

Background: Helicobacter pylori VacA receptor(s) responsible for apoptotic cell death and autophagy has not been identified.
Results: VacA-induced autophagy via low-density lipoprotein receptor-related protein-1 (LRP-1) binding precedes apoptosis.
Conclusion: LRP1 mediates VacA-induced autophagy and apoptosis.
Significance: This study identified LRP1 as a VacA receptor associated with toxin-induced autophagy and apoptosis and demonstrated its importance in the processes.
Keywords: Apoptosis, Autophagy, Cell Biology, Receptors, Toxins, Helicobacter pylori, LRP1, Vacuolating Cytotoxin (VacA)
Abstract
In Helicobacter pylori infection, vacuolating cytotoxin (VacA)-induced mitochondrial damage leading to apoptosis is believed to be a major cause of cell death. It has also been proposed that VacA-induced autophagy serves as a host mechanism to limit toxin-induced cellular damage. Apoptosis and autophagy are two dynamic and opposing processes that must be balanced to regulate cell death and survival. Here we identify the low-density lipoprotein receptor-related protein-1 (LRP1) as the VacA receptor for toxin-induced autophagy in the gastric epithelial cell line AZ-521, and show that VacA internalization through binding to LRP1 regulates the autophagic process including generation of LC3-II from LC3-I, which is involved in formation of autophagosomes and autolysosomes. Knockdown of LRP1 and Atg5 inhibited generation of LC3-II as well as cleavage of PARP, a marker of apoptosis, in response to VacA, whereas caspase inhibitor, benzyloxycarbonyl-VAD-fluoromethylketone (Z-VAD-fmk), and necroptosis inhibitor, Necrostatin-1, did not inhibit VacA-induced autophagy, suggesting that VacA-induced autophagy via LRP1 binding precedes apoptosis. Other VacA receptors such as RPTPα, RPTPβ, and fibronectin did not affect VacA-induced autophagy or apoptosis. Therefore, we propose that the cell surface receptor, LRP1, mediates VacA-induced autophagy and apoptosis.
Introduction
Helicobacter pylori colonizes more than half the world's population. Although persistent infection by H. pylori is accepted as a major cause of gastroduodenal diseases (e.g. peptic ulcer disease, gastric lymphoma, gastric adenocarcinoma), the responsible cellular pathways have not been defined. Variation in manifestations of H. pylori infection in different populations suggests differences in virulence of strains, host genetic susceptibility, and responses to environmental factors. Many H. pylori strains isolated from patients contain the cagA gene (cytotoxin-associated gene A) as well as produce the vacuolating cytotoxin, VacA. Additional H. pylori products, including urease, OipA, adhesins, heat-shock protein, and lipopolysaccharide appear to be involved in virulence (1, 2).
Interestingly, VacA causes epithelial damage in mouse models both when given orally as a single agent (3) and when delivered by a toxigenic strain of H. pylori during gastric infection (4, 5). In vitro, VacA is internalized by endocytosis (6), which is inhibited by CagA (7, 8), and exerts multiple effects on susceptible cells, including vacuolation and mitochondrial damage, leading eventually to apoptosis (9–13). In addition, VacA forms hexametric pores, followed by endocytosis and processing into late-endosomal compartments (14), which then undergo osmotic swelling to become large acidic vacuoles. Although vacuolation is the most obvious effect of VacA in vitro, it is not as obvious in vivo. The pleiotropic effects of VacA appear to result from activation of different signal transduction pathways through binding to several epithelial cell receptors, e.g. receptor protein-tyrosine phosphatase (RPTP)3 β and α (15, 16), fibronectin (FN) (17), sphingomyelin (18).
VacA enhanced tyrosine phosphorylation of the G protein-coupled receptor kinase-interactor 1 (Git1) as did pleiotrophin, an endogenous ligand of RPTPβ (19). Oral administration of VacA to wild-type mice, but not to RPTPβ knock-out mice, resulted in gastric ulcer. However, cells lacking RPTPβ were able to internalize VacA and undergo vacuolation (20), suggesting that other VacA receptors were responsible for vacuolation. Recent interest has focused on the immunosuppressive effects of VacA, i.e. VacA inhibited proliferation of T cells due to down-regulation of interleukin-2 (IL-2) transcription (21, 22). Through interactions with the β2-integrin subunit CD18 of the leukocyte-specific integrin LFA-1 (23), VacA plays an important role in inhibition of interleukin-2 (IL-2) gene expression after clathrin-independent endocytosis via PKC-dependent phosphorylation of the cytoplasmic tail of CD18 (24). Thus, VacA has effects on both epithelial cells (25) as well as inflammatory cells (26).
Over the last 10 years, studies have focused on the mechanism of cell death resulting from mitochondrial damage caused by VacA (10, 12, 13, 27). Additional recent studies have shown that VacA induces autophagy, but the pathway has not been identified (28, 29). Autophagy can promote the survival of dying cells (30). However, increased autophagic activity can also lead to cell death (31–35), suggesting that autophagy can be responsible for both cytoprotective and cytotoxic activities, depending on the specific cellular conditions.
Here we purified from AZ-521 cells, a human gastric epithelial cell line, a surface membrane protein, p500, which binds VacA, and identified it as low-density lipoprotein receptor-related protein-1 (LRP1). LRP1 binding of VacA was shown to be specifically responsible for VacA-induced autophagy and apoptosis. Similar to RPTPα and RPTPβ, LRP1 mediates VacA internalization in AZ-521 cells, but in contrast to RPTPα and RPTPβ, LRP1 targeted downstream pathways leading to autophagy and apoptosis.
EXPERIMENTAL PROCEDURES
Antibodies and Other Reagents
Anti-LC3B, anti-cleaved caspase-7, anti-cleaved PARP, anti-Beclin-1, and anti-mammalian target of rapamycin antibodies were from Cell Signaling. Mouse monoclonal antibodies reactive with LRP1 (8G1) were from Santa Cruz Biotechnologies; those reactive with RPTPβ were from BD Biosciences; and those reactive with LC3 (clone 1703) were from Cosmo Bio. Anti-RPTPβ antibody was raised against its extracellular domain, corresponding to the N-terminal amino acids of the human protein (36). Anti-RPTPα rabbit polyclonal antibodies for immunoblotting were provided by Dr. Jan Sap and anti-RPTPα rabbit polyclonal antibodies for immunofluorescence experiments were raised against its extracellular domain, corresponding to the N-terminal amino acids of the human protein; mouse monoclonal antibodies reactive with α-tubulin, necrostatin-1, and 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB) were from Sigma. Diamidino-2-phenylindole dihydrochloride (DAPI) and 4,4′-diisothiocyanostibene-2,2′-disulfonic acid (DIDS) were from Invitrogen. A general caspase inhibitor, Z-VAD-fmk was from BD Pharmingen. 3-Methyladenine was from MP Biomedicals.
Cell Culture and Gene Silencing
AZ-521 cells, a human gastric cancer cell line obtained from the Japan Health Sciences Foundation, were cultured in Earle's minimal essential medium (Sigma) containing 10% fetal calf serum. AGS cells, a human gastric cancer cell line, were cultured in RPMI1640 (Sigma) containing 10% fetal calf serum. Cells were plated into 24-well dishes (5 × 104 cells/well) or 12-well dishes (1 × 105 cells/well) in Earle's minimal essential medium containing 10% FCS. RNA interference-mediated gene knockdown was performed using validated Qiagen HP small-interfering RNAs (siRNAs) for mammalian target of rapamycin (SI00300244). The validated LRP1 siRNA was purchased from Ambion. Beclin-1 siRNA was designed and validated as described by Høyer-Hansen et al. (37). Atg5 siRNAs (Atg5-1, agugaacaucugagcuacccggaua; Atg5-2, caaucccauccagaguugcuuguga) were designed and validated as described by Yang et al. (38). RPTPβ siRNA (5′-gcacaagaaucgauaacaua-3′) and RPTPα siRNA (5′-cgaagagaauacagacuau-3′) were synthesized by B-Bridge. Negative-control siRNAs were purchased from Sigma. AZ-521 cells were transfected with 100 nm of the indicated siRNAs for 48–72 h using LipofectamineTM RNAiMax transfection reagent (Invitrogen) according to the manufacturer's protocol. Knockdown of the target proteins was confirmed by immunoblotting with the indicated antibodies.
RPTPα shRNA Expression Vector Construction and Transfection
The three highest scoring shRNA sequences targeted for human RPTPα were chosen by B-Bridge International, Inc.: RPTPα siRNA1, 5′-cggcagaaccagttaaaga-3′; RPTPα siRNA2, 5′-gcaccaacattcagcccaa-3′; RPTPα siRNA3, 5′-ggagaatggcagacgacaa-3′. The shRNA negative control, obtained from B-Bridge International, Inc. (Tokyo, Japan), has no homology to any human mRNA sequences in the NCBI Reference Sequence Database. We used the pSH1-H1-H1-Puro shRNA Lentiviral Expression System (SBI Inc.) to generate lentivirus supernatants from HEK293FT cells. In brief, HEK293FT cells were seeded in 10-cm dishes at 5 × 106 cells/dish. After cells reached 90–95% confluence, the constructed shRNA expression vector (3 μg/dish) in ViraPower Packaging Mix (9 μg/dish) with Lipofectamine 2000 (Invitrogen Inc.) was transfected into HEK293FT cells. Twelve hours after initiating transfection, the plasmid/Lipofectamine solution was removed, and cell growth medium without antibiotics was added. The lentivirus-containing supernatants were harvested 48 and 72 h post-transfection. The AZ-521 cells were plated to 30–50% confluence and transfected with appropriate dilutions of lentivirus supernatants. 24 h after transfection, the cells were cultured in cell growth medium containing puromycin (0.5 μg/ml) to obtain the stable, transfected AZ-521 cells. After several selections, we isolated AZ-521 cells with knockdown of endogenous RPTPα.
Purification of VacA
The toxin-producing H. pylori strain ATCC 49503 was the source of VacA for purification as previously described (36).
Assay for Vacuolating Activity
Vacuolating activity was assessed using AZ-521 cells as previously described (36). Briefly, cells (1 × 104 cells/well, 100 μl) were grown as monolayers in 96-well culture plates for 24 h in a 5% CO2 atmosphere at 37 °C. VacA was added, and cells were incubated at 37 °C for the indicated times. To quantify vacuolating activity, the uptake of neutral red into vacuoles was determined.
Preparation of Alexa 555-labeled VacA
To investigate VacA binding to cells and co-localization with other proteins in cells, VacA was labeled using the Alexa Fluor 555 Protein Labeling Kit (Molecular Probes), according to instructions provided by the manufacturer. In brief, 50 μl of 1 m sodium bicarbonate buffer (pH 8.5) were added to 500 μl (500 μg in phosphate-buffered saline (PBS)) of VacA, followed by incubation with the reactive dye in the vial for 15 min at room temperature. To remove excess dye, the reaction mixture was applied to a PD-10 column (Amersham Biosciences). Alexa 555-labeled VacA (100 μg/ml) was stored at −20 °C.
Purification and Identification of p500
To purify p500 using affinity columns, AZ521 cells (5 × 107 cells) were washed twice with PBS, and suspended in 10 ml of Sol buffer containing 50 mm Tris-HCl (pH 7.5), 100 mm NaCl, 10% glycerol, 1% Triton X-100, with protease inhibitor mixture (Roche Diagnostics)) for 15 min on ice. After centrifugation (20 min at 17,400 × g), the supernatant was filtered (0.45 μm, Millipore) and the filtrate (10 ml) applied to a Maackia amurensis (MAA)-agarose column (2 ml bed volume, Seikagaku Corporation). After washing the column, Sol buffer containing 50 mm ethylenediamine was used to elute the carbohydrate-containing proteins in 1-ml fractions. To confirm the presence of p500 in the eluted fractions, proteins in effluents were detected by lectin blotting using MAA as described previously (15, 16). To identify p500, proteins in effluents were precipitated with chloroform/methanol, then heated at 100 °C for 10 min in 1× SDS-PAGE sample buffer, separated in 6% gels, and transferred to PVDF membranes, which were stained with Coomassie Brilliant Blue. The stained bands were used for LC-MS/MS analysis.
Immunoprecipitation
Immunoprecipitation of VacA-binding proteins from AZ521 cells was performed as described previously. In brief, biotinylated AZ521 cell lysates (100 μg/200 μl) were incubated at 4 °C for 1 h with 1 μg of native VacA or heat-inactivated VacA (100 °C, 10 min), followed by incubation overnight at 4 °C with 1 μl of rabbit anti-VacA antibodies. Antibody-bound proteins were collected after addition of 20 μl of rProtein G-agarose (Invitrogen), 50% (v/v) in Sol buffer, and incubated at 4 °C for 1.5 h. After the beads were washed three times with Sol buffer, proteins were solubilized in SDS-PAGE sample buffer, resolved by SDS-PAGE, and transferred to PVDF membranes (Millipore; Immobilon-P membranes), which were incubated with streptavidin-HRP (Amersham Biosciences). Biotinylated proteins were detected using the enhanced chemiluminescence system (Pierce).
Immunofluorescence Confocal Microscopy
For immunofluorescence analysis of VacA co-localization with LRP1, RPTPα, RPTPβ, or LC3B, AZ-521 cells (1 × 105 cells) on coverglass (Matsunami) were incubated with 120 nm Alexa 555-labeled VacA for the indicated times, cells were fixed with 4% paraformaldehyde (PFA) at room temperature for 15 min, washed with PBS twice, and then immediately permeabilized with ice-cold 100% methanol for 10 min at −20 °C. The cells are then rinsed three times with PBS and incubated with blocking buffer (5% goat serum, 0.3% Triton X-100 in PBS) at room temperature for 1 h. To visualize LRP1 (8G1 antibody, 1:50), RPTPα (antibody provided by Jan Sap, 1:100), RPTPβ (polyclonal, 1:250), or LC3B (D11, 1:200), cells were further incubated with the primary antibodies in 1% BSA/PBS buffer overnight at 4 °C, washed twice with PBS and incubated with anti-rabbit Alexa 488 (Molecular Probes), anti-mouse 488 (Molecular Probes), or anti-mouse Cy5 (Jackson ImmunoResearch Laboratories Inc.) antibodies at room temperature for 1 h in the dark. After washing with PBS three times, cells were mounted on glass slides using Prolong Gold Antifade reagent with DAPI. For staining the lysosomal compartment in VacA-treated cells, cells were incubated with 100 nm LysoTracker Red DND-99 (Molecular Probes) according to the instruction manual, before fixation with 4% paraformaldehyde. Colocalization of VacA and the indicated proteins was analyzed by FV10i-LIV confocal microscopy (Olympus). The images were arranged with Adobe Photoshop CS4.
Statistics
Densitometric analysis on the immunoblots was done by Image Gauge software (FUJI FILM). The p values for densitometric analysis and vacuolating assay were determined by Student's t test with GraphPad Prism software (GraphPad, San Diego, CA). p values of <0.05 were considered statistically significant.
RESULTS
Purification and Identification of p500
Our analysis of membrane proteins that bind VacA revealed three proteins, i.e. RPTPα, RPTPβ, and an unidentified p500. The latter protein had a molecular mass higher than RPTPβ and reacted with MAA lectin (15, 16). In the present study, we purified p500 using MAA-agarose column chromatography and identified it by LC-MS/MS as LRP1 (Fig. 1). We confirmed its association with native VacA by immunoprecipitation (Fig. 1).
FIGURE 1.
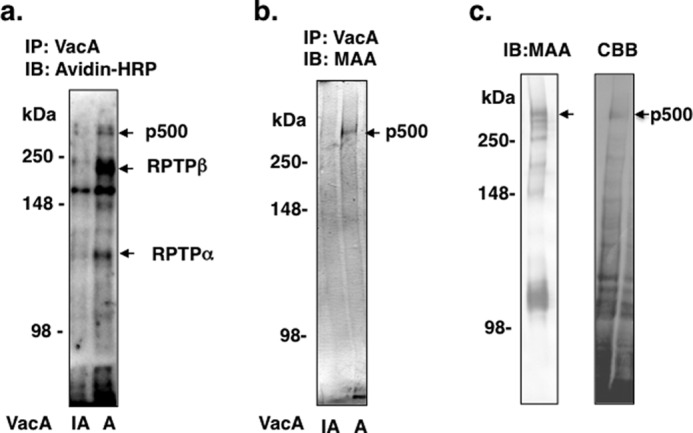
Purification of p500 from AZ-521 cells by MAA-agarose column. a, after biotinylation of surface proteins, AZ-521 cells were solubilized and immunoprecipitated with heat-inactivated (IA) or wild-type VacA (A) as described under “Experimental Procedures.” Immunocomplexes were separated by SDS-PAGE in 6% gels and transferred to PVDF membranes. VacA-binding proteins were detected with streptavidin-HRP. b, proteins immunoprecipitated (IP) with heat-inactivated or wild-type VacA were separated by SDS-PAGE in 6% gels and transferred to PVDF membranes, which were incubated with MAA-lectin conjugated to digoxigenin and then with anti-digoxigenin Fab fragments conjugated to alkaline phosphatase, followed by reaction with 4-nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate. c, biotinylated AZ-521 cell lysates were incubated overnight with a MAA-agarose column (2 ml bed volume), which was washed with 20 ml of Sol buffer. Bound proteins were eluted, concentrated, and separated by SDS-PAGE as described under “Experimental Procedures.” MAA-lectin blotting is shown in the left panel and Coomassie Brilliant Blue (CBB) staining in the right panel. The stained p500 protein band was hydrolyzed with trypsin and subjected to LC-MS/MS analysis. The procedures described in a-c were repeated at least three times with similar results. IB, immunoblot.
LRP1 Mediates VacA Binding and Internalization in AZ-521 Cells
Confocal microscopy analysis revealed that in AZ-521 cells VacA colocalized with LRP1 on cell membranes, and was internalized, whereas heat-inactivated VacA did not show colocalization and internalization with LRP1 (data not shown) (Fig. 2A). Furthermore, AZ-521 cells transfected with siRNA of LRP1 did not show significant toxin binding resulting in internalization, suggesting that LRP1 mediates VacA binding to the cell surface and facilitates its internalization. In agreement with these data, silencing of the p500 gene inhibited vacuole formation caused by VacA (Fig. 2B). These results suggest that LRP1 is associated with toxin internalization.
FIGURE 2.

LRP1-dependent VacA internalization and vacuolation in AZ-521 cells. A, confocal microscopic analysis of VacA binding to AZ-521 cells via LRP1. Nontargeting (NC) or LRP1 siRNA-transfected AZ-521 cells were incubated with Alexa 555-labeled VacA (red) for 30 min at 4 °C or for 1 h at 37 °C, fixed with 4% paraformaldehyde, and reacted with anti-LRP1 antibodies (green) as described under “Experimental Procedures.” The nuclei were stained with DAPI. A merged picture shows co-localization of VacA and LRP1 in AZ-521 cells. Bars represent 20 μm. Experiments were repeated two times with similar results. B, silencing of LRP1 gene inhibited VacA-induced vacuolation. The indicated siRNA-transfected AZ-521 cells were incubated with 120 nm heat-inactivated (IA) or wild-type VacA (A) for 18 h at 37 °C. Vacuolating activity was evaluated by neutral red uptake assay as described under “Experimental Procedures.” Data are presented as mean ± S.D. and significance is (*) p < 0.01 (n = 3). Experiments were repeated three times with similar results.
VacA Induced Generation of LC3-II in an LRP1-dependent Manner
Based on the prior reports (28, 29) that VacA induced autophagy in AGS cells, we determined whether VacA induced LC3-II generation from LC3-I in AZ-521 cells. Consistent with previous findings, Western blot analysis showed that VacA induced LC3-II generation from LC3-I in a time-dependent manner (Fig. 3a). As expected, immunoblots of VacA-treated cells transfected with control siRNA indicated a progressive conversion over 10 h of LC3-I to LC3-II. In LRP1 siRNA-transfected cells, LRP1 expression was down-regulated after 4 h with VacA and conversion of LC3-II from LC3-I was suppressed (Fig. 3b and supplemental Fig. S1). These data suggest an important role of LRP1 in mediating autophagy in AZ-521 cells in response to VacA.
FIGURE 3.

VacA induced generation of LC3-II in an LRP1-dependent manner. a, AZ-521 cells were incubated with 120 nm heat-inactivated (IA) or wild-type VacA (A) for the indicated time points and harvested for immunoblotting (IB) with the indicated antibodies. Quantification of VacA-induced LC3-II levels in AZ-521 cells was performed by densitometry (bottom panel). Data are presented as mean ± S.D. of values from two experiments. Experiments were repeated two times with similar results. b, the indicated siRNA-transfected AZ-521 cells were incubated with 120 nm heat-inactivated or wild-type VacA for 4–5 h at 37 °C and the cell lysates were subjected to immunoblotting with the indicated antibodies. α-Tubulin served as a loading control. Quantification of VacA-induced LC3-II levels in AZ-521 cells was performed by densitometry (bottom panel). Data are presented as mean ± S.D. and significance is (*) p < 0.01 (n = 4). Experiments were repeated four times with similar results.
VacA Induced Formation of Autophagosomes and Autolysosomes in AZ-521 Cells
To determine whether VacA induces autophagic vacuoles, AZ-521 cells were incubated with 120 nm VacA. We microscopically observed that active VacA (A) is sufficient to trigger autophagic vacuoles such as autophagosomes containing LC3-II after a 4-h incubation, followed after by 12 h incubation by formation of autolysosomes as detected by LysoTracker (Fig. 4A). Cells incubated with heat-inactivated VacA (IA) showed low or undetectable levels of these autophagic vacuoles after 12 h incubation. Furthermore, confocal microscopy analysis showed that intracellular VacA partially co-localized with LC3-II and LRP1, consistent with the conclusion that LRP1 plays an important role in VacA-induced autophagosome formation. However, LRP1 knockdown with siRNA suppressed VacA co-localization with LC3-II, suggesting that LRP1 is essential for formation of autophagosomes in response to VacA (Fig. 4B).
FIGURE 4.

VacA induced formation of autophagic vacuoles in AZ-521 cells via LRP1. A, VacA-induced formation of autophagosomes and autolysosomes in AZ-521 cells. AZ-521 cells were incubated with 120 nm heat-inactivated (IA) or wild-type VacA (A) for the indicated time points and fixed for immunofluorescence staining with LC3B (green) antibodies as described under “Experimental Procedures.” The acidic autophagolysosomes were stained by LysoTracker, as described under “Experimental Procedures.” A merged picture shows co-localization in AZ-521 cells. The nuclei were stained with DAPI. Bars represent 20 μm. Experiments were repeated two times with similar results. B, induction of autophagy by VacA in an LRP1-dependent manner. The indicated siRNA-transfected AZ-521 cells were incubated with 120 nm Alexa 555-labeled VacA (red) for 10 h at 37 °C and fixed for immunofluorescence staining with anti-LC3B (green) or anti-LRP1 (blue) antibodies as described under “Experimental Procedures.” A merged picture shows co-localization in AZ-521 cells. The nuclei were stained with DAPI. Bars represent 20 μm. Experiments were repeated two times with similar results.
Vacuoles Caused by VacA Are Characterized as Autophagosomes and Autophagolysosomes
Confocal microscope visualization of LC3-II, VacA, and LRP1 revealed that vacuoles caused by VacA are of at least two different types; one type consists of autophagic vacuoles such as autophagosomes and autophagolysosomes and the second type lacks LC3-II (Fig. 5A). These observations support previous findings that VacA-dependent autophagosomes and large vacuoles are distinct intracellular compartments and autophagy is independent of the formation of large vacuoles by VacA (29). Interestingly, some vacuoles observed with RPTPβ revealed small light vacuoles without LC3-II (Fig. 5B) and dense vacuoles with RPTPα were devoid of LC3-II (Fig. 5C). Although little is known about the physiological importance of the autophagy-dependent degradation of mitochondria (mitophagy) (39), several studies have suggested that PINK1/parkin-dependent mitophagy selectively degrades mitochondria (40), implying that mitophagy contributes to mitochondrial quality control. As shown in Fig. 5D, after 10 h incubation mitochondria were not observed in vacuoles with LC3-II. Furthermore, recent studies revealed that p62 binds to LC3 on the autophagosome membrane to target aggregates to autophagosomes for degradation (41). After 24 h incubation, VacA, not heat-inactivated VacA, induced formation of puncta, which were colocalized with LC3-II and p62 (Fig. 5E).
FIGURE 5.


Various vacuoles formed by VacA. A, small autophagic vacuoles induced by VacA contain LC3-II, LRP1, and toxin: AZ-521 cells were incubated with 120 nm Alexa 555-labeled VacA (red) for 10 h at 37 °C and fixed for immunofluorescence staining with anti-LC3B (green), or anti-LRP1 (blue) antibodies or the nuclei were stained with DAPI as described under “Experimental Procedures.” A merged picture shows co-localization in AZ-521 cells. Solid arrows show VacA, LC3B, and LRP1 colocalization to puncta. Bars represent 20 μm. Experiments were repeated two times with similar results. B, VacA-induced light vacuoles contain toxin and RPTPβ, and are different from autophagic vacuoles: AZ-521 cells were treated with 120 nm Alexa 555-labeled VacA (red) as similar to above. Cells were fixed and stained for anti-LC3B (blue), anti-RPTPβ (green), and with DAPI. A merged picture shows co-localization in AZ-521 cells. Solid arrows show VacA and RPTPβ colocalization to puncta. Bars represent 20 μm. Experiments were repeated two times with similar results. C, VacA-induced dense vacuoles contain toxin and RPTPα, and are different from autophagic vacuoles: AZ-521 cells were treated with 120 nm Alexa 555-labeled VacA (red) as similar to above. Cells were fixed and stained for anti-LC3B (blue), anti-RPTPα (green), and with DAPI. A merged picture shows co-localization in AZ-521 cells. Solid arrows show VacA and RPTPα colocalization to puncta. Bars represent 20 μm. Experiments were repeated two times with similar results. D, VacA-induced autophagic vacuoles do not contain functional mitochondria: AZ-521 cells were treated with 120 nm heat-inactivated (IA) or native VacA (A) for 10 h at 37 °C and 100 nm MitoTracker (red) was added to cells before fixation as described under “Experimental Procedures.” Cells were stained for anti-LC3B (green), anti-p62 (green), and with DAPI. Bars represent 20 μm. Experiments were repeated two times with similar results. E, VacA induced p62 generation in a time-dependent manner. AZ-521 cells were treated with 120 nm heat-inactivated or native VacA for the indicated time points at 37 °C. Cells were fixed and stained for anti-LC3B (red) and with DAPI. Merged and higher magnification images of the outlined areas are shown. Bars represent 20 μm. Experiments were repeated two times with similar results.
Among VacA-binding Proteins, LRP1, but Not RPTPs and FN, Mediates VacA-dependent Autophagy
To assess which VacA-binding proteins were responsible for VacA-induced autophagy, we examined the effect of silencing and knockout of the genes for RPTPβ, RPTPα, and fibronectin. Although LRP1 silencing blocked VacA-stimulated generation of LC3-II as shown in Fig. 3b, silencing these other genes did not show a similar effect, suggesting that only LRP1 may be critical for VacA-induced autophagy (Fig. 6).
FIGURE 6.

Among VacA-binding proteins, LRP1, but not RPTPs and FN, mediates VacA-dependent autophagy. a, the indicated siRNA-transfected AZ-521 cells were incubated with 120 nm heat-inactivated (IA) or wild-type VacA (A) for 4–5 h at 37 °C and the cell lysates were subjected to immunoblotting (IB) with the indicated antibodies. The knockdown levels of RPTPβ or RPTPα were detected by the antibodies (right panel). α-Tubulin served as a loading control. b, the indicated NC or RPTPβ siRNA-transfected AZ-521 cells were incubated with 120 nm heat-inactivated or wild-type VacA for 4–5 h at 37 °C and the cell lysates were subjected to immunoblotting with anti-LC3B or anti-FN antibodies. α-Tubulin served as a loading control. c, AZ-521 cells were transfected with NC or FN siRNA and treated with heat-inactivated or wild-type VacA for 4–5 h at 37 °C. Cell lysates were subjected to immunoblotting with the indicated antibodies. α-Tubulin served as a loading control. A blot representative of at least three separate experiments is shown.
LRP-1, but Not RPTPs, Mediates Cleavage of Caspase-7 and PARP Caused by VacA
Excessive autophagy can cause cell death (34, 42). Furthermore, VacA-induced cell death may occur through a programmed necrosis pathway in a caspase-independent process in AZ-521 cells (27). Therefore, we examined whether VacA-induced cell death resulted from autophagy via an LRP1-dependent pathway. Western blot analysis showed that LRP1 silencing blocked VacA-induced generation of LC3-II as well as cleavages of effector caspase-7 and PARP, suggesting that VacA binding to LRP-1 is responsible for not only autophagy but also for apoptosis in AZ-521 cells (Fig. 7).
FIGURE 7.

LRP1, but not RPTPs, mediates VacA-dependent cleavage of caspase-7 and PARP. a, NC or LRP1 siRNA-transfected AZ-521 cells were incubated with 120 nm heat-inactivated (IA) or wild-type VacA (A) for 6 h at 37 °C and the cell lysates were subjected to immunoblotting (IB) with anti-LC3B, anti-cleaved PARP, anti-cleaved caspase-7, or anti-LRP1 antibodies. α-Tubulin served as a loading control. A blot representative of four separate experiments is shown. Quantification of VacA-induced cleavage of PARP (cPARP) or caspase-7 (cCas7) levels in the indicated siRNA-transfected AZ-521 cells was performed by densitometry (bottom panel). Data are presented as mean ± S.D. and significance is (*) p < 0.01 (n = 4) and (**) p < 0.03 (n = 4). b, the indicated siRNA-transfected AZ-521 cells were incubated with 120 nm heat-inactivated or wild-type VacA for 6 h at 37 °C and the cell lysates were subjected to immunoblotting with anti-LC3B, anti-cleaved PARP, anti-RPTPα, or anti-RPTPβ antibodies. α-Tubulin served as a loading control. A blot representative of three separate experiments is shown.
Effects of Atg5 Silencing, Z-VAD-fmk and Necrostatin-1 on VacA-induced LC3-II Production and Cleavage of PARP
To further examine the link between autophagy and apoptosis, the effects of Atg5 silencing with siRNA, general caspase inhibitor (Z-VAD-fmk) and RIPK inhibitor (Necrostatin-1) on LC3-II generation, and PARP cleavage was evaluated. Silencing of the Atg5 gene inhibited generation of LC3-II as well as PARP cleavages in response to VacA (Fig. 8), whereas both inhibitors, Z-VAD-fmk and Necrostatin-1, which interfere with apoptosis (43), did not inhibit VacA-induced autophagy, suggesting that VacA-induced autophagy precedes apoptosis in AZ-521 cells. Necrostatin-1, which inhibits necroptosis (44), did not interfere with VacA-induced generation of LC3-II and PARP cleavage.
FIGURE 8.

Effects of Atg5 silencing, Z-VAD-fmk, and Necrostatin-1 on VacA-induced LC3-II generation and PARP cleavage. a, the indicated siRNA-transfected AZ-521 cells were incubated with 120 nm heat-inactivated (IA) or wild-type VacA (A) for 8–10 h at 37 °C and the cell lysates were subjected to immunoblotting (IB) with anti-LC3B, anti-cleaved PARP, or anti-Atg5 antibodies. α-Tubulin, as a loading control. A blot representative of three separate experiments is shown. Quantification of VacA-induced LC3-II or PARP cleavage (cPARP) in the indicated siRNA-transfected AZ-521 cells was performed by densitometry (bottom panel). Data are presented as mean ± S.D. and significance is (*) p < 0.05 (n = 5) and (**) p < 0.01(n = 5). b, AZ-521 cells were pretreated with 50 μm Z-VAD-fmk (Z-VAD) or 50 μm Necrostatin-1 (Necrostatin) for 30 min, and then 120 nm heat-inactivated or wild-type VacA were added to cells. After a 10-h incubation at 37 °C, cell lysates were analyzed by Western blotting using antibodies against LC3B and cleaved PARP. α-Tubulin served as a loading control. Data are representative of three separate experiments. Quantification of VacA-induced PARP cleavage (cPARP) levels in the indicated siRNA-transfected AZ-521 cells was performed by densitometry (bottom panel). Data are presented as mean ± S.D. and significance is (*) p < 0.01 (n = 3).
Effect of Anion Channel Blockers, NPPB and DIDS, on VacA-induced LC3-II Production
To assess whether membrane channels formed by VacA may also be involved in autophagy (29), we tested the effects of pretreating AZ-521 or AGS cells with chloride channel blockers, NPPB and DIDS, which are known to block both VacA-mediated channel activity and cellular vacuolation (45). AZ-521 cells were pretreated for 30 min with 100 μm NPPB or 100 μm DIDS prior to incubation with VacA for 6 h. Both NPPB and DIDS inhibited VacA-induced LC3-II generation in AZ-521 cells (Fig. 9a), but not in AGS cells under these conditions (Fig. 9b).
FIGURE 9.

Effect of anion-channel inhibitor on VacA-induced LC3-II generation. AZ-521 (a) or AGS (b) cells were pretreated with 100 μm NPPB or DIDS for 30 min at 37 °C and then incubated with 120 nm heat-inactivated (IA) or wild-type VacA (A) for 4 h at 37 °C. Cell lysates were subjected to immunoblotting (IB) with anti-LC3B antibody or anti-α-tubulin antibody as a loading control. Quantification of VacA-induced LC3-II levels in the cells was performed by densitometry (bottom panel). Data are mean ± S.D. of values from two independent experiments.
DISCUSSION
VacA has two functional domains, an N-terminal 33.4-kDa domain (named p33, p34 or p37, comprising residues 1–311) and a C-terminal domain of 54.8 kDa (named p55 or p58, comprising residues 312–821) (10, 46, 47). Vacuolization of epithelial cells by VacA is strictly dependent on the formation of anion-selective membrane channels, which are targeted to late endosomes after internalization of the toxin (45, 48). The pore and channel forming by the N-terminal p33 domain alone drives pleiotropic cellular activities of VacA; i.e. vacuolation, mitochondria damage, apoptosis (10, 47), autophagy (28, 29), and programmed necrosis (27), suggesting that VacA may be characterized as a pore-forming toxin (47). Another study has indicated that both p33 and p55 are required to form a functional channel in the inner mitochondria membrane and trigger apoptosis (49). In addition, it is now widely accepted that the C-terminal p55 domain of VacA plays an essential role in its binding to target cells (50, 51).
The present study defines a novel role for VacA signaling through LRP1 in AZ-521 cells, inducing autophagy and apoptosis (Fig. 7). LRP1 is a large endocytic receptor belonging to the LDL receptor family. This membrane protein consists of a 515-kDa heavy chain containing the extracellular ligand-binding domains and a noncovalently associated 85-kDa light chain, which consists of a transmembrane domain and a short cytoplasmic tail. LRP1 functions as a clearance receptor mediating the uptake and catabolism of various ligands from the pericellular environment, e.g. LRP1 binds to apolipoprotein E-rich lipoproteins, lipoprotein lipase, α2-macroglobulin, lactoferrin, and tissue plasminogen activator; it functions in lipoprotein metabolism, degradation of proteases and proteinase/inhibitor complexes, activation of lysosomal enzymes and cellular entry of viruses, and bacterial toxin such as Pseudomonas exotoxin A (52). LRP1 has also been shown to function in the turnover of fibronectin (53).
This is the first study to provide evidence that LRP1 mediates autophagy. In AZ-521 cells, VacA triggered formation of autophagosomes, followed by autolysosome formation, consistent with the observations in AGS cells (29). Because LRP1 knockdown with siRNA resulted in inhibition of VacA-induced LC3-II generation and cleavage of both caspase 7 and PARP, induction by VacA of both autophagy and apoptosis occurred via, at least in part, association with LRP1. VacA also promoted formation of vacuoles containing RPTPβ and RPTPα, which were characterized as light and dense vacuoles, respectively, by confocal microscopy, and large vacuoles (Fig. 5). We observed the presence in AZ-521 cells of large vesicles without autophagosome markers in wild-type and Atg12 knockdown AGS cells (29). In general, vacuole formation caused by VacA is required for VacA channel activity (54). Our studies using chloride channel blockers, NPPB and DIDS, to address the relationship between VacA-induced autophagy and channel activity of VacA in AZ-521 cells treated with VacA revealed that these channel blockers inhibited LC3-II generation in response to VacA (Fig. 9), suggesting that channel activity may be required for LRP1-dependent autophagy. More interestingly, VacA-induced autophagy was not blocked by caspase inhibitor and RIPK inhibitor, suggesting that VacA-induced autophagy via LRP1 binding precedes apoptosis.
Autophagy is a degradation process that involves formation of autophagosomes, which engulf cytoplasmic components, and fuse with the lysosome/vacuole for degradation of contents. This process is considered cytoprotective but in certain settings excessive autophagy can cause cell death (34, 42). Little information exists concerning the molecular mechanisms underlying the regulation of apoptosis by autophagy. Our data indicate that autophagy induced by VacA does not involve the canonical pathway in which Beclin-1 initiates the generation of autophagosomes by forming a multiprotein complex with class III PI3K, because 3MA, a class III PI3K inhibitor, and silencing of Beclin-1 did not inhibit autophagy induced by VacA (supplemental Fig. S2). The detailed mechanism by which VacA induces autophagy and apoptosis via LRP1 is not clear. Within VacA-intoxicated cells that provoke death signaling via mitochondrial damage, cells attempt to limit damage by seeking what catabolic benefits may be found in autophagy as indicated by Terebiznik et al. (29). Therefore, once the stress-provoking, death-signaling response to VacA is overwhelming, autophagy is futile, and it is beneficial to induce apoptosis. Future studies involving mouse models and human specimens will help to determine whether LRP1 plays a critical role in the pathobiology of H. pylori infection in vivo.
Supplementary Material
Acknowledgments
We thank K. Maeda for skillful assistance and P. I. Padilla (University of the Philippines Visayas, Philippines) and B. De Guzman (St. Luke's Medical Center, Philippines) for helpful discussions.
This work was supported by Grants-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan and Improvement of Research Environment for Young Researchers from the Japan Science and Technology Agency.

This article contains supplemental Figs. S1 and S2.
- RPTP
- receptor protein-tyrosine phosphatase
- LRP1
- low-density lipoprotein receptor-related protein
- NPPB
- 5-nitro-2-(3-phenylpropylamino)benzoic acid
- DIDS
- 4,4′-diisothiocyanostibene-2,2′-disulfonic acid
- MAA
- Maackia amurensis
- FN
- fibronectin
- Z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- PARP
- poly(ADP-ribose) polymerase.
REFERENCES
- 1. Wroblewski L. E., Peek R. M., Jr., Wilson K. T. (2010) Helicobacter pylori and gastric cancer. Factors that modulate disease risk. Clin. Microbiol. Rev. 23, 713–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yamaoka Y. (2010) Mechanisms of disease. Helicobacter pylori virulence factors. Nat. Rev. Gastroenterol. Hepatol. 7, 629–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Telford J. L., Ghiara P., Dell'Orco M., Comanducci M., Burroni D., Bugnoli M., Tecce M. F., Censini S., Covacci A., Xiang Z. (1994) Gene structure of the Helicobacter pylori cytotoxin and evidence of its key role in gastric disease. J. Exp. Med. 179, 1653–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marchetti M., Aricò B., Burroni D., Figura N., Rappuoli R., Ghiara P. (1995) Development of a mouse model of Helicobacter pylori infection that mimics human disease. Science 267, 1655–1658 [DOI] [PubMed] [Google Scholar]
- 5. Ghiara P., Marchetti M., Blaser M. J., Tummuru M. K., Cover T. L., Segal E. D., Tompkins L. S., Rappuoli R. (1995) Role of the Helicobacter pylori virulence factors vacuolating cytotoxin, CagA, and urease in a mouse model of disease. Infect. Immun. 63, 4154–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuo C. H., Wang W. C. (2003) Binding and internalization of Helicobacter pylori VacA via cellular lipid rafts in epithelial cells. Biochem. Biophys. Res. Commun. 303, 640–644 [DOI] [PubMed] [Google Scholar]
- 7. Oldani A., Cormont M., Hofman V., Chiozzi V., Oregioni O., Canonici A., Sciullo A., Sommi P., Fabbri A., Ricci V., Boquet P. (2009) Helicobacter pylori counteracts the apoptotic action of its VacA toxin by injecting the CagA protein into gastric epithelial cells. PLoS Pathog. 5, e1000603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Akada J. K., Aoki H., Torigoe Y., Kitagawa T., Kurazono H., Hoshida H., Nishikawa J., Terai S., Matsuzaki M., Hirayama T., Nakazawa T., Akada R., Nakamura K. (2010) Helicobacter pylori CagA inhibits endocytosis of cytotoxin VacA in host cells. Dis. Model Mech. 3, 605–617 [DOI] [PubMed] [Google Scholar]
- 9. Kuck D., Kolmerer B., Iking-Konert C., Krammer P. H., Stremmel W., Rudi J. (2001) Vacuolating cytotoxin of Helicobacter pylori induces apoptosis in the human gastric epithelial cell line AGS. Infect. Immun. 69, 5080–5087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cover T. L., Blanke S. R. (2005) Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat. Rev. Microbiol. 3, 320–332 [DOI] [PubMed] [Google Scholar]
- 11. Yamasaki E., Wada A., Kumatori A., Nakagawa I., Funao J., Nakayama M., Hisatsune J., Kimura M., Moss J., Hirayama T. (2006) Helicobacter pylori vacuolating cytotoxin induces activation of the proapoptotic proteins Bax and Bak, leading to cytochrome c release and cell death, independent of vacuolation. J. Biol. Chem. 281, 11250–11259 [DOI] [PubMed] [Google Scholar]
- 12. Calore F., Genisset C., Casellato A., Rossato M., Codolo G., Esposti M. D., Scorrano L., de Bernard M. (2010) Endosome-mitochondria juxtaposition during apoptosis induced by H. pylori VacA. Cell Death Differ. 17, 1707–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jain P., Luo Z. Q., Blanke S. R. (2011) Helicobacter pylori vacuolating cytotoxin A (VacA) engages the mitochondrial fission machinery to induce host cell death. Proc. Natl. Acad. Sci. U.S.A. 108, 16032–16037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Papini E., de Bernard M., Milia E., Bugnoli M., Zerial M., Rappuoli R., Montecucco C. (1994) Cellular vacuoles induced by Helicobacter pylori originate from late endosomal compartments. Proc. Natl. Acad. Sci. U.S.A. 91, 9720–9724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yahiro K., Niidome T., Kimura M., Hatakeyama T., Aoyagi H., Kurazono H., Imagawa K., Wada A., Moss J., Hirayama T. (1999) Activation of Helicobacter pylori VacA toxin by alkaline or acid conditions increases its binding to a 250-kDa receptor protein-tyrosine phosphatase β. J. Biol. Chem. 274, 36693–36699 [DOI] [PubMed] [Google Scholar]
- 16. Yahiro K., Wada A., Nakayama M., Kimura T., Ogushi K., Niidome T., Aoyagi H., Yoshino K., Yonezawa K., Moss J., Hirayama T. (2003) Protein-tyrosine phosphatase α, RPTP α, is a Helicobacter pylori VacA receptor. J. Biol. Chem. 278, 19183–19189 [DOI] [PubMed] [Google Scholar]
- 17. Hennig E. E., Godlewski M. M., Butruk E., Ostrowski J. (2005) Helicobacter pylori VacA cytotoxin interacts with fibronectin and alters HeLa cell adhesion and cytoskeletal organization in vitro. FEMS Immunol. Med. Microbiol. 44, 143–150 [DOI] [PubMed] [Google Scholar]
- 18. Gupta V. R., Patel H. K., Kostolansky S. S., Ballivian R. A., Eichberg J., Blanke S. R. (2008) Sphingomyelin functions as a novel receptor for Helicobacter pylori VacA. PLoS Pathog. 4, e1000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Papadimitriou E., Mikelis C., Lampropoulou E., Koutsioumpa M., Theochari K., Tsirmoula S., Theodoropoulou C., Lamprou M., Sfaelou E., Vourtsis D., Boudouris P. (2009) Roles of pleiotrophin in tumor growth and angiogenesis. Eur. Cytokine Netw. 20, 180–190 [DOI] [PubMed] [Google Scholar]
- 20. Fujikawa A., Shirasaka D., Yamamoto S., Ota H., Yahiro K., Fukada M., Shintani T., Wada A., Aoyama N., Hirayama T., Fukamachi H., Noda M. (2003) Mice deficient in protein-tyrosine phosphatase receptor type Z are resistant to gastric ulcer induction by VacA of Helicobacter pylori. Nat. Genet. 33, 375–381 [DOI] [PubMed] [Google Scholar]
- 21. Gebert B., Fischer W., Weiss E., Hoffmann R., Haas R. (2003) Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 301, 1099–1102 [DOI] [PubMed] [Google Scholar]
- 22. Boncristiano M., Paccani S. R., Barone S., Ulivieri C., Patrussi L., Ilver D., Amedei A., D'Elios M. M., Telford J. L., Baldari C. T. (2003) The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J. Exp. Med. 198, 1887–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sewald X., Gebert-Vogl B., Prassl S., Barwig I., Weiss E., Fabbri M., Osicka R., Schiemann M., Busch D. H., Semmrich M., Holzmann B., Sebo P., Haas R. (2008) Integrin subunit CD18 Is the T-lymphocyte receptor for the Helicobacter pylori vacuolating cytotoxin. Cell Host Microbe 3, 20–29 [DOI] [PubMed] [Google Scholar]
- 24. Sewald X., Jimenez-Soto L., Haas R. (2011) PKC-dependent endocytosis of the Helicobacter pylori vacuolating cytotoxin in primary T lymphocytes. Cell Microbiol. 13, 482–496 [DOI] [PubMed] [Google Scholar]
- 25. Cover T. L., Krishna U. S., Israel D. A., Peek R. M., Jr. (2003) Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 63, 951–957 [PubMed] [Google Scholar]
- 26. Singh M., Prasad K. N., Saxena A., Yachha S. K. (2006) Helicobacter pylori induces apoptosis of T- and B-cell lines and translocates mitochondrial apoptosis-inducing factor to nucleus. Curr. Microbiol. 52, 254–260 [DOI] [PubMed] [Google Scholar]
- 27. Radin J. N., Gonzalez-Rivera C., Ivie S. E., McClain M. S., Cover T. L. (2011) Helicobacter pylori VacA induces programmed necrosis in gastric epithelial cells. Infect. Immun. 79, 2535–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Raju D., Hussey S., Ang M., Terebiznik M. R., Sibony M., Galindo-Mata E., Gupta V., Blanke S. R., Delgado A., Romero-Gallo J., Ramjeet M. S., Mascarenhas H., Peek R. M., Correa P., Streutker C., Hold G., Kunstmann E., Yoshimori T., Silverberg M. S., Girardin S. E., Philpott D. J., El Omar E., Jones N. L. (2012) Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterology 142, 1160–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Terebiznik M. R., Raju D., Vázquez C. L., Torbricki K., Kulkarni R., Blanke S. R., Yoshimori T., Colombo M. I., Jones N. L. (2009) Effect of Helicobacter pylori's vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy 5, 370–379 [DOI] [PubMed] [Google Scholar]
- 30. Lum J. J., Bauer D. E., Kong M., Harris M. H., Li C., Lindsten T., Thompson C. B. (2005) Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120, 237–248 [DOI] [PubMed] [Google Scholar]
- 31. Gozuacik D., Kimchi A. (2004) Autophagy as a cell death and tumor suppressor mechanism. Oncogene 23, 2891–2906 [DOI] [PubMed] [Google Scholar]
- 32. McPhee C. K., Logan M. A., Freeman M. R., Baehrecke E. H. (2010) Activation of autophagy during cell death requires the engulfment receptor Draper. Nature 465, 1093–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wirawan E., Vande Walle L., Kersse K., Cornelis S., Claerhout S., Vanoverberghe I., Roelandt R., De Rycke R., Verspurten J., Declercq W., Agostinis P., Vanden Berghe T., Lippens S., Vandenabeele P. (2010) Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 1, e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maiuri M. C., Criollo A., Kroemer G. (2010) Cross-talk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J. 29, 515–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rubinstein A. D., Eisenstein M., Ber Y., Bialik S., Kimchi A. (2011) The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol. Cell 44, 698–709 [DOI] [PubMed] [Google Scholar]
- 36. Nakayama M., Hisatsune J., Yamasaki E., Nishi Y., Wada A., Kurazono H., Sap J., Yahiro K., Moss J., Hirayama T. (2006) Clustering of Helicobacter pylori VacA in lipid rafts, mediated by its receptor, receptor-like protein-tyrosine phosphatase β, is required for intoxication in AZ-521 cells. Infect. Immun. 74, 6571–6580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Høyer-Hansen M., Bastholm L., Mathiasen I. S., Elling F., Jäättelä M. (2005) Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ. 12, 1297–1309 [DOI] [PubMed] [Google Scholar]
- 38. Yang S., Wang X., Contino G., Liesa M., Sahin E., Ying H., Bause A., Li Y., Stommel J. M., Dell'antonio G., Mautner J., Tonon G., Haigis M., Shirihai O. S., Doglioni C., Bardeesy N., Kimmelman A. C. (2011) Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25, 717–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Youle R. J., Narendra D. P. (2011) Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vives-Bauza C., Przedborski S. (2011) Mitophagy. The latest problem for Parkinson disease. Trends Mol. Med. 17, 158–165 [DOI] [PubMed] [Google Scholar]
- 41. Kirkin V., McEwan D. G., Novak I., Dikic I. (2009) A role for ubiquitin in selective autophagy. Mol. Cell 34, 259–269 [DOI] [PubMed] [Google Scholar]
- 42. Yoshimori T. (2007) Autophagy. Paying Charon's toll. Cell 128, 833–836 [DOI] [PubMed] [Google Scholar]
- 43. Zhu H., Fearnhead H. O., Cohen G. M. (1995) An ICE-like protease is a common mediator of apoptosis induced by diverse stimuli in human monocytic THP.1 cells. FEBS Lett. 374, 303–308 [DOI] [PubMed] [Google Scholar]
- 44. Degterev A., Huang Z., Boyce M., Li Y., Jagtap P., Mizushima N., Cuny G. D., Mitchison T. J., Moskowitz M. A., Yuan J. (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119 [DOI] [PubMed] [Google Scholar]
- 45. Tombola F., Carlesso C., Szabò I., de Bernard M., Reyrat J. M., Telford J. L., Rappuoli R., Montecucco C., Papini E., Zoratti M. (1999) Helicobacter pylori vacuolating toxin forms anion-selective channels in planar lipid bilayers. Possible implications for the mechanism of cellular vacuolation. Biophys. J. 76, 1401–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Montecucco C., Rappuoli R. (2001) Living dangerously. How Helicobacter pylori survives in the human stomach. Nat. Rev. Mol. Cell Biol. 2, 457–466 [DOI] [PubMed] [Google Scholar]
- 47. Boquet P., Ricci V. (2012) Intoxication strategy of Helicobacter pylori VacA toxin. Trends Microbiol. 20, 165–174 [DOI] [PubMed] [Google Scholar]
- 48. Szabò I., Brutsche S., Tombola F., Moschioni M., Satin B., Telford J. L., Rappuoli R., Montecucco C., Papini E., Zoratti M. (1999) Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO J. 18, 5517–5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Foo J. H., Culvenor J. G., Ferrero R. L., Kwok T., Lithgow T., Gabriel K. (2010) Both the p33 and p55 subunits of the Helicobacter pylori VacA toxin are targeted to mammalian mitochondria. J. Mol. Biol. 401, 792–798 [DOI] [PubMed] [Google Scholar]
- 50. Torres V. J., Ivie S. E., McClain M. S., Cover T. L. (2005) Functional properties of the p33 and p55 domains of the Helicobacter pylori vacuolating cytotoxin. J. Biol. Chem. 280, 21107–21114 [DOI] [PubMed] [Google Scholar]
- 51. Isomoto H., Moss J., Hirayama T. (2010) Pleiotropic actions of Helicobacter pylori vacuolating cytotoxin, VacA. Tohoku J. Exp. Med. 220, 3–14 [DOI] [PubMed] [Google Scholar]
- 52. Herz J., Strickland D. K. (2001) LRP. A multifunctional scavenger and signaling receptor. J. Clin. Invest. 108, 779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Salicioni A. M., Gaultier A., Brownlee C., Cheezum M. K., Gonias S. L. (2004) Low density lipoprotein receptor-related protein-1 promotes beta1 integrin maturation and transport to the cell surface. J. Biol. Chem. 279, 10005–10012 [DOI] [PubMed] [Google Scholar]
- 54. Genisset C., Puhar A., Calore F., de Bernard M., Dell'Antone P., Montecucco C. (2007) The concerted action of the Helicobacter pylori cytotoxin VacA and of the v-ATPase proton pump induces swelling of isolated endosomes. Cell Microbiol. 9, 1481–1490 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.