

Background: De novo DNA methyltransferase 3A is mutated in acute myeloid leukemia (AML) patients.
Results: AML mutations disrupt DNMT3A tetramerization and the processive methylation of human promoters in vitro.
Conclusion: DNMT3A oligomeric interfaces control processivity, which may alter methylation patterns in AML patients.
Significance: DNMT3A provides a plausible explanation for changes in epigenetic regulation in AML patients.
Keywords: DNA Methylation, Enzyme Catalysis, Epigenetics, Leukemia, Protein Complexes
Abstract
DNA methylation is a key regulator of gene expression and changes in DNA methylation occur early in tumorigenesis. Mutations in the de novo DNA methyltransferase gene, DNMT3A, frequently occur in adult acute myeloid leukemia patients with poor prognoses. Most of the mutations occur within the dimer or tetramer interface, including Arg-882. We have identified that the most prevalent mutation, R882H, and three additional mutants along the tetramer interface disrupt tetramerization. The processive methylation of multiple CpG sites is disrupted when tetramerization is eliminated. Our results provide a possible mechanism that accounts for how DNMT3A mutations may contribute to oncogenesis and its progression.
Introduction
Covalent modification of DNA by cytosine methylation is a heritable and reversible epigenetic process that regulates a diverse range of biological processes. In mammals, DNA methylation is essential to cellular differentiation, X-inactivation, imprinting, silencing of transposable elements, and gene regulation (1, 2). Cancer progression involves a mosaic of hypo- and hypermethylated genomic regions, resulting in dramatic alterations in gene expression patterns (3). DNA methylation is initially (de novo) established by DNMT3A and the closely related DNMT3B in a spatially and temporally dependent fashion (4–8). A fundamental, yet unresolved aspect of mammalian DNA methylation is the mechanisms regulating de novo methylation, including the misregulation that occurs in cancer cells.
Many proteins bind DNMT3A, the most well characterized is DNMT3-like (DNMT3L)2 (9–11). DNMT3L is an essential but inactive homolog (12, 13) and acts as a processivity factor by regulating the oligomeric state of DNMT3A (14). DNMT3L binds to DNMT3A along a surface with high structural conservation among cytosine methyltransferases to form a DNMT3L-DNMT3A-DNMT3A-DNMT3L complex (15). This interface also supports DNMT3A homotetramerization in vitro (the tetramer interface) in the absence of DNMT3L (14). DNMT3A can even further oligomerize (14, 15). Disruption of the tetramer interface has modest effects on the catalytic function of the enzyme but greatly reduces the number of methyl groups transferred by DNMT3A before it dissociates from the substrate (14).
At the opposite side of the DNMT3A catalytic domain, some 30 angstroms away in the recognition domain, the symmetrical DNMT3A homodimer interface (dimer interface) is composed of buried hydrophobic residues surrounded by electrostatic interactions with low crystallographic B-factors (see Fig. 1A) (14). The orientation of the dimer interface results in two enzyme active sites separated by approximately one helical turn in B-form DNA. One particular residue Arg-882, located in the dimer interface is mutated in 12% of adult acute myeloid leukemia (AML) patients, whereas 22% of all AML patients have mutations throughout DNMT3A, a strikingly high correlation (16). DNMT3A mutations are also found in the preform of AML, myelodysplastic syndrome (MDS). AML patients with DNMT3A mutations show poor prognoses (16), but they are more responsive to treatment with the methyltransferase inhibitor decitabine (17). The mutations in DNMT3A result in normal global in vivo methylation levels, but patients have some altered methylation patterns (16, 18). Although reports have suggested the dimer interface is essential for catalytic activity (15, 19), in vitro examination of the most common AML mutation, R882H, showed only a 2-fold reduction in the rate of methylation (20). As many of the AML mutations are found at either interface, we sought to investigate the significance of DNMT3A oligomerization and the consequences of AML mutants on the oligomerization and function of the enzyme.
FIGURE 1.

DNMT3A homodimer interface. A, DNMT3A-DNMT3L heterotetramer based on the crystal structure (Protein Data Bank code 2QRV), showing the dimer and tetramer interface; the boxed region is enlarged below. B, the dimer interface is symmetrical; shown is a close-up of half the interface and all of the center interactions. Underlined residues were mutated in this study. The interface has many ionic interactions and a few aromatic residues. Residues are colored based upon their predicted energetic contribution to the dimer interface, in ΔΔG, compared with alanine as determined using the Rosetta interface alanine scanning module. Bright yellow residues provide the greatest contribution to the DNMT3A-DNMT3A interface. Boxed is the full dimer interface showing both symmetrical sides of the dimer interface.
All interface mutants, including AML mutations at the tetramer and dimer interfaces, disrupted oligomerization. Disruption of the dimer interface decreased activity and all DNMT3A dimers show altered catalytic properties. The homotetramers carried out multiple cycles of methylation on the same piece of DNA (processive catalysis), whereas dimers had faster product release, resulting in non-processive catalysis. Our results show mutations identified in AML patients regulate the oligomeric state and thus the catalytic properties of DNMT3A. We also provide a possible mechanism for the altered methylation patterns observed in AML patients with DNMT3A mutations.
EXPERIMENTAL PROCEDURES
Cloning and Protein Purification
The catalytic domain of DNMT3A has similar kinetic parameters as the full-length enzyme, including kcat, KmDNA, KmAdoMet, processivity, and DNMT3L activation (8, 13), and was used for the DNMT3A/DNMT3L co-crystal structure (9). Both the homo- and DNMT3L hetero-oligomerization interfaces are located on the catalytic domain. The catalytic domain of DNMT3A and full-length DNMT3L were purified as stated in Holz-Schietinger et al. (21). Plasmids used for protein expression and site-directed mutagenesis include codon-optimized pET28a-hDNMT3A_CD (Δ1–611) (22), and pTYB1-3L for hDNMT3L (11). M.HhaI was purified as stated in Matje et al. (23), and EcoRV was prepared as described in Hiller et al. (24).
Mutagenesis Design and Computational Modeling
The DNMT3A dimer Protein Data Bank code 2QRV was submitted to the Rosetta computational alanine scanning server (25, 26) to evaluate the contribution of individual residues to the tetramer interface.
DNA Sequences
The DNA used as substrates include duplex poly-dIdC (∼1000 bp) from Sigma-Aldrich, linearized plasmid pCpGL (non-CpG substrate) (27), and the human promoter (RASSF1A) in plasmid pCpGL. The RASSF1A human promoter (GenBank accession number NM_007182) was amplified using primers 5′-CGCGTAGCAGTGTGAGGTAA-3′ and 5′-GAGTCCGAGTCCTCTTGGCT-3′ and then cloned into pCpGL and linearized. Lastly, fluorescent DNA (5′-fluorescently labeled deoxyoligonucleotide; GCbox30) with fluorescein (6-FAM) on the 5′ end of the top strand, (5′/6-FAM/TGGATATCTAGGGGCGCTATGATATCT-3′) duplex was purchased from Integrated DNA Technologies and HPLC-purified.
Size-exclusion Chromatography Coupled to Multiangle Light Scattering
Experiments were done as described in Holz-Schietinger et al. (14).
Native Gel Mobility DNA Shift Assays
Experiments were done as described in Holz-Schietinger et al. (14), other than samples were run on native 6% polyacrylamide gels in 0.25 × TBE, pH 7.8, at 250 V for 35 min. Gel was visualized for fluorescein using a Typhoon scanner, and data were analyzed using ImageJ software (28).
Methylation Assays
DNMT3A methylation assays measured the amount of tritiated methyl groups transferred from cofactor AdoMet to the DNA by the enzyme. Reactions were carried out at 37 °C in reaction buffer (50 mm KH2PO4/K2HPO4, 1 mm EDTA, 1 mm DTT, 0.2 mg/ml BSA, and 20 mm NaCl) at pH 7.8.
Concentration-dependent activity assays were carried out with the multiple site substrate poly-dIdC at either 500 nm base pairs or 2 μm base pairs, AdoMet was at saturation (5 μm), and DNMT3A was varied as indicated. Data were fit to a dose-response curve (Y = plateau/(1 + 10 ((logEC50 − X)*Hill coefficient))) using Prism (version 5.0, GraphPad).
All other methylation assays had a total enzyme concentration of 150 nm, which corresponds to 27 nm active enzyme (determined previously in Purdy et al. (22)). In reactions, DNA was the multiple site substrate poly-dIdC or RASSF1A promoter used at saturation (40 μm) unless indicated. AdoMet was used at saturation (5 μm) unless indicated. Processivity assays (chase assay), DNMT3L activation assays, and mathematical modeling were carried out as described in Holz-Schietinger et al. (21). DNMT3L assays had a 1:1 ratio of DNMT3A:DNMT3L with a 1-h preincubation with AdoMet.
The chase assay determines the length of time and the number of turnovers the enzyme stays associated with the multiple site DNA substrate. The chase assay begins with DNMT3A carrying out 1 to 2 turnovers for ∼20 min on poly-dIdC DNA (repeated recognition site) or the human promoter RASSFIA (1000 bp with 106 CpG sites). After this time, a 25-fold excess of pCpGL, a 3872-bp plasmid lacking recognition site (CG) for DNMT3A, is added to capture enzyme that dissociates from the substrate after carrying out catalysis. A processive enzyme will continue to methylate the original, multisite substrate after chase DNA is added, and the addition of chase DNA has little, or a delayed impact, on the rate of methylation (wild type DNMT3A, see Fig. 5A). A non-processive enzyme will dissociate after one methyl transfer; thus, after the addition of chase DNA, the enzyme will bind the excess chase DNA and cause an immediate decrease in product formation (see H873A in Fig. 5C).
FIGURE 5.

Dimer interface disrupting mutants eliminate processive catalysis. Chase assays show DNMT3A homotetramers (WT) are processive and dimers (H873A and R882H) are non-processive, A, WT; B, H873A; and C, R882H. ●, only substrate (20 μm bp RASSF1A); ■, substrate and then 400 μm bp chase (pCpGL) at 20 min; ▴, substrate and pCpGL at the start of the reaction. Minimal methylation is detected after addition of chase DNA with the dimer mutant (H873A and R882H), unlike tetramer (WT), which shows less than 10% change in activity. D, homodimers that were formed by disrupting the dimer interface resulted in enzymes that bind DNA followed by methylation and then fast dissociation.
The data were fit to a nonlinear regression (one phase decay). Data fit separately before the experimental reaction had chaser DNA added (time 0–20 min) and after the chaser DNA was added, using Prism (version 5.0). Error bars represent the mean S.D. of at least three replicates. The number of turnovers is calculated by product formed (nm) divided by catalytic active sites (nm).
For Km values, data were fit to the Michaelis-Menten equation, kcat, and processivity data were fit to either linear regression or a fit to a nonlinear regression using Prism (version 5). Error bars are S.E. from three reactions. Bar charts of kinetic values compared mutants to wild type using one-way analysis of variance to determine the p value using Prism. Mathematical modeling was conducted as described in Holz-Schietinger et al. (21).
All reactions were quenched by the addition of 500 μm AdoMet and 50 μg/ml proteinase K. Samples were spotted onto Whatman DE81 filters then washed, dried, and counted as described previously (22).
Fluorescence Anisotropy
koff values were determined as described in Holz-Schietinger et al. (14) with enzyme at 250 nm and 5′ 6-FAM-labeled GCbox30 duplex DNA at 20 nm to reach maximum anisotropy, followed by adding chase DNA (unlabeled GCbox30) at 100× concentration of labeled DNA. The decrease in anisotropy was measured with time on a PerkinElmer LS 55 fluorescence spectrometer using Fl Winlab software. Data were fit to a one-phase exponential decay (Y = span e−kt + plateau) using GraphPad, from two independent experiments.
RESULTS
Computational Assessment of Dimer Interface of DNMT3A
As a preliminary prediction of the energetic contribution of individual residues to dimerization, the DNMT3A dimer interface was analyzed using Rosetta computational interface alanine scanning to obtain approximations for the energetic contributions of individual residues to the stabilization of this interface (ΔΔG). Three residues that form a well defined network at the center of the interface, Arg-885, Asp-876, and His-873, and two residues that comprise the hydrophobic pockets, Asn-879 and Trp-860, were predicted to have the greatest contribution to the stability of the dimer interface (Fig. 1B). On the basis of our computational results, four residues were mutated to alanine and two mutants homologous to DNMT3B immunodeficiency centromeric instability and facial anomalies syndromes mutants (D876G and H873R) were prepared to evaluate their ability to disrupt the dimer interface. In addition, Arg-882 located at the dimer interface was mutated to histidine, as this mutation accounts for ∼50% of DNMT3A mutations found in AML patients. Multiple mutations were made and tested in DNMT3A to clarify the consequence of disrupting the interface from pleiotropic effects of a particular residue. The dimer interface is within the DNA binding domain so it is highly possible some mutants could have large effects on the enzyme beyond the disruption of the interface. Mutations at the dimer interface are highly correlated with disease states, so it is necessary to understand how these mutants regulate the function of DNMT3A.
Concentration-dependent Activation
The importance of oligomerization on the activity of DNMT3A was first tested by varying the concentration of enzyme and measuring the amount of product formed on the multiple site substrate (poly-dIdC); this was monitored by the transfer of tritiated methyl groups from cofactor AdoMet onto DNA. The data were fitted to a sigmoidal curve resulting in a Hill coefficient of 2.02 ± 0.22 (Table 1). A Hill coefficient greater than 1 indicates positive cooperativity (Fig. 2A). Thus, the concentration of DNMT3A (tested from 5 to 300 nm) controls the rate of methylation, implying that the rate of methylation is dependent upon the oligomerization state of the protein. This likely corresponds to the transition from either monomers and/or dimers to tetramers and higher order structures.
TABLE 1.
DNMT3A cooperativity
There is a non-linear relationship between enzyme activity and enzyme concentration at low DNA concentrations.
| [DNA]/enzyme | Hill coefficient | EC50 | R2 |
|---|---|---|---|
| 500 nm | |||
| DNMT3A | 2.02 ± 0.22 | 89.5 | 0.97 |
| DNMT3A/DNMT3L | 2.08 ± 0.18 | 80.6 | 0.98 |
| 2 μm | |||
| DNMT3A | 1.09 ± 0.17 | NA | 0.98 |
FIGURE 2.
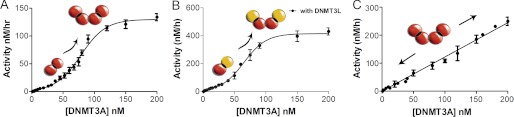
Concentration-dependent DNMT3A activation. A, the activity of DNMT3A shows a sigmoidal relationship with protein concentration. DNMT3A is more active at higher enzyme concentrations suggesting that DNMT3A oligomerization is concentration-dependent. The DNA is below the Km at 500 nm bp. B, DNMT3A with DNMT3L is more active at higher DNMT3A concentrations (DNMT3L is kept constant at saturating concentrations), suggesting that the concentration-dependent oligomerization takes place at the dimer interface (the interface that does not bind DNMT3L). The DNA is below the Km at 500 nm bp. C, the activity of DNMT3A shows a linear relationship when DNA is at over above the Km, data shown are when DNA is at 2 μm bp. Reactions took place on poly-dIdC, a multiple site substrate.
As expected, the sigmoidal curvature is observed only at low DNA concentrations below the KmDNA (500 nm). At or above the KmDNA (2 μm plus), there is a linear relationship between enzyme concentration and product formed with a Hill coefficient of 1.09 ± 0.17 (Fig. 2C); this is most likely due to the DNA affecting the oligomeric equilibria. In fact, it was shown previously that DNA facilitates the formation of the dimer interface of DNMT3A (14).
The concentration-dependent activity of DNMT3A was also tested with DNMT3L. The sigmoidal activity was seen with DNMT3A in complex with DNMT3L, resulting in a Hill coefficient of 2.08 ± 0.18 (Fig. 2B, Table 1). DNMT3L binds at the tetramer interface and activates DNMT3A (14, 15). It was observed that at low DNMT3A concentrations, DNMT3L still activates DNMT3A, demonstrating that the formation of the DNMT3A-DNMT3L tetramer interface is not controlled by protein concentration (tested from 3–400 nm). The DNMT3A-DNMT3L complex shows the same sigmoidal behavior of DNMT3A, providing strong evidence that increasing the concentration of DNMT3A promotes oligomerization along the dimer interface (where DNMT3L does not bind). The concentration-dependent formation of the dimer interface is also supported by the fact that DNA facilitates oligomerization at the dimer interface (14).
All Mutants Disrupt Tetramerization
We combined size exclusion chromatography with multiangle light scattering to evaluate the oligomeric state of the wild type DNMT3A and the interface mutants. As shown previously, our preparations of wild type DNMT3A catalytic domain have a weight-average molecular mass of 127 kDa in solution at pH 7.8, in agreement with four 36-kDa monomers forming a tetramer (Fig. 3A) (14). The light scattering peak is broad, and tailing suggests the tetrameric form is in equilibrium with both the dimeric and monomeric forms of the enzyme in the absence of DNA. Surprisingly, all mutations at the dimer interface disrupted the oligomeric state of the enzyme observed by gel filtration and multi-angle light scattering (Fig. 3A). All of the mutants tested in this study (H873A, H873R, R885A, W860A, N879A, D876G, and R882H) had weight-average molecular weights close to the theoretical dimer mass of 72 kDa (82–66 kDa) (Table 2). These dimers also eluted from the column slower than the wild type tetramers.
FIGURE 3.

Dimer interface mutants disrupt the interface resulting in dimers on DNA. A, size-exclusion chromatography of light scattering traces of tetrameric wild type catalytic domain (black trace) and representative dimeric H873A (blue trace). Molecular weights were determined from the amount of scattered light, in relation to protein concentration determined by A280. B, diagram of oligomeric mutants with and without DNA. C, EMSA of size markers, DNA (GCbox30) has binding sites for size standards, one site for M. HhaI (37 kDa), a known monomer and two binding sites for EcoRV (29 kDa), a known dimer, which creates dimer and tetramer bands. D, EMSA showed mutations at the dimer interface of DNMT3A disrupt oligomerization resulting in dimers on DNA (200 nm enzyme, 300 nm DNA). E, the wild type DNMT3A at varying concentrations (20–500 nm) migrates as a tetramer or larger on DNA (200 nm). F, the H873A mutant (20–500 nm) migrates as a dimer on DNA (200 nm) at varying concentrations.
TABLE 2.
Biophysical characterization of dimer interface mutants
Molecular weights were determined by light scattering. Light scattering data show that mutants in solution are either mostly monomers or dimers, unlike wild type enzyme, which is mostly tetrameric. The form of the enzyme on DNA was determined by gel shifts showing mutants are dimers on DNA or tetrameric and even larger structures like the wild type enzyme. Computational ΔΔG values were determined by Rosetta alanine scanning of the generated homotetramer model.
| Enzyme | Molecular mass | Gel shift | Gel shift with 3L | ΔΔG value | AML | ICF |
|---|---|---|---|---|---|---|
| kDa | ||||||
| WT | 127.1 ± 2.1 | Tetramer + | Tetramer | |||
| H873A | 68.0 ± 4.3 | Dimer | Dimer | 1.71, 1.71 | ||
| H873R | 73.8 ± 2.1 | Dimer | Dimer | Yes | ||
| W860A | 66.5 ± 2.0 | Dimer | Dimer | 2.77, 2.96 | ||
| N879A | 74.0 ± 1.4 | Dimer | Dimer | 1.23, 1.78 | ||
| R882H | 71.9 ± 2.5 | Dimer | Tetramer | Yes | ||
| D876G | 70.0 ± 1.0 | Dimer | Tetramer | 0.75, 1.71 | Yes | |
| R885A | 82.1 ± 1.2 | No binding | No binding | 0.92, 0.38 |
Electrophoretic mobility gel shift assays were used to resolve the oligomeric state of the mutants bound to DNA at concentrations closer to cellular conditions and our in vitro activity assay conditions. The gel shifts used a 30-base pair (bp) GCbox30 that contains binding sites for two well-behaved enzymes; a central GCGC site bound by the monomeric M.HhaI methyltransferase (37 kDa) and two GATATC sites bound by the homodimeric EcoRV endonuclease (29 kDa) at either end (Fig. 3C). At equimolar concentrations of protein and DNA, one or two EcoRV dimers bind to the DNA, resulting in approximate standards for dimers and tetramers, respectively, whereas M.HhaI on DNA serves as the monomer standard. The binding site for M.HhaI contains the CG sequence that is the major recognition site of DNMT3A. Fig. 3D shows the formation of discrete bands when wild type and mutant DNMT3A enzymes are incubated with fluorescently labeled GCbox30. Oligomeric mutants at the dimer interface (H873A, H873R, W860A, N879A, D876G, and R882H) shifted the DNA closest to the dimer size standard (EcoRV dimer, 58 kDa; DNMT3A dimer, 68 kDa) (Fig. 3D). The dimer interface mutants show a faint binding band, most likely due to an increase in the rate of dissociation and/or a decrease in DNA affinity (see below). No binding of R885A to DNA was detectable. Varying the concentration of wild type DNMT3A shows the wild type enzyme bound to DNA as a tetramer and higher oligomeric forms (Fig. 3E), indicated by the smear in the higher molecular mass region, creating a faint band over a large area. Fig. 3F shows that titration of H873A from 40 to 300 nm in the presence of 200 nm substrate resulted in the presence of dimeric enzyme bound to the DNA. Fig. 3B diagrams the oligomeric state of the mutants.
Disruption of Dimer Interface Reduces Activity and Results in Altered Catalytic Properties
Mutants with a disrupted dimer interface were examined for changes in their steady-state kinetic parameters by monitoring the transfer of tritiated methyl groups from cofactor AdoMet onto DNA. kcat, KmDNA, KmAdoMet, and processivity values were obtained for all the mutants and compared with the wild type enzyme (Table 3). kcat values were obtained at saturating DNA and AdoMet concentrations after determining the respective Km values.
TABLE 3.
Mutations that disrupt the dimer interface result in a loss of processivity
Dimer interface disrupting mutants have a decrease in activity (kcat), an increase in Km for DNA and AdoMet, a large increase in the rate of DNA dissociation, and a loss of processivity. kcat and Km were determined by monitoring the ability of the enzyme to incorporate tritiated methyl groups transferred from cofactor AdoMet onto DNA (poly-dIdC). koff values were determined by binding excess enzyme to 5′ FAM-6 labeled GCbox30 duplex and measuring the rate of change in anisotropy upon the addition of excess unlabeled GCbox30 (100-fold labeled DNA). Processivity n½ was determined by mathematical modeling of the activity curve.
| Substrate | poly-dIdC |
RASSF1A |
poly-dIdC |
poly-dIdC |
GCbox30 |
poly-dIdC |
RASSF1A |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| kcat | Fold ↓ | kcat | Fold ↓ | KmAdoMet | Fold ↑ | KmDNA | Fold ↑ | koff | Fold ↑ | Processivity | Fold ↓ | Processivity | Fold ↓ | |
| h−1 | h−1 | μm | μm | min−1 | ||||||||||
| WT | 3.5 ± 0.27 | 1.0 | 1.39 ± 0.06 | 1.0 | 0.20 ± 0.02 | 1.0 | 1.2 ± 0.1 | 1.0 | 0.21 ± 0.01 | 1.0 | 30 ± 4.5 | 1.0 | 15 ± 5.0 | 1.0 |
| H873A | 1.02 ± 0.16 | 3.4 | 0.60 ± 0.16 | 2.3 | 0.90 ± 0.07 | 4.4 | 4.9 ± 0.7 | 3.9 | 3.48 ± 0.29 | 16 | 2.4 ± 1.5 | 13 | 1.7 ± 0.5 | 9.1 |
| H873R | 0.26 ± 0.08 | 13 | 0.22 ± 0.09 | 6.3 | 1.17 ± 0.18 | 5.8 | 5.0 ± 0.8 | 4.0 | 4.17 ± 0.78 | 20 | 1.4 ± 0.7 | 21 | 0.9 ± 0.4 | 17 |
| W860A | 0.34 ± 0.08 | 10 | 0.31 ± 0.05 | 4.4 | 1.41 ± 0.28 | 6.9 | 4.8 ± 0.6 | 3.9 | 3.07 ± 0.19 | 14 | 2.0 ± 1.1 | 15 | 0.9 ± 0.3 | 17 |
| N879A | 0.73 ± 0.15 | 4.7 | 0.71 ± 0.04 | 1.9 | 0.81 ± 0.06 | 4.0 | 3.7 ± 0.7 | 3.0 | 1.52 ± 0.13 | 7.2 | 2.4 ± 1.7 | 12 | 2.1 ± 0.5 | 7.3 |
| R882H | 1.41 ± 0.15 | 2.5 | 0.75 ± 0.09 | 1.8 | 1.30 ± 0.14 | 6.4 | 6.6 ± 0.6 | 5.3 | 2.36 ± 0.04 | 11 | 2.2 ± 1.9 | 14 | 2.1 ± 0.7 | 7.3 |
| D876G | 1.43 ± 0.46 | 2.4 | 0.78 ± 0.14 | 1.8 | 0.57 ± 0.02 | 2.8 | 3.4 ± 0.3 | 2.7 | 1.35 ± 0.10 | 6.4 | 3.8 ± 2.2 | 7.9 | 1.9 ± 0.3 | 8.2 |
| R885A | No activity | No activity | NA | NA | 11.2 ± 2.32 | 53 | NA | NA | ||||||
The mutants with a disrupted dimer interface (H873A, H873R, W860A, N879A, D876G, and R882H) had a 2–13-fold decrease in activity (Fig. 4, A and B, and Table 3) on the human promoter RASSF1A and poly-dIdC. We observed no difference between background levels and the activity of R885A even with large amounts of enzyme, DNA, and AdoMet, consistent with previous reports. Arg-885 lies within the DNA binding domain, suggesting a possible role in DNA recognition. The AML mutant R882H had a 2.5-fold reduction in activity, also in agreement with previous studies (20). These results illustrate the need to determine the function of oligomerization along the dimer interface. The decrease in kcat for the dimer mutants is consistent with the enzyme titration curve shown in Fig. 2 that indicates smaller oligomeric states have lower activity. The KmAdoMet for wild type DNMT3A is 210 ± 20 nm (14), and dimer mutants have a 3 to 7-fold increase in KmAdoMet (Table 3). H814R of DNMT3B, homologous to H873R in DNMT3A, has significant disruption in AdoMet affinity (29).
FIGURE 4.
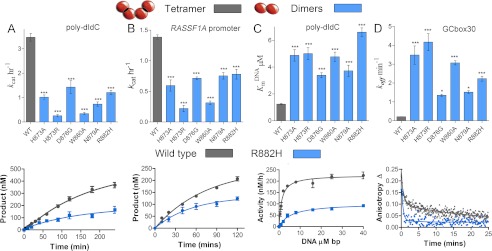
DNMT3A homodimers and homotetramers are active with different mechanisms. Disruption of the dimer interface reduces activity, but the dimers are still active with increased off-rates. The wild type tetramers are labeled gray and the dimer mutants are labeled blue. A and B, kcat values of DNMT3A wild type and oligomeric mutants were determined on multisite substrates, poly-dIdC (A), and the human promoter RASSF1A (B.). Below are the wild type (gray) and dimer mutant R882H (blue) time course trace. C, eliminating tetramer formation in DNMT3A increased KmDNA (substrate poly-dIdC), above is the bar chart of KmDNA values, and below are the kinetic fits. D, DNMT3A homodimers have an increase in off-rate compared with homotetramers, above is the bar chart of koff values determined on GCbox30, and below are the kinetic fits. All error bars were determined from at least three experiments given as S.E.; one-way analysis of variance was used to compare wild values to each mutant. *, p > 0.05; **, p > 0.01; ***, p > 0.001.
Dimers Rapidly Dissociate from DNA
Wild type DNMT3A has a KmDNAof 1.2 μm base pairs (bp) on poly-dIdC (22). Disruption of the dimer interface results in an increase in the KmDNAby 3- to 5-fold (Fig. 4C and Table 3). This observed increase in KmDNA indicates a perturbation to substrate binding and/or product dissociation in the dimer mutants. To characterize the effect of oligomerization on dissociation from DNA, dissociation rates constant (koff) were determined by binding excess enzyme to 6-FAM-labeled single sites duplex (GCbox30) and measuring the rate of change in anisotropy upon the addition of 100-fold excess unlabeled GCbox30.
Wild type DNMT3A has a koff of 0.20 min−1 as reported previously (14). The dimeric mutants dissociate 6- to 20-fold faster than the wild type enzyme (Fig. 4D and Table 3), with R882H dissociating 11-fold faster then the wild type enzyme. The Arg-882 equivalent in DNMT3B Arg-823 was previously mutated to Gly, which had a defect in dissociating from DNA but did not disrupt oligomerization. R885A, the inactive mutant, showed a 53-fold increase in koff, providing a basis for the large decrease in activity (Table 3). The large increase in koff for the dimer mutants, demonstrates that DNMT3A tetramers are productively bound to the substrate due to the increased surface area to the DNA. To directly relate the oligomeric form of the enzyme to changes in processivity, we determined their processivity on multisite substrates.
The Dimer Interface Controls Processivity
Both full-length and the catalytic domain of wild type DNMT3A act processively on several DNA substrates, including human promoters, methylating an average of 30 ± 6 cytosines before dissociating from poly-dIdC substrate (Table 4) (21). The processivity of DNMT3A was assessed on both poly-dIdC and a linear fragment of the human promoter RASSF1A using both mathematical modeling and chase experiments. The RASSF1A promoter construct is 1000-bp-long with 104 CpG sites spread throughout the sequence. Wild type DNMT3A methylates an average of 15 ± 3 cytosines on the RASSF1A promoter before dissociating (Table 3). The kinetic modeling has been used previously for DNMT1 (30) and DNMT3A (21). The processivity model was used to calculate that the dimer mutants all carry out an average of one to three turnovers before dissociating from their substrate (Table 3). The loss of processivity is due to a 4–7-fold increase in the off rate along with a decrease (2–6-fold) in the rate of methylation (Table 4), consistent with our measured kinetic parameters.
TABLE 4.
Processivity values for DNMT3A dimer and tetramer interface mutants
Disruption of either the dimer or tetramer interface eliminated processivity in DNMT3A as observed on two different substrates. The addition of DNMT3L restored processivity in mutants that become heterotetrameric; heterodimers still had a large reduction in processivity. Values were determined by modeling as described above in the “Experimental Procedures.”
| k | koff | p | n½ | |
|---|---|---|---|---|
| h−1 | h−1 | |||
| poly-dIdC | ||||
| Wild type | 11 ± 0.8 | 0.3 ± 0.0 | 0.98 | 30 ± 6.2 |
| H873A | 4.8 ± 0.1 | 1.7 ± 0.1 | 0.76 | 2.4 ± 1.3 |
| H873R | 1.7 ± 0.3 | 1.1 ± 0.3 | 0.62 | 1.4 ± 1.0 |
| W860A | 2.7 ± 0.8 | 1.1 ± 0.5 | 0.71 | 2.0 ± 1.2 |
| N879A | 4.7 ± 0.6 | 1.7 ± 0.3 | 0.75 | 2.4 ± 1.3 |
| D876G | 4.0 ± 0.3 | 1.0 ± 0.2 | 0.83 | 3.8 ± 1.5 |
| R882H | 5.6 ± 0.5 | 2.1 ± 0.2 | 0.73 | 2.2 ± 1.5 |
| RASSF1A | ||||
| Wild type | 5.1 ± 0.5 | 0.2 ± 0.1 | 0.96 | 16 ± 5.0 |
| H873A | 3.4 ± 0.4 | 1.7 ± 0.2 | 0.66 | 1.7 ± 0.5 |
| H873R | 1.4 ± 0.5 | 1.6 ± 0.8 | 0.47 | 0.9 ± 0.4 |
| W860A | 0.8 ± 0.1 | 1.2 ± 0.2 | 0.46 | 0.9 ± 0.3 |
| N879A | 2.7 ± 0.3 | 1.0 ± 0.2 | 0.72 | 2.1 ± 0.5 |
| D876G | 2.1 ± 0.3 | 0.8 ± 0.3 | 0.69 | 2.1 ± 0.7 |
| R882H | 3.9 ± 0.3 | 1.7 ± 0.2 | 0.72 | 1.9 ± 0.3 |
| WT + 3L | 18 ± 0.3 | 0.1 ± 0.0 | 1.00 | 154 ± 45 |
| H873A + 3L | 10 ± 0.7 | 1.2 ± 0.2 | 0.90 | 6.4 ± 2.2 |
| R882H + 3L | 15 ± 0.4 | 0.3 ± 0.1 | 0.98 | 54 ± 8.6 |
| R729W + 3L | 20 ± 0.9 | 0.1 ± 0.1 | 0.99 | 132 ± 35 |
We also used the chase processivity assay (21). As shown previously (21), the wild type DNMT3A had unaltered methylation activity for 90 min after the chase DNA was added, indicating the enzyme is continuing to methylate the same substrate for multiple rounds of methylation. The wild type enzyme is shown here to be processive both on poly-dIdC and the human promoter RASSF1A (Fig. 5A). The chase assay was carried out on two of the mutants that disrupt the dimer interface on both poly-dIdC and the RASSF1A promoter (R882H and H873A); they both show an immediate decrease in activity upon the addition of the chase DNA on both substrates (Fig. 5, B and C). The chase assay and the kinetic modeling show that the dimer mutants are no longer processive on either substrate (diagrammed in Fig. 5D).
DNMT3L Restores Processivity in R882H Dimer Interface Mutant
The wild type DNMT3L-DNMT3A-DNMT3A-DNMT3L heterotetramer complex forms a discrete band at the expected molecular weight when bound to the fluorescently labeled GCbox30 substrate in the electrophoretic mobility assay. As shown previously, an equal concentration of DNMT3L eliminates any wild type DNMT3A homotetramer band and results in a distinct heterotetramer band (Fig. 6A). Incubation of DNMT3L with all of the dimer interface mutants, except R882H, results in the formation of a dimeric DNMT3A-DNMT3L complex at the appropriate location on the gel (Fig. 6A). These results provide clear evidence that the dimer interface is disrupted, and DNMT3L that binds at the tetramer interface is replacing the DNMT3A at the tetramer interface. However, the addition of DNMT3L results unexpectedly in a heterotetramer with R882H, indicating that DNMT3L facilities the formation of the dimer interface, which is only disrupted weakly by R882H (Fig. 6A).
FIGURE 6.

DNMT3L and the formation of heterotetramers restores processivity to R882H DNMT3A. A, the EMSA shows that DNMT3L binds to DNMT3A tetramers to form heterotetramers. R882H binds to DNMT3L; all other mutants at the dimer interface become heterodimers. B, DNMT3L (1:1 ratio) activates DNMT3A tetramers at ∼5-fold. Dimer interface mutants show a 2–4-fold activation other than R882H, which show an 8.8-fold stimulation. DNMT3A heterotetramers (C, WT; D, R882H) are processive, and heterodimers (E, H873A) are non-processive as demonstrated by the processive chase assay. ●, only substrate (20 μm bp RASSF1A); ■, substrate and then 400 μm bp chase (pCpGL at 20 min; ▴, substrate and pCpGL at the start of the reaction.
DNMT3L activates the catalytic activity of DNMT3A 5-fold at a 1:1 ratio in vitro (11, 21) and in vivo (31) and increases processivity 3-fold (21). We observed the homodimers had a 2–5-fold increase in activity when incubated with equimolar or greater concentrations of DNMT3L (Fig. 6B and Table 5). R885A is not activated by DNMT3L. In contrast, DNMT3L activated R882H 7-fold, most likely by stabilizing the heterodimeric form (Table 5).
TABLE 5.
Functional characterization of DNMT3A dimer interface mutants with DNMT3L, showing DNMT3L restores processivity in the R882H mutant
Values were determined as stated in Table 1 but with DNMT3L preincubated with enzyme for 1 h at 37 °C in reaction buffer with AdoMet at a 1:1 ratio of DNMT3A to DNMT3L. Fold stimulation were determined by the fold change in methyl group transferred in 1 h comparing with and without DNMT3L.
| Activation | Fold ↑ | koff | Fold ↑ | Processivity | Fold ↓ | Processive | |
|---|---|---|---|---|---|---|---|
| min−1 | n½ | ||||||
| WT + 3L | 5.4 ± 0.2 | 1 | 0.17 ± 0.01 | 1.0 | 154 ± 45.2 | 1.0 | Yes |
| H873A + 3L | 3.7 ± 0.5 | 0.68 | 2.25 ± 0.25 | 13.4 | 6.4 ± 2.19 | 24.0 | Some |
| H873R + 3L | 3.1 ± 0.6 | 0.58 | |||||
| W860A + 3L | 2.9 ± 0.5 | 0.54 | 2.16 ± 0.34 | 12.9 | |||
| N879A + 3L | 2.2 ± 0.2 | 0.41 | |||||
| R882H + 3L | 8.9 ± 0.8 | 1.65 | 0.43 ± 0.07 | 2.6 | 54 ± 8.6 | 2.9 | Yes |
| D876G + 3L | 3.7 ± 0.2 | 0.68 | |||||
| R885A + 3L | No activity |
As shown above, the DNMT3A dimer interface mutants have a large increase in koff relative to wild type enzyme. Previously, we showed that the addition of DNMT3L to wild type DNMT3A caused the koff to be decreased 25%, from 0.2 min−1 to 0.15 min−1 (Table 5) (14). DNMT3L also decreased the dissociation rate for the dimer interface mutants by 1.5-fold, other than R882H, which showed a 5.5-fold reduction (Table 5). The large reduction in the dissociation rate for R882H can be explained by DNMT3L partially facilitating the formation of the dimer interface.
We directly tested whether DNMT3L could restore processivity by carrying out the processivity assay with the dimer interface mutants H873A and R882H with DNMT3L. DNMT3L increases wild type DNMT3A processivity 3-fold, shown with the chase assay (Fig. 6C) and modeling the activity curve (Table 4). R882H, which forms a heterotetramer with DNMT3L, had its processivity restored with the addition of DNMT3L, as shown with the chase assay (Fig. 6D compared with Fig. 5B). Processivity modeling indicates R882H with DNMT3L is 4-fold less processive than the wild type enzyme with DNMT3L (Table 4). On the other hand, H873A with DNMT3L show an immediate decrease in activity when chase DNA was added, indicating DNMT3L does not restore processivity in the dimer interface mutant (Fig. 6E compared with Fig. 5C). These results were expected as DNMT3L binds to the tetramer interface, and the gel shifts indicate the dimer interface is most likely still disrupted. Processivity modeling of H873A with DNMT3L indicates that the enzyme is slightly processive, with six methyl groups transferred per binding event, but still a 5-fold reduction compared with wild type DNMT3A alone and a 25-fold reduction compared with the wild type DNMT3A with DNMT3L (Table 4). Our processivity results with DNMT3L are consistent with the change in koff measured with the addition of DNMT3L and the formation of tetramers.
AML/MDS Mutations at Tetramer Interface also Disrupt Processivity
The majority of the AML and MDS patients have a mutation at the dimer interface, but multiple mutations also occur at the tetramer interface (Fig. 7A). We tested three AML or MDS mutants, R771L, R729W, and R736H, at the tetramer interface to see whether there were underlying functional or structural similarities between AML oligomeric mutants. We found all three of these AML mutants disrupt the tetramer interface, as observed by forming discrete dimers in gel shifts (Fig. 7B). R771L had a 1.8-fold increase in activity, and R729W had a 1.2-fold decrease in activity compared with wild type enzyme; these are relatively small deviations from the wild type enzyme (Fig. 7C and Table 6). They both show an 8- to 15-fold increase in the Km for DNA and 2-fold increase in KmAdoMet (Table 6). R736H is a highly conserved residue and showed a large reduction in activity (8-fold), (Fig. 7C), a 7-fold increase in KmDNA, and a 7-fold increase in KmAdoMet (Table 6). The equivalent residue (Arg-106) in the bacterial DNA methyltransferase (M.HhhaI) when mutated to alanine reduced activity by 4-fold (32). When the equivalent residue of Arg-736 in the bacterial DNA methyltransferase (M.HhaI-Arg-106) was mutated to alanine, there was a 4-fold reduction in activity.
FIGURE 7.

AML mutants disrupt the tetramer interface and eliminate DNMT3A processivity. A, mutants identified in AML and MDS patients located along DNMT3A tetramer interface. B, AML mutants at the tetramer interface disrupt the homotetramer and form heterotetramer with DNMT3L. C, the rate of catalysis is minimally changed for two of the tetramer interface disrupting mutants (R771L and R729W). D, disruption of the tetramer interface increases the off-rate. E and F, disruption of the tetramer interface also eliminates processivity, as demonstrated by the processive chase assay (E, R771A; F, R729W). G and H, DNMT3L restores processivity by forming a heterotetramer (G, R771A; H, R729W). The chase assay was done as follows: ●, only substrate (20 μm bp RASSF1A); ■, substrate and then 400 μm bp chase (pCpGL) at 20 min; ▴, substrate and pCpGL at the start of the reaction.
TABLE 6.
Tetramer interface disrupting mutants found in AML and MDS patients show full activity but altered processivity
kcat and Km values were determined by monitoring the ability of the enzyme to incorporate tritiated methyl groups transferred from cofactor AdoMet onto DNA (poly-dIdC). koff values were determined by binding excess enzyme to 5′ FAM-6-labeled GCbox30 duplex and measuring the rate of change in anisotropy upon the addition of excess unlabeled GCbox30 (100-fold labeled DNA). Processivity was determined from the chase assay.
| kcat | Fold ↑ | KmAdoMet | Fold ↑ | KmDNA | Fold ↑ | koff | Fold ↑ | Processive | |
|---|---|---|---|---|---|---|---|---|---|
| h−1 | μm | μm bp | min−1 | ||||||
| WT | 3.5 ± 0.3 | 1.0 | 0.20 ± 0.02 | 1.0 | 1.2 ± 0.1 | 1.0 | 0.21 ± 0.01 | 1.0 | Yes |
| R771L | 6 ± 0.5 | 1.7 | 0.62 ± 0.09 | 3.1 | 16.3 ± 1.4 | 13.1 | 0.96 ± 0.05 | 4.5 | No |
| R729W | 2.9 ± 0.35 | 0.8 | 0.58 ± 0.09 | 2.8 | 8.4 ± 1.2 | 6.8 | 1.87 ± 0.20 | 8.8 | No |
| R736H | 0.68 ± 0.04 | 0.2 | 1.40 ± 0.40 | 6.9 | 21.5 ± 6.7 | 17.4 | 1.22 ± 0.10 | 5.8 | No |
| R882H | 1.41 ± 0.15 | 0.4 | 1.30 ± 0.14 | 6.4 | 6.6 ± 0.6 | 5.3 | 2.36 ± 0.04 | 11.1 | No |
Mutations at the tetramer interface were also tested to determine whether disruption of processivity is a common alteration for the AML mutants. All the tetramer interface disrupting mutants had a 4.5–8.8-fold increase in the dissociation rate (Table 6 and Fig. 7D). The chase assay showed R771L and R729W (R736H was not tested) lost the ability to carry out processive catalysis (Fig. 7, E and F). The tetramer interface mutants with DNMT3L all result in heterotetramer formation (Fig. 7B). R771L and R729W showed a 7–9-fold activation by DNMT3L and, oddly, R736H shows minimal activation (Table 7). When DNMT3L was added to R771L and R729W the dissociation rates were restored to 1.4–2.3-fold wild type levels (Table 7). DNMT3L also resulted in a restoration of processivity for R771L and R729W as shown with the chase assay (Fig. 7, G and H). Our data indicate that oligomeric mutants at either the dimer or tetrameric interface retain activity but lose the ability to carry out processive catalysis.
TABLE 7.
AML and MDS mutants including tetramer interface mutants with DNMT3L
| Activation | Fold ↑ | koff | Fold ↑ | Processivity | Fold ↓ | Processive | |
|---|---|---|---|---|---|---|---|
| min−1 | n½ | ||||||
| WT + 3L | 5.4 ± 0.2 | 1.0 | 0.2 ± 0.01 | 1.0 | 154 ± 45.2 | 1.0 | Yes |
| R771L + 3L | 7.5 ± 0.1 | 1.4 | 0.5 ± 0.03 | 2.2 | 132 ± 35.2 | 1.2 | Yes |
| R729W + 3L | 6.9 ± 0.8 | 1.3 | 0.3 ± 0.02 | 1.4 | 112 ± 22.3 | 1.4 | Yes |
| R736H + 3L | 1.5 ± 0.1 | 0.3 | 0.5 ± 0.03 | 2.3 | Not tested | Not tested | |
| R882H + 3L | 8.9 ± 0.8 | 1.6 | 0.4 ± 2.57 | 2.6 | 53.9 ± 8.6 | 2.9 | Yes |
DISCUSSION
DNA methylation patterning plays an essential role in transcriptional regulation and is known to change during cancer progression (33, 34). However, the mechanisms that control the enzymatic activity and specificity of the human DNA methyltransferases are not understood. We identified a novel function associated with higher-order DNMT3A complexes that provides a plausible explanation for how mutations at the two oligomeric interfaces of DNMT3A lead to adverse outcomes in AML patients (16, 18, 20). Oligomerization at both the DNMT3A homo- and hetero- interfaces control the catalytic properties of DNMT3A. Disruption of the inner dimer interface decreases activity, but the resultant dimers are still active and dissociate from DNA more rapidly. These dimers lose the ability to processively methylate CpG sites in human promoter sequences, which is observed with the wild type homotetramer and DNMT3A:DNMT3L heterotetramer.
Recent reports identified mutations in DNMT3A in adult AML patients with significantly worse outcome (16). R882, located at the dimer interface is a mutational hotspot and another ∼10% are dispersed throughout the tetramer interface (16, 35). It is not understood how these mutations alter DNMT3A function, although it is currently thought to be a loss of function phenotype (16, 18, 20, 35–37). However, recurrent mutations at a single amino acid position suggest an alteration to enzyme function with selective advantage for the cancer cell, while widely divergent mutations at many positions in a gene generally suggest loss-of-function, a pattern seen for many classic tumor genes, including p53 (16, 38). Our results show the AML and MDS mutations disrupt DNMT3A oligomerization, which is consistent with the model that the oligomeric state of DNMT3A is important to tumorigenesis. Disruptions of either the dimer or tetramer interface result in active enzymes with altered catalytic properties. These results are consistent with the findings that patients with DNMT3A mutations have unchanged global DNA methylation levels (16). Overexpression of DNMT3A has been reported in AML patients (39), and knock-out of DNMT3A inhibits tumor cell proliferation and metastasis (40, 41). DNA methyltransferase inhibitors are used successfully to treat MDS and AML patients (42–44). In fact, AML patients with DNMT3A mutations have significantly better response rate to the DNA methyltransferase inhibitor (decitabine) (17) than patients lacking these mutations, making it hard to understand how the loss of DNMT3A function would decrease patient survival, as these inhibitors slow tumor growth. Fundamentally, all clinical evidence suggests DNMT3A mutations are affecting cancer progression by a mechanism other than loss of function.
The AML patients with DNMT3A mutations show alterations in methylation and gene expression patterns, including a loss of methylation at clustered CpG sites found in CpG islands (16, 20). We show that all mutations that result in DNMT3A dimers cause a ∼10-fold increase in the rate at which the mutants dissociate from DNA, eliminating the ability of the enzyme to stay associated with the DNA to methylate multiple sites. We propose that DNMT3A mutations alter methylation patterns by eliminating the ability of the enzyme to processively methylate clustered sites. Processive catalysis and its regulation by accessory proteins are critical to the action of several DNA modifying enzymes (45, 46). Processivity is controlled in DNMT3A by self-oligomerization at both the dimer and tetramer interface (14). The dimer interface is at the DNA target-recognition domain, and thus oligomerization doubles the DNA binding surface (15). It is well established that increasing the surface area of the enzyme bound to the substrate can enhance processivity in other DNA modifying enzymes by decreasing product release and facilitating translocation (47–49).
The control of the processivity of DNMT3A is relevant to the establishment of DNA methylation, as the degree of 5mC content in promoter regions or coding regions has important consequences for transcriptional regulation (1, 50). Methylation of CpG sites in human promoters controls transcriptional activation via binary, all-or-nothing patterning (51, 52) that would be expected because DNMT1 (30, 53, 54) and both DNMT3A and DNMT3B (18, 21, 31) are processive enzymes. Disruption of enzyme processivity would be expected to disrupt promoter patterning and subsequent transcriptional regulation.
It also is likely that the DNMT3A mutations disrupt the interactions with binding partners that are needed for DNTM3A genomic localization and/or regulation of enzyme activity. DNMT3L binding at the tetramer interface increases DNMT3A processivity and is essential in early development (12, 15, 21). We show that DNMT3L restores processivity in the AML mutants, including the R882H dimer interface mutant, by forming heterotetramers. All other mutations at the dimer interface result in DNMT3A-DNMT3L heterodimers. Although DNMT3L is only present in early development, the oligomeric state of DNMT3A may be modulated by the many known binding proteins, including thymine-DNA glycosylase (55) and EVI1 (ecotropic viral integration site 1) (56). These partners bind DNMT3A at unknown interfaces within the catalytic domain and misregulation of both EVI1 and thymine-DNA glycosylase play key roles in cancer progression (57–60). DNMT3B also binds DNMT3A, possibly at either interface with unknown affects on patients with DNMT3A mutations. Furthermore, the binding of large homo-DNMT3A complexes to nucleosomes could be modulated by these AML mutations (19). Because both interfaces of DNMT3A are necessary for heterochromatic localization, AML patients with DNMT3A mutations may have increased methylation in euchromatic targets (19). In addition, 20% of the DNMT3A AML mutations are outside the catalytic domain, in regions known to interact with other proteins (16).
The mutations found in DNMT3A highlight the importance of higher-order complexes in the regulation of DNMT3A activity and illustrate our need to better understand the consequences and regulation of DNMT3A homo- and heterocomplexes. Our data suggest the loss-of-function model cannot explain the observed correlations between DNMT3A mutations and carcinoma. It is likely that multiple mechanisms are causing altered DNA methylation patterns by DNMT3A, including changing the catalytic properties of the enzyme, its processivity, and disrupting binding partner interactions. The dimer interface of DNMT3A is dynamic and changes with protein concentration, DNA, tetramer interface occupation, DNMT3L, pH, and somatic mutations. Further work is needed to identify and understand the mechanisms by which binding partners (including possibly regulatory RNAs), mutations, and cellular conditions drive oligomerization of DNMT3A and the resultant cellular phenotypes.
Footnotes
- DNMT3L
- DNMT3-like
- AML
- acute myeloid leukemia
- MDS
- myelodysplastic syndrome
- AdoMet
- S-adenosylmethionine
- poly-dIdC
- poly(deoxyinosinic-deoxycytidylic).
REFERENCES
- 1. Bird A. (2002) DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21 [DOI] [PubMed] [Google Scholar]
- 2. Reik W., Dean W., Walter J. (2001) Epigenetic reprogramming in mammalian development. Science 293, 1089–1093 [DOI] [PubMed] [Google Scholar]
- 3. Robertson K. D. (2005) DNA methylation and human disease. Nat. Rev. Genet. 6, 597–610 [DOI] [PubMed] [Google Scholar]
- 4. Okano M., Bell D. W., Haber D. A., Li E. (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257 [DOI] [PubMed] [Google Scholar]
- 5. Kaneda M., Okano M., Hata K., Sado T., Tsujimoto N., Li E., Sasaki H. (2004) Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429, 900–903 [DOI] [PubMed] [Google Scholar]
- 6. Hsieh C. L. (1999) In vivo activity of murine de novo methyltransferases, Dnmt3a and Dnmt3b. Mol. Cell. Biol. 19, 8211–8218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hsieh C. L. (2005) The de novo methylation activity of Dnmt3a is distinctly different than that of Dnmt1. BMC Biochem. 6, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen T., Ueda Y., Dodge J. E., Wang Z., Li E. (2003) Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 23, 5594–5605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chedin F., Lieber M. R., Hsieh C. L. (2002) The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc. Natl. Acad. Sci. U.S.A. 99, 16916–16921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suetake I., Shinozaki F., Miyagawa J., Takeshima H., Tajima S. (2004) DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J. Biol. Chem. 279, 27816–27823 [DOI] [PubMed] [Google Scholar]
- 11. Kareta M. S., Botello Z. M., Ennis J. J., Chou C., Chédin F. (2006) Reconstitution and mechanism of the stimulation of de novo methylation by human DNMT3L. J. Biol. Chem. 281, 25893–25902 [DOI] [PubMed] [Google Scholar]
- 12. Bourc'his D., Xu G. L., Lin C. S., Bollman B., Bestor T. H. (2001) Dnmt3L and the establishment of maternal genomic imprints. Science 294, 2536–2539 [DOI] [PubMed] [Google Scholar]
- 13. Hata K., Okano M., Lei H., Li E. (2002) Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 129, 1983–1993 [DOI] [PubMed] [Google Scholar]
- 14. Holz-Schietinger C., Matje D. M., Harrison M. F., Reich N. O. (2011) Oligomerization of DNMT3A controls the mechanism of de novo DNA methylation. J. Biol. Chem. 286, 41479–41488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jia D., Jurkowska R. Z., Zhang X., Jeltsch A., Cheng X. (2007) Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 449, 248–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ley T. J., Ding L., Walter M. J., McLellan M. D., Lamprecht T., Larson D. E., Kandoth C, Payton J. E., Baty J., Welch J., Harris C. C., Lichti C. F., Townsend R. R., Fulton R. S., Dooling D. J., Koboldt D. C., Schmidt H., Zhang Q., Osborne J. R., Lin L., O'Laughlin M., McMichael J. F., Delehaunty K. D., McGrath S. D., Fulton L. A., Magrini V. J., Vickery T. L., Hundal J., Cook L. L., Conyers J. J., Swift G. W., Reed J. P., Alldredge P. A., Wylie T., Walker J., Kalicki J., Watson M. A., Heath S., Shannon W. D., Varghese N., Nagarajan R., Westervelt P., Tomasson M. H., Link D. C., Graubert T. A., DiPersio J. F., Mardis E. R., Wilson R. K. (2010) DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 363, 2424–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Metzeler K. H., Walker A., Geyer S., Garzon R., Klisovic R. B., Bloomfield C. D., Blum W., Marcucci G. (2012) DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia 26, 128–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shah M. Y., Licht J. D. (2011) DNMT3A mutations in acute myeloid leukemia. Nat. Genet. 43, 289–290 [DOI] [PubMed] [Google Scholar]
- 19. Jurkowska R. Z., Rajavelu A., Anspach N., Urbanke C., Jankevicius G., Ragozin S., Nellen W., Jeltsch A. (2011) Oligomerization and binding of the Dnmt3a DNA methyltransferase to parallel DNA molecules: Heterochromatic localization and role of Dnmt3L. J. Biol. Chem. 286, 24200–24207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan X. J., Xu J., Gu Z. H., Pan C. M., Lu G., Shen Y., Shi J. Y., Zhu Y. M., Tang L., Zhang X. W., Liang W. X., Mi J. Q., Song H. D., Li K. Q., Chen Z., Chen S. J. (2011) Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 43, 309–315 [DOI] [PubMed] [Google Scholar]
- 21. Holz-Schietinger C., Reich N. O. (2010) The inherent processivity of the human de novo methyltransferase 3A (DNMT3A) is enhanced by DNMT3L. J. Biol. Chem. 285, 29091–29100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Purdy M. M., Holz-Schietinger C., Reich N. O. (2010) Identification of a second DNA binding site in human DNA methyltransferase 3A by substrate inhibition and domain deletion. Arch. Biochem. Biophys. 498, 13–22 [DOI] [PubMed] [Google Scholar]
- 23. Matje D. M., Coughlin D. F., Connolly B. A., Dahlquist F. W., Reich N. O. (2011) Determinants of precatalytic conformational transitions in the DNA cytosine methyltransferase M.HhaI. Biochemistry 50, 1465–1473 [DOI] [PubMed] [Google Scholar]
- 24. Hiller D. A., Fogg J. M., Martin A. M., Beechem J. M., Reich N. O., Perona J. J. (2003) Simultaneous DNA binding and bending by EcoRV endonuclease observed by real-time fluorescence. Biochemistry 42, 14375–14385 [DOI] [PubMed] [Google Scholar]
- 25. Kortemme T., Baker D. (2002) A simple physical model for binding energy hot spots in protein-protein complexes. Proc. Natl. Acad. Sci. U.S.A. 99, 14116–14121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kortemme T., Kim D. E., Baker D. (2004) Computational alanine scanning of protein-protein interfaces. Sci. STKE 2004, pl2. [DOI] [PubMed] [Google Scholar]
- 27. Klug M., Rehli M. (2006) Functional analysis of promoter CpG methylation using a CpG-free luciferase reporter vector. Epigenetics 1, 127–130 [DOI] [PubMed] [Google Scholar]
- 28. Abramoff M. D., Magelhaes P. J., Ram S. J. (2004) Biophotonics International 11, 36–42 [Google Scholar]
- 29. Moarefi A. H., Chédin F. (2011) ICF syndrome mutations cause a broad spectrum of biochemical defects in DNMT3B-mediated de novo DNA methylation. J. Mol. Biol. 409, 758–772 [DOI] [PubMed] [Google Scholar]
- 30. Svedruzic Z. M., Reich N. O. (2005) Mechanism of allosteric regulation of Dnmt1's processivity. Biochemistry 44, 14977–14988 [DOI] [PubMed] [Google Scholar]
- 31. Wienholz B. L., Kareta M. S., Moarefi A. H., Gordon C. A., Ginno P. A., Chédin F. (2010) DNMT3L modulates significant and distinct flanking sequence preference for DNA methylation by DNMT3A and DNMT3B in vivo. PLOS Genet. 6, e1001106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sharma V., Youngblood B., Reich N. (2005) Residues distal from the active site that alter enzyme function in M.HhaI DNA cytosine methyltransferase. J. Biomol. Struct. Dyn. 22, 533–543 [DOI] [PubMed] [Google Scholar]
- 33. Robertson K. D. (2001) DNA methylation, methyltransferases, and cancer. Oncogene 20, 3139–3155 [DOI] [PubMed] [Google Scholar]
- 34. Bestor T. H. (2000) The DNA methyltransferases of mammals. Hum. Mol. Genet. 9, 2395–2402 [DOI] [PubMed] [Google Scholar]
- 35. Walter M. J., Ding L., Shen D., Shao J., Grillot M., McLellan M., Fulton R., Schmidt H., Kalicki-Veizer J., O'Laughlin M., Kandoth C., Baty J., Westervelt P., DiPersio J. F., Mardis E. R., Wilson R. K., Ley T. J., Graubert T. A. (2011) Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 25, 1153–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Markova J., Michkova P., Burckova K., Brezinova J., Michalova K., Dohnalova A., Maaloufova J. S., Soukup P., Vitek A., Cetkovsky P., Schwarz J. (2012) Prognostic impact of DNMT3A mutations in patients with intermediate cytogenetic risk profile acute myeloid leukemia. Eur. J. Haematol. 88, 128–135 [DOI] [PubMed] [Google Scholar]
- 37. Thol F., Heuser M., Damm F., Klusmann J. H., Reinhardt K., Reinhardt D. (2011) DNMT3A mutations are rare in childhood acute myeloid leukemia. Haematologica 96, 1238–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hollstein M., Sidransky D., Vogelstein B., Harris C. C. (1991) p53 mutations in human cancers. Science 253, 49–53 [DOI] [PubMed] [Google Scholar]
- 39. Mizuno S., Chijiwa T., Okamura T., Akashi K., Fukumaki Y., Niho Y., Sasaki H. (2001) Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood 97, 1172–1179 [DOI] [PubMed] [Google Scholar]
- 40. Deng T., Kuang Y., Wang L., Li J., Wang Z., Fei J. (2009) An essential role for DNA methyltransferase 3a in melanoma tumorigenesis. Biochem. Biophys. Res. Commun. 387, 611–616 [DOI] [PubMed] [Google Scholar]
- 41. Zhao Z., Wu Q., Cheng J., Qiu X., Zhang J., Fan H. (2010) Depletion of DNMT3A suppressed cell proliferation and restored PTEN in hepatocellular carcinoma cell. J. Biomed. Biotechnol. 2010, 737535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cashen A. F., Schiller G. J., O'Donnell M. R., DiPersio J. F. (2010) Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J. Clin. Oncol. 28, 556–561 [DOI] [PubMed] [Google Scholar]
- 43. Chitambar C. R., Libnoch J. A., Matthaeus W. G., Ash R. C., Ritch P. S., Anderson T. (1991) Evaluation of continuous infusion low-dose 5-azacytidine in the treatment of myelodysplastic syndromes. Am. J. Hematol. 37, 100–104 [DOI] [PubMed] [Google Scholar]
- 44. Kantarjian H., Issa J. P., Rosenfeld C. S., Bennett J. M., Albitar M., DiPersio J., Klimek V., Slack J., de Castro C., Ravandi F., Helmer R., 3rd, Shen L., Nimer S. D., Leavitt R., Raza A., Saba H. (2006) Decitabine improves patient outcomes in myelodysplastic syndromes: Results of a phase III randomized study. Cancer 106, 1794–1803 [DOI] [PubMed] [Google Scholar]
- 45. Chelico L., Pham P., Calabrese P., Goodman M. F. (2006) APOBEC3G DNA deaminase acts processively 3′ → 5′ on single-stranded DNA. Nat. Struct. Mol. Biol. 13, 392–399 [DOI] [PubMed] [Google Scholar]
- 46. Walstrom K. M., Dozono J. M., von Hippel P. H. (1997) Kinetics of the RNA-DNA helicase activity of Escherichia coli transcription termination factor rho. 2. Processivity, ATP consumption, and RNA binding. Biochemistry 36, 7993–8004 [DOI] [PubMed] [Google Scholar]
- 47. Serebrov V., Beran R. K., Pyle A. M. (2009) Establishing a mechanistic basis for the large kinetic steps of the NS3 helicase. J. Biol. Chem. 284, 2512–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Symmons M. F., Jones G. H., Luisi B. F. (2000) A duplicated fold is the structural basis for polynucleotide phosphorylase catalytic activity, processivity, and regulation. Structure 8, 1215–1226 [DOI] [PubMed] [Google Scholar]
- 49. Nudler E., Avetissova E., Markovtsov V., Goldfarb A. (1996) Transcription processivity: Protein-DNA interactions holding together the elongation complex. Science 273, 211–217 [DOI] [PubMed] [Google Scholar]
- 50. Ball M. P., Li J. B., Gao Y., Lee J. H., LeProust E. M., Park I. H., Xie B., Daley G. Q., Church G. M. (2009) Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 27, 361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Grunau C., Hindermann W., Rosenthal A. (2000) Large-scale methylation analysis of human genomic DNA reveals tissue-specific differences between the methylation profiles of genes and pseudogenes. Hum. Mol. Genet. 9, 2651–2663 [DOI] [PubMed] [Google Scholar]
- 52. Strichman-Almashanu L. Z., Lee R. S., Onyango P. O., Perlman E., Flam F., Frieman M. B., Feinberg A. P. (2002) A genome-wide screen for normally methylated human CpG islands that can identify novel imprinted genes. Genome Res. 12, 543–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vilkaitis G., Suetake I., Klimasauskas S., Tajima S. (2005) Processive methylation of hemimethylated CpG sites by mouse Dnmt1 DNA methyltransferase. J. Biol. Chem. 280, 64–72 [DOI] [PubMed] [Google Scholar]
- 54. Svedruži Ž. M. (2011) Dnmt1 structure and function. Prog Mol. Biol. Transl. Sci. 101, 221–254 [DOI] [PubMed] [Google Scholar]
- 55. Li Y. Q., Zhou P. Z., Zheng X. D., Walsh C. P., Xu G. L. (2007) Association of Dnmt3a and thymine DNA glycosylase links DNA methylation with base-excision repair. Nucleic Acids Res. 35, 390–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Senyuk V., Premanand K., Xu P., Qian Z., Nucifora G. (2011) The oncoprotein EVI1 and the DNA methyltransferase Dnmt3 cooperate in binding and de novo methylation of target DNA. PloS One 6, e20793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Barjesteh van Waalwijk van Doorn-Khosrovani S., Erpelinck C., van Putten W. L., Valk P. J., van der Poel-van de Luytgaarde S., Hack R., Slater R., Smit E. M., Beverloo H. B., Verhoef G., Verdonck L. F., Ossenkoppele G. J., Sonneveld P., de Greef G. E., Löwenberg B., Delwel R. (2003) High EVI1 expression predicts poor survival in acute myeloid leukemia: A study of 319 de novo AML patients. Blood 101, 837–845 [DOI] [PubMed] [Google Scholar]
- 58. Kennedy R. D., D'Andrea A. D. (2006) DNA repair pathways in clinical practice: Lessons from pediatric cancer susceptibility syndromes. J. Clin. Oncol. 24, 3799–3808 [DOI] [PubMed] [Google Scholar]
- 59. Lugthart S., Figueroa M. E., Bindels E., Skrabanek L., Valk P. J., Li Y., Meyer S., Erpelinck-Verschueren C., Greally J., Löwenberg B., Melnick A., Delwel R. (2011) Aberrant DNA hypermethylation signature in acute myeloid leukemia directed by EVI1. Blood 117, 234–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tadokoro Y., Ema H., Okano M., Li E., Nakauchi H. (2007) De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J. Exp. Med. 204, 715–722 [DOI] [PMC free article] [PubMed] [Google Scholar]