data. Bioinformatics 27, 428–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Price M. A., Rogers A. E., Treisman R. (1995) Comparative analysis of the ternary complex factors Elk-1, SAP-1a, and SAP-2 (ERP/NET). EMBO J. 14, 2589–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Riechmann J. L., Krizek B. A., Meyerowitz E. M. (1996) Dimerization specificity of Arabidopsis MADS domain homeotic proteins APETALA1, APETALA3, PISTILLATA, and AGAMOUS. Proc. Natl. Acad. Sci. U.S.A. 93, 4793–4798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mead J., Bruning A. R., Gill M. K., Steiner A. M., Acton T. B., Vershon A. K. (2002) Interactions of the Mcm1 MADS box protein with cofactors that regulate mating in yeast. Mol. Cell. Biol. 22, 4607–4621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhou Q., Wong W. H. (2004) CisModule: de novo discovery of cis-regulatory modules by hierarchical mixture modeling. Proc. Natl. Acad. Sci. U.S.A. 101, 12114–12119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wingender E., Chen X., Hehl R., Karas H., Liebich I., Matys V., Meinhardt T., Prüss M., Reuter I., Schacherer F. (2000) TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 28, 316–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Blanchette M., Bataille A. R., Chen X., Poitras C., Laganière J., Lefèbvre C., Deblois G., Giguère V., Ferretti V., Bergeron D., Coulombe B., Robert F. (2006) Genome-wide computational prediction of transcriptional regulatory modules reveals new insights into human gene expression. Genome Res. 16, 656–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cai X., Hou L., Su N., Hu H., Deng M., Li X. (2010) Systematic identification of conserved motif modules in the human genome. BMC Genomics 11, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ding J., Hu H., Li X. (2012) Thousands of cis-regulatory sequence combinations are shared by Arabidopsis and poplar. Plant Physiol. 158, 145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. King D. C., Taylor J., Elnitski L., Chiaromonte F., Miller W., Hardison R. C. (2005) Evaluation of regulatory potential and conservation scores for detecting cis-regulatory modules in aligned mammalian genome sequences. Genome Res. 15, 1051–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jin V. X., Rabinovich A., Squazzo S. L., Green R., Farnham P. J. (2006) A computational genomics approach to identify cis-regulatory modules from chromatin immunoprecipitation microarray data–a case study using E2F1. Genome Res. 16, 1585–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu T., Ortiz J. A., Taing L., Meyer C. A., Lee B., Zhang Y., Shin H., Wong S. S., Ma J., Lei Y., Pape U. J., Poidinger M., Chen Y., Yeung K., Brown M., Turpaz Y., Liu X. S. (2011) Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 12, R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Joseph R., Orlov Y. L., Huss M., Sun W., Kong S. L., Ukil L., Pan Y. F., Li G., Lim M., Thomsen J. S., Ruan Y., Clarke N. D., Prabhakar S., Cheung E., Liu E. T. (2010) Integrative model of genomic factors for determining binding site selection by estrogen receptor-α. Mol. Syst. Biol. 6, 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lan X., Witt H., Katsumura K., Ye Z., Wang Q., Bresnick E. H., Farnham P. J., Jin V. X. (2012) Integration of Hi-C and ChIP-seq data reveals distinct types of chromatin linkages. Nucleic Acids Res. 10.1093/nar/gks501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yaffe E., Tanay A. (2011) Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nat. Genet. 43, 1059–1065 [DOI] [PubMed] [Google Scholar]
- 67. Tolhuis B., Palstra R. J., Splinter E., Grosveld F., de Laat W. (2002) Looping and interaction between hypersensitive sites in the active β-globin locus. Mol. Cell 10, 1453–1465 [DOI] [PubMed] [Google Scholar]
- 68. Chen Z., Zhang C., Wu D., Chen H., Rorick A., Zhang X., Wang Q. (2011) Phospho-MED1-enhanced UBE2C locus looping drives castration-resistant prostate cancer growth. EMBO J. 30, 2405–2419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hsu P. Y., Hsu H. K., Singer G. A., Yan P. S., Rodriguez B. A., Liu J. C., Weng Y. I., Deatherage D. E., Chen Z., Pereira J. S., Lopez R., Russo J., Wang Q., Lamartiniere C. A., Nephew K. P., Huang T. H. (2010) Estrogen-mediated epigenetic repression of large chromosomal regions through DNA looping. Genome Res. 20, 733–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Botta M., Haider S., Leung I. X., Lio P., Mozziconacci J. (2010) Intra- and interchromosomal interactions correlate with CTCF binding genome wide. Mol. Syst. Biol. 6, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Agrawal R., Srikant R. (1994) Fast algorithms for mining association rules in large databases. The 20th International Conference on Very Large Data Bases, pp. 487–499, Morgan Kaufmann, Los Altos, CA [Google Scholar]
- 72. Wada Y., Ohta Y., Xu M., Tsutsumi S., Minami T., Inoue K., Komura D., Kitakami J., Oshida N., Papantonis A., Izumi A., Kobayashi M., Meguro H., Kanki Y., Mimura I., Yamamoto K., Mataki C., Hamakubo T., Shirahige K., Aburatani H., Kimura H., Kodama T., Cook P. R., Ihara S. (2009) A wave of nascent transcription on activated human genes. Proc. Natl. Acad. Sci. U.S.A. 106, 18357–18361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bieda M., Xu X., Singer M. A., Green R., Farnham P. J. (2006) Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res. 16, 595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pearl J. (2000) Causality: Models, Reasoning, and Inference, Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 75. Eaton D., Ghahramani Z. (2009) Proceedings of the Twelfth International Workshop on Artificial Intelligence and Statistics, April 16–18, 2009, Clearwater Beach, FL, Vol. 5, pp. 145–152, JMLR Inc., Lafayette, IN [Google Scholar]
- 76. Tian J., Pearl J. (2001) Proceedings of the Seventeenth Conference on Uncertainty in Artificial Intelligence, August 2–5, 2001, Seattle, WA, pp. 512–521, Morgan Kaufmann Publishers, San Francisco, CA [Google Scholar]
- 77. Korb K. B., Hope L. R., Nicholson A. E., Axnick K. (2004) PRICAI 2004: Trends in Artificial Intelligence, 8th Pacific Rim International Conference on Artificial Intelligence, Auckland, New Zealand, August 9–13, 2004: Proceedings, Vol. 8, pp. 322–331, Springer, New York [Google Scholar]
- 78. Friedman N., Murphy K., Russell S. (1998) Proceedings of the Fourteenth Conference on Uncertainty in Artificial Intelligence, July 24–26, 1998, Madison, WI, pp. 139–147, Morgan Kaufmann Publishers, San Francisco, CA [Google Scholar]
- 79. Shang Y., Hu X., DiRenzo J., Lazar M. A., Brown M. (2000) Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103, 843–852 [DOI] [PubMed] [Google Scholar]
- 80. Métivier R., Penot G., Hübner M. R., Reid G., Brand H., Kos M., Gannon F. (2003) Estrogen receptor-α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115, 751–763 [DOI] [PubMed] [Google Scholar]