

Background: ErbB2 is the preferred dimerization partner for the epidermal growth factor (EGF) receptor.
Results: Heterodimerization with ErbB2 increases the affinity of the EGFR for EGF and increases the level of dimers maintained at any given concentration of EGF.
Conclusion: ErbB2 modulates EGF receptor affinity and dimer stability.
Significance: This study elucidates the molecular basis for the enhanced binding of EGF to EGFR/ErbB2 heterodimers.
Keywords: Cooperativity, Growth Factors, Ligand-binding Protein, Receptor Tyrosine Kinase, Tyrosine-protein Kinase (Tyrosine Kinase), EGF Receptor, ErbB2, Negative Cooperativity
Abstract
The epidermal growth factor (EGF) receptor is a member of the ErbB family of receptors that also includes ErbB2, ErbB3, and ErbB4. These receptors form homo- and heterodimers in response to ligand with ErbB2 being the preferred dimerization partner. Here we use 125I-EGF binding to quantitate the interaction of the EGF receptor with ErbB2. We show that the EGFR/ErbB2 heterodimer binds EGF with a 7-fold higher affinity than the EGFR homodimer. Because it cannot bind a second ligand, the EGFR/ErbB2 heterodimer is not subject to ligand-induced dissociation caused by the negatively cooperative binding of EGF to the second site on the EGFR homodimer. This increases the stability of the heterodimer relative to the homodimer and is associated with enhanced and prolonged EGF receptor autophosphorylation. These effects are independent of the kinase activity of ErbB2 but require back-to-back dimerization of the EGF receptor with ErbB2. Back-to-back dimerization is also required for phosphorylation of ErbB2. These findings provide a molecular explanation for the apparent preference of the EGF receptor for dimerizing with ErbB2 and suggest that the phosphorylation of ErbB2 occurs largely in the context of the EGFR/ErbB2 heterodimer, rather than through lateral phosphorylation of isolated ErbB2 subunits.
Introduction
The EGF receptor is the founding member of the ErbB family of growth factor receptors. This family also includes ErbB2, ErbB3, and ErbB4 (1). All members of the ErbB family share a common structure that includes a large extracellular domain, a single α helical transmembrane segment, an intracellular tyrosine kinase domain, and an unstructured C-terminal tail (2, 3). Although the EGF receptor, ErbB3, and ErbB4 each bind several members of a family of homologous ligands (4), ErbB2 has no known ligand (5).
ErbB receptors appear to exist in the membrane as monomers and inactive pre-dimers (6–10). Binding of ligand induces dimerization of ErbB receptors (11). The ErbB receptors form both homodimers and heterodimers. However, the ligand-less ErbB2 appears to be the preferred dimerization partner for the EGF receptor, ErbB3 and ErbB4 (12, 13).
Ligand-induced dimerization of the extracellular domains of ErbB receptors leads to the formation of an activating asymmetric dimer of the intracellular kinase domains (14). Only one kinase domain can be activated at a time within this asymmetric dimer and dissociation of the dimer terminates kinase activation. Activation of the tyrosine kinase promotes autophosphorylation of the receptors on their C-terminal tails and leads to the binding of SH2 and PTB domain-containing proteins and the activation of numerous downstream signaling pathways.
X-ray crystallography studies have shown that the extracellular domain of the EGF receptor consists of four subdomains, numbered I through IV. In the absence of ligand, the extracellular domain of the EGF receptor exists in a closed, tethered conformation in which the dimerization arm from subdomain II makes contact with the tethering arm in subdomain IV (15). Binding of ligand releases this intramolecular tether allowing the receptor to open into an extended conformation in which the dimerization arm is available for inter-molecular interactions. Back-to-back dimers form between two ligand-occupied receptors, mediated by the dimerization arm in subdomain II (16, 17). The tethering arm in subdomain IV may also participate in inter-molecular interactions (18, 19). Although the dimerization interface is homologous in all four ErbB receptors, it is not identical. Thus, it is possible that the properties of a given ErbB receptor could differ, depending on the nature of its dimerization partner.
The binding of EGF to its receptor shows multiple affinity states. We have recently used radioligand binding studies to explore the basis for this heterogeneity in ligand binding by the EGF receptor (18, 20, 21). These studies characterized the interaction between EGF receptor subunits in EGFR homodimers and defined the binding properties of the various monomeric and dimeric species of the receptor. Our studies demonstrated that ligand binding to the EGF receptor is best explained by a model involving negative cooperativity in an aggregating system (20). The negative cooperativity is strong enough so that EGF receptor homodimers tend to dissociate following the binding of EGF to the second site on the dimer.
Heterodimerization of the EGF receptor with ErbB2 affects EGF binding affinity (13, 22), suggesting that subunit-subunit interactions are indeed different in the EGFR homodimer and the EGFR/ErbB2 heterodimer. To better understand the nature of EGF receptor/ErbB2 interactions, we have expanded our radioligand binding studies to quantitate the interactions between the EGF receptor and ErbB2 and between the heterodimer and the EGF ligand. Our data indicate that EGFR/ErbB2 heterodimers are “preferred” not only because they exhibit a higher affinity for EGF but also because, in the absence of a second binding site, they lack negative cooperativity and cannot be dissociated by the binding of a second ligand. As a result, a higher concentration of heterodimers than homodimers can be maintained at all concentrations of EGF. The effect of ErbB2 expression on EGF receptor function requires dimerization of the extracellular domains but is independent of ErbB2 kinase activity. Dimerization of the EGF receptor and ErbB2 is also required for phosphorylation of ErbB2. Together, these data suggest that the nature of the subunit-subunit interface affects the ligand binding properties of the EGF receptor and that the ligand-less nature of ErbB2 provides a structural mechanism for controlling the stability of EGF receptor-containing dimers. They also suggest that phosphorylation of ErbB2 occurs mostly in the context of heterodimers rather than through lateral phosphorylation of isolated subunits.
MATERIALS AND METHODS
Mutagenesis, Cells, and Tissue Culture
CHO-K1 Tet-on cells (Clontech) were stably transfected with pcDNA3.1-Zeo constitutively expressing the EGF receptor from a CMV promoter. The cells expressed ∼300,000 EGF receptors per cell. A cDNA encoding wild type ErbB2 was ligated into the pBI Tet vector (Clontech) between the NheI and EcoRV sites. K732M-ErbB2 and Y253D-ErbB2 were generated in the pBI-tet vector using a QuikChange mutagenesis kit (Stratagene). The appropriate pBI-tet plasmid was then co-transfected with pTK-Hyg into the tet-on cells constitutively expressing the EGF receptor using Lipofectamine 2000 (Invitrogen). Stable clones were isolated by selection in 500 μg/ml of hygromycin (Invitrogen). Cells were grown in Dulbecco's modified Eagle's medium containing 10% Fetalplex (Gemini Bio-Products), 100 μg/ml of hygromycin, and 100 μg/ml of Zeocin. The generation of the ΔC-EGFR-NLuc/ΔC-EGFR-CLuc and ΔC-EGFR-CLuc/ΔC-ErbB2-NLuc cell lines expressing the truncated EGF receptor and/or ErbB2 fused to the NLuc or CLuc fragments of firefly luciferase has been described previously (23). The generation of the full-length EGFR-CLuc/ErbB2-NLuc cell lines has also been described (23).
125I-EGF and 125I-Trastuzumab Synthesis and Binding Analyses
Murine EGF (Biomedical Technologies, Inc.) and trastuzumab (Barnes Hospital pharmacy) were radiolabeled with 125I using the ICl method (24). For binding, cells were plated into 6-well dishes 2 days prior to the experiment. Binding of 125I-EGF was carried out as described previously (20). Binding of 125I-trastuzumab was performed in the same manner except that 30 pm 125I-trastuzumab was added to each well in the presence of increasing concentrations of unlabeled trastuzumab ranging from 0 to 60 nm. The total number of ErbB2 receptors was determined by analyzing the data using GraphPad Prism 4.0. All binding data points were done in triplicate.
An Origin C program was developed to fit the experimental data globally using Origin's nonlinear least squares advanced fitting tool. MT, NT, L20, K11, K21, and K22 were provided as constant values. MT and NT were determined directly from the binding of 125I-EGF and 125I-trastuzumab, respectively. L20, K11, K21, and K22 were set to values derived from the analysis of a total of 18 separate saturation binding isotherms for 125I-EGF binding to CHO cells expressing only EGF receptors (18, 20, 21). These were: L20, 7.6e11 D−1; K11, 4.8e9 m−1; K21, 1.9e9 m−1; and K22, 1.7e8 m−1. All experimental curves were fitted simultaneously to Equation 1, sharing J20 and H21 among all curves. Each data point was weighted by the inverse of its standard deviation squared. Because the EGF receptors and ErbB2 receptors are present as a two-dimensional density rather than a three dimension concentration, the values for receptor concentration are expressed in units of (mol/dm2)−1, which we designate as D−1 (20). As a result, L20 and J20 are also expressed in units of D−1. All other association constants are in units of m−1.
Simulations
A Matlab program was developed to simulate the binding isotherms and calculate the concentrations of the different EGF receptor or ErbB2 species. The independent variable, the free [EGF], was varied from 5.0e-13 to 1.80e-7 m. All the equilibrium association constants L20, K11, K21, K22, J20, and H21, as well as MT and NT, the two-dimensional density of the EGF receptors and ErbB2 receptors, were provided as constants. Similar to the Origin C fitting program, the Matlab program first solves the cubic equation analytically as described in the supplemental Materials to calculate Mf, the concentration of free EGF receptor monomers. Once Mf is known for each given free EGF concentration, the concentration of the other species in the systems can be readily calculated.
Autophosphorylation and Down-regulation
Cells were plated in Dulbecco's modified Eagle's medium containing 10% Fetalplex in 6-well dishes and grown for 48 h. Cells expressing wild type or K732M-ErbB2 for a tet-inducible promoter were plated without or with 100 ng/ml of doxycycline. Cells expressing Y253D-ErbB2 from a tet-inducible promoter were plated without or with 250 ng/ml of doxycycline. For assay, the medium was removed and replaced with Ham's F-12 medium containing 1 mg/ml of bovine serum albumin and the indicated concentration of EGF. Plates were incubated at 37 °C for the indicated times and the medium was aspirated. The reaction was stopped by washing the cells twice in ice-cold phosphate-buffered saline and the cells were scraped into RIPA buffer containing phosphatase and protease inhibitors (25). Equal amounts of protein were analyzed by SDS-polyacrylamide gel electrophoresis. Gels were transferred to polyvinylidene difluoride (Millipore) membranes and probed by Western blotting with the indicated antibodies.
For down-regulation assays, cells were incubated with 10 nm EGF for the indicated times and then washed once with ice-cold phosphate-buffered saline, twice for 2 min with acid wash (50 mm glycine, 100 mm NaCl), and again with phosphate-buffered saline. The cells were then incubated with 0.5 nm 125I-EGF overnight at 4 °C. At the end of the incubation, the binding medium was aspirated and the plates were washed 3 times with phosphate-buffered saline. Monolayers were dissolved in 1 n NaOH and counted for 125I.
Luciferase Complementation Assays
Assays were done using cells stably expressing ΔC-EGFR-NLuc and ΔC-EGFR-CLuc or ΔC-ErbB2-NLuc and ΔC-EGFR-CLuc. The ΔC designation indicates that these constructs are C terminally truncated and contain only the extracellular and transmembrane domains of the receptors fused to the luciferase splits. The generation of these lines has been described previously (23).
Cells were plated in 96-well black-walled dishes 48 h prior to use. Cultures were transferred to Dulbecco's modified Eagle's medium without phenol red but with 1 mg/ml of bovine serum albumin. d-Luciferin (0.6 mg/ml) was incubated with the cells for 20 min at 37 °C prior to assay. EGF was then added at the concentration indicated and cell radiance (photons/s/cm2/sr) was measured at 30-s intervals over a 25-min time course using a cooled CCD camera and imaging system (IVIS 50). Points were done in quadruplicate and the data were analyzed as described previously (26).
RESULTS
Effect of ErbB2 on the Binding of EGF
To determine how the expression of ErbB2 affects the binding properties of the EGF receptor, a line of CHO cells was established that constitutively expressed ∼300,000 EGF receptors/cell but expressed ErbB2 from a tet-inducible promoter. The binding of 125I-EGF to this cell line was then analyzed in cells induced to express levels of ErbB2 expression ranging from 35,000 receptors/cell to 2.1 million receptors/cell, roughly 10-fold below to 10-fold above the level of EGF receptors. ErbB2 receptor levels were measured by 125I-trastuzumab binding (27). The binding data are shown in Fig. 1A. As can be seen from the figure, increasing the level of ErbB2 resulted in a leftward shift of the 125I-EGF binding isotherms. This suggests that expression of ErbB2 enhances the affinity of EGF for its receptor.
FIGURE 1.

Binding of 125I-EGF to cells expressing EGF receptors and ErbB2. A, 125I-EGF binding to cells expressing ∼300,000 EGF receptors and the indicated number of ErbB2 molecules. The level of ErbB2 was measured by the binding of 125I-trastuzumab. B, model for the binding of EGF to EGFR homodimers and EGFR/ErbB2 heterodimers. Open circles represent EGF receptor molecules. Hatched circles represent ErbB2 molecules. E is a bound EGF molecule. The term that refers to the equilibrium association constant for each interaction is indicated over the arrows. Fitted values for the equilibrium association constants are shown. Values in red indicate the values that were fitted to Equation 1 based on the binding data in panel A. Values in black were set as constants during the fitting and are based on previous studies of EGF binding to only the EGF receptor. Values in gray were calculated based on the principle of microscopic equilibrium.
To quantitate the changes in EGF binding properties, the data were fit to the equation (Equation 1) for the model shown in Fig. 1B. In this model, the open circles represent EGF receptor monomers. The hatched circles represent ErbB2 monomers. ErbB2 is thought to dimerize only at very high receptor concentrations (28, 29). Thus, this species was not included in the model.
For this model, ¯Y, the fractional saturation of the EGF receptor, is given by the equation,
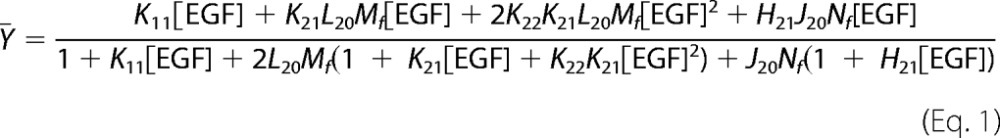 |
where Mf is the concentration of free EGF receptor monomers in the system and Nf is the concentration of free ErbB2 monomers. This equation is cubic in Mf but can be solved analytically (see supplemental Materials) permitting us to globally fit the 125I-EGF binding data to this equation.
In this analysis, the four parameters, K11, L20, K21, and K22, that had been previously determined in cells expressing only EGF receptors were held constant at the values indicated in Fig. 1B (shown in black in the figure). These values were determined by globally fitting a compilation of all previously reported wild type binding isotherms (18, 20, 21, 30). This was necessary as the fitting did not converge if all six variables were allowed to float. This approach allowed us to determine the values for J20 and H21 (shown in red in Fig. 1B).
J20 is the association constant for the heterodimerization of the unoccupied EGF receptor with ErbB2. Based on the global fit of the data, this dimerization constant was determined to be 2.8 × 1011 D−1. As L20, the association constant for EGF receptor homodimerization is 7.6 × 1011 D−1, this leads to the surprising conclusion that in terms of the formation of pre-dimers, EGF receptor homodimers are slightly preferred over EGFR/ErbB2 heterodimers.
As predicted from the leftward shift of the 125I-EGF binding isotherms with increasing expression of ErbB2, the analysis yielded a fitted value of 1.3 × 1010 m−1 for H21, the association constant for the binding of EGF to the heterodimer. This is ∼7-fold higher than the value for K21, the affinity of EGF for binding to the first site on the EGF receptor homodimer. In terms of dissociation constants, the Kd for EGF binding to the EGFR/ErbB2 heterodimer is 77 pm, whereas the Kd for EGF binding to the first site on the EGF receptor homodimer is 525 pm. Thus, dimerization of the EGF receptor with ErbB2 increases the affinity of the receptor for EGF.
Using the principle of microscopic equilibrium, one can calculate the association constants for the dimerization of an occupied EGF receptor monomer with either an unoccupied EGF receptor monomer (L21) or an ErbB2 monomer (J21). This calculation yields a value of 3.0 × 1011 D−1 for L21 and a value of 7.6 × 1011 D−1 for J21. Thus, if EGF binds to a receptor monomer, the occupied EGF receptor monomer has a weak preference for heterodimerization with ErbB2 over homodimerization with the EGF receptor.
These fitted parameters were used in a set of simulations to predict the distribution of the various receptor species at increasing concentrations of EGF. These simulations are shown in Fig. 2. Fig. 2A shows the predicted distribution of EGF receptors between monomeric and dimeric species in cells expressing 50,000 EGF receptors/cell. Dotted lines represent unliganded species. Solid lines indicate species with bound EGF. In the absence of EGF, a significant level of EGF receptor “pre-dimers” is predicted based on the value of L20. A substantial body of biochemical evidence supports the existence of such inactive pre-dimers (10, 31–33). Upon addition of EGF, singly ligated homodimers (B1/B1 + E) form, peaking at ∼0.6 nm. As the concentration of EGF is increased, the concentration of the singly ligated EGF receptor dimers declines. At saturating concentrations of EGF, the major species formed is the occupied EGF receptor monomer (B1 + E) with only a very low level of doubly occupied dimers (B1/B1 + 2E). This distribution is observed because, although binding of EGF to the first site on the dimer stabilizes the dimer, binding of EGF to the second site on the dimer is negatively cooperative and destabilizes the dimer. The affinity of EGF for binding to the second site on the dimer is actually lower than that for binding of EGF to the monomer (K22 < K11), so the dimer tends to dissociate.
FIGURE 2.

Simulation of the binding of EGF to EGF receptor homodimers or EGFR/ErbB2 heterodimers. A, distribution of EGF receptor species in cells expressing 50,000 EGF receptors at increasing concentrations of EGF. B, distribution of EGF receptor and ErbB2 species in cells expressing 25,000 EGF receptors plus 25,000 ErbB2 molecules. C, distribution of EGF receptor species in cells expressing 50,000 EGF receptors lacking negative cooperativity (K22 = K21 = 1.9e9) at increasing concentrations of EGF.
Fig. 2B shows how the presence of ErbB2 modifies this distribution of the receptor between monomeric and dimeric species. In this simulation, the total number of receptor subunits has been maintained at 50,000 per cell. However, there are 25,000 EGF receptor subunits and 25,000 ErbB2 subunits. In the absence of EGF, the concentration of EGFR/EGFR pre-dimers (B1/B1) is slightly greater than that of EGFR/ErbB2 pre-dimers (B1/B2). This is because L20, the association constant for homodimers, is higher than J20, the association constant for heterodimers. As the concentration of EGF rises, the level of occupied EGF receptor monomers markedly increases. However, in the presence of ErbB2, the concentration of occupied EGFR/ErbB2 heterodimers (B1/B2 + E) also rises and, at saturating levels of EGF, a significant number of heterodimers are maintained. Thus, expression of ErbB2 results in a system in which substantially higher levels of dimeric receptor species are formed at all concentrations of EGF than when EGF receptors are expressed alone.
Although the enhanced affinity of EGF for the heterodimer would play a role in the preference for ErbB2 as a dimerization partner, another major contributor to this aspect of receptor biology is the absence of negative cooperativity from the EGFR/ErbB2 heterodimer. The effect of negative cooperativity is illustrated by the simulation shown in Fig. 2C.
In this simulation, there were 50,000 EGF receptors but negative cooperativity was abolished by setting K21 = K22, that is EGF binds with the same affinity to the second site as to the first site on the dimer. In this scenario, the extent of homodimer formation is similar to that seen when ErbB2 is present. In particular, significant levels of doubly occupied homodimers are present at all concentrations of EGF and are maintained at saturating doses of the growth factor. Thus, the existence of negative cooperativity in the EGF receptor homodimer, but not in the EGFR/ErbB2 heterodimer, is the major contributor to the apparent preference of the EGF receptor for dimerization with ErbB2.
Effect of ErbB2 Expression on the Dimerization of the EGF Receptor
Our binding data indicate that EGF binds with a higher affinity to EGFR/ErbB2 heterodimers than to EGF receptor homodimers. To determine whether this difference in ligand binding affinity led to a difference in the ability of EGF to induce receptor dimerization, we used luciferase fragment complementation imaging to monitor the dimerization of the EGF receptor and ErbB2 (23, 26). In this system, firefly luciferase is split into an N-terminal fragment (NLuc) and a C-terminal fragment (CLuc). Individually, the fragments are inactive but when brought into proximity, they complement each other and form an active luciferase enzyme that generates light upon incubation with luciferin (34).
The NLuc and CLuc fragments were fused to EGF receptors and/or ErbB2 that had been C terminally truncated to remove the entire intracellular domain (ΔC-EGFR and ΔC-ErbB2, respectively). Therefore, this assay measures only dimerization mediated by the extracellular domains of the receptors. The constructs were stably coexpressed in CHO cells and the studies were done in intact cells in real time.
As shown in Fig. 3A, in cells co-expressing ΔC-EGFR-NLuc and ΔC-EGFR-CLuc, addition of increasing doses of EGF resulted in an increase in the rate as well as the extent of light production. The curves could be fit to a single exponential, yielding kobs values for the rate of dimerization. These rate constants were plotted against the concentration of EGF in Fig. 3C, yielding an EC50 value of 5.1 nm for homodimerization of the EGF receptor. This is similar to the affinity of EGF for binding to this truncated receptor (21).
FIGURE 3.

Effect of ErbB2 on EGF receptor dimerization. A, CHO cells stably expressing ΔC-EGFR-NLuc and ΔC-EGFR-CLuc were stimulated with the indicated concentrations of EGF and receptor dimerization was monitored via luciferase activity. B, CHO cells stably expressing ΔC-ErbB2-NLuc and ΔC-EGFR-CLuc were stimulated with the indicated concentrations of EGF and receptor dimerization was monitored via luciferase activity. C, the data in panels A and B were fit to a single exponential association model using Prism 4.0 and the derived kobs values were plotted against the [EGF]. These data were then fit to a sigmoidal dose-response curve using Prism 4.0. The fitted EC50 values are reported in the text.
Addition of increasing doses of EGF to cells co-expressing ΔC-EGFR-CLuc and ΔC-ErbB2-NLuc also led to an increase in the rate and extent of heterodimer formation (Fig. 3B). It should be noted that this form of the assay only detects EGFR/ErbB2 heterodimers. Although EGFR homodimers can form, they do not give a signal in this assay. Fitting of the data to a single exponential association model yielded kobs values, which, when plotted versus the concentration of EGF, gave an EC50 of 1.4 nm for heterodimerization. This is ∼4-fold lower than the EC50 determined for the homodimerization reaction. Thus, the increase in EGF binding affinity of the EGFR/ErbB2 heterodimer is mirrored by an increase in sensitivity to EGF for the formation of EGFR/ErbB2 heterodimers.
The above luciferase assays were done using EGF receptor and ErbB2 constructs that lacked the entire intracellular domain. The observation that the expression of truncated ErbB2 enhanced the affinity for EGF in this assay suggests that the intracellular domain, and in particular the kinase activity, is not required for the effects of ErbB2 on EGF receptor binding affinity. However, this does not address the requirement for extracellular domain dimerization to observe the functional effects of ErbB2 on the EGF receptor.
To more specifically assess the structural requirements for ErbB2 modulation of EGF receptor function, we generated two mutants of ErbB2: K732M-ErbB2 and Y253D-ErbB2. K753M-ErbB2 is the kinase-dead version of this receptor (23, 35). Tyr-253 is at the tip of the dimerization arm of ErbB2 and mutation to Asp is predicted to block the ability of ErbB2 to form ligand-induced, back-to-back dimers with the EGF receptor (16, 17, 28, 29).
We first tested the ability of these two mutant ErbB2 proteins to interact with the EGF receptor using the luciferase fragment complementation assay (Fig. 4). Stable cell lines were generated that expressed wild type EGF receptor fused to CLuc and wild type, kinase-dead, or Y253D-ErbB2 fused to NLuc. These experiments utilized the full-length forms of the EGF receptor and ErbB2. As we have reported previously and as seen in Fig. 4A (black line), stimulation of cells expressing WT-EGFR-CLuc and WT-ErbB2-NLuc with EGF led to a biphasic response in the luciferase assay. There was an initial decrease in light production, followed by a recovery back toward baseline levels. The basal light production is due to the presence of pre-dimers that allow complementation of the luciferase fragments. These pre-dimers are disrupted or re-organized by the addition of EGF, leading to the decrease in light production (23, 25, 26). However, ligand-stimulated dimerization of receptor monomers results in new complementation pairs, leading to the increase in light production.
FIGURE 4.

Y253D-ErbB2 does not undergo EGF-dependent dimerization with the EGF receptor and is not phosphorylated. Cells stably expressing full-length EGFR-CLuc and transiently transfected with wild type, kinase-dead, or Y253D-ErbB2 fused to NLuc were stimulated with 10 nm EGF and assayed in the luciferase complementation assay. A, WT-EGFR/WT-ErbB2 (black line) compared with WT-EGFR/kinase-dead ErbB2 (gray line). B, WT-EGFR/WT-ErbB2 (black line) compared with WT-EGFR/Y253D-ErbB2 (gray line). C, dose-response to EGF for the phosphorylation of ErbB2 in cells co-expressing wild type EGF receptor with wild type, kinase-dead, or dimerization-defective Y253D-ErbB2.
When the EGFR-CLuc was co-expressed with the kinase-dead K732M-ErbB2-NLuc, a similar biphasic pattern of light production was observed (Fig. 4A, gray line). This indicates that the kinase-dead version of ErbB2 interacts with the EGF receptor in a manner similar to WT-ErbB2. The data are consistent with the conclusion that the ability of ErbB2 to interact with the EGF receptor does not require the kinase activity of ErbB2.
When EGFR-CLuc was co-expressed with Y253D-ErbB2, stimulation with EGF led to a persistent decline in light production (Fig. 4B, gray line). It appears that, as with wild type ErbB2, pre-dimers of the EGF receptor and Y253D-ErbB2 can form and they are disrupted by the addition of EGF. However, due to the Y253D mutation, EGF cannot promote the dimerization of monomeric EGF receptors and Y253D-ErbB2 and thus there is no recovery of luciferase activity. These data demonstrate that the Y253D mutation severely impairs the ability of ErbB2 to undergo ligand-stimulated dimerization with the EGF receptor.
Assays of the EGF-stimulated phosphorylation of ErbB2 in cells expressing these mutants are consistent with these findings (Fig. 4C). As expected, wild type ErbB2 exhibited robust EGF-stimulated tyrosine phosphorylation when it was co-expressed with wild type ErbB2. Similarly, kinase-dead ErbB2 was heavily phosphorylated when cells co-expressing the wild type EGF receptor were stimulated with EGF. By contrast, stimulation of cells expressing wild type EGF receptors and Y253D-ErbB2 with EGF did not lead to a significant increase in the phosphorylation of the Y253D-ErbB2. Thus, optimal phosphorylation of ErbB2 appears to require formation of back-to-back EGFR/ErbB2 heterodimers.
Structural Requirements for the Effects of ErbB2 on EGF Receptor Function
To determine the effect of these two mutations on the ability of ErbB2 to modulate EGF receptor function, we used stable cell lines that expressed the wild type EGF receptor from a constitutive promoter and also expressed wild type ErbB2, K732M-ErbB2, or Y253D-ErbB2 from a tet-inducible promoter. The expression levels of ErbB2 in the absence and presence of doxycycline are shown in Fig. 5A.
FIGURE 5.

Y253D-ErbB2 does not modulate EGF receptor binding affinity. Cells constitutively expressing the EGF receptor were stably transfected with wild type ErbB2 (panel A), K732M-ErbB2 (panel B), or Y253D-ErbB2 (panel C) expressed from a tet-inducible promoter. For assay, cells were grown in the absence (open circles) or presence (filled circles) of doxycycline to induce the expression of ErbB2. All lines express ∼300,000 EGF receptors/cell. Relative levels of ErbB2 expressed in each line are shown in the inset of panel A. 125I-EGF binding isotherms were then generated in cells expressing only the EGF receptor or the same number of EGF receptors plus the different forms of ErbB2.
These lines were then used to test the ability of the mutated forms of ErbB2 to increase the affinity of EGF for its receptor. When wild type EGF receptors were expressed alone at a level of ∼300,000 receptors/cell, the 125I-EGF saturation binding isotherm exhibited a KD of ∼1 nm. Upon addition of doxycycline to induce the expression of a similar level of wild type ErbB2, the binding isotherm shifted ∼3-fold to the left (Fig. 5A). This is the expected result because the EGFR/ErbB2 heterodimer exhibits a higher affinity for EGF than does the EGFR homodimer. As shown in Fig. 5B, expression of a similar level of kinase-dead ErbB2 in cells constitutively expressing the wild type EGF receptor also resulted in a leftward shift in the 125I-EGF binding isotherm. This indicates that the ability of ErbB2 to elicit an increase in the affinity of EGF for the EGFR/ErbB2 heterodimer is independent of the kinase activity of ErbB2. By contrast, expression of the Y253D-ErbB2 mutant that cannot dimerize with the EGF receptor failed to shift the 125I-EGF binding isotherm (Fig. 5C), indicating that the ability of ErbB2 to enhance the affinity of EGF for its receptor is dependent on the formation of a back-to-back EGFR·ErbB2 complex.
The expression of ErbB2 has been reported to prolong the autophosphorylation of the EGF receptor and reduce or delay down-regulation of the EGF receptor (35, 36). To determine whether the structural features required for modulation of ligand binding affinity also pertain to these effects of ErbB2, we assessed the autophosphorylation and down-regulation of the EGF receptor in the absence or presence of wild type, K732M-, or Y253D-ErbB2.
Co-expression of the EGF receptor with wild type ErbB2 led to a marked increase in the extent and duration of EGF receptor phosphorylation (Fig. 6A). This observation is consistent with the predictions from our simulations (Fig. 2) that indicate that expression of ErbB2 leads to the formation and maintenance of higher levels of signaling-competent receptor dimers. The expression of kinase-dead K721M-ErbB2 also enhanced the extent and duration of EGF receptor autophosphorylation (Fig. 6B). By contrast, expression of the nondimerizing Y253D-ErbB2 failed to alter the pattern of EGF receptor tyrosine phosphorylation (Fig. 6C). These data indicate that the ability of ErbB2 to modulate EGF receptor phosphorylation requires the interaction of its extracellular domain with the EGF receptor but is independent of its kinase activity.
FIGURE 6.

Effect of ErbB2 expression on EGF receptor phosphorylation. Cells constitutively expressing ∼300,000 EGF receptors/cell were stably transfected with wild type ErbB2 (panel A), K732M-ErbB2 (panel B), or Y253D-ErbB2 (panel C) expressed from a tet-inducible promoter. For assay, cells were grown in the absence (open circles) or presence (filled circles) of doxycycline to induce the expression of ErbB2. Cells were stimulated with 10 nm EGF for the times indicated and the autophosphorylation of the EGF receptor was analyzed by Western blotting with an antibody against phosphotyrosine 1068 in the EGF receptor. The results are quantitated in the graphs below.
As shown in Fig. 7, these same structural requirements were observed for the ability of ErbB2 to blunt EGF receptor down-regulation. Treatment of cells expressing only the wild type EGF receptor with 10 nm EGF for increasing lengths of time resulted in a rapid reduction in the number of cell surface EGF receptors as assessed by 125I-EGF binding (Fig. 7A). Induction of the expression of wild type ErbB2 in these cells reduced the extent of receptor down-regulation by approximately half. Similarly, co-expression of kinase-dead ErbB2 with the EGF receptor reduced the extent of receptor down-regulation induced by EGF (Fig. 7B). However, expression of Y253D-ErbB2 failed to alter the rate at which the EGF receptor was removed from the cell surface following exposure to EGF (Fig. 7C).
FIGURE 7.

Effect of ErbB2 expression on down-regulation of the EGF receptor. Cells constitutively expressing ∼300,000 EGF receptors/cell were stably transfected with wild type ErbB2 (panel A), K732M-ErbB2 (panel B), or Y253D-ErbB2 (panel C) expressed from a tet-inducible promoter. For assay, cells were grown in the absence (open circles) or presence (filled circles) of doxycycline to induce the expression of ErbB2. Cells were treated with 10 nm EGF for the indicated times then washed to remove surface bound EGF. Subsequently, binding of 125I-EGF to the cultures was measured as described under “Materials and Methods.”
DISCUSSION
ErbB2 appears to be the preferred dimerization partner among ErbB receptors (12, 13) and its expression has been reported to change the binding properties of EGF in cells (13, 22, 37). However, given the complexity introduced by the presence of both EGF receptor homodimers and EGFR/ErbB2 heterodimers in cells expressing both receptors, it has not been possible to explicitly define the effect of ErbB2 on EGF binding affinity or to understand why ErbB2 appears to be the preferred dimerization partner of all ErbB receptors, including the EGF receptor. The work reported here addresses these issues. By measuring the binding of 125I-EGF to the EGF receptor in cells containing progressively increasing levels of ErbB2, we were able to determine the equilibrium association constants for the homo- and heterodimerization of the EGF receptor and ErbB2, as well as the affinity of EGF for the EGF receptor homodimer versus the EGFR/ErbB2 heterodimer.
Our data indicate that for the unoccupied EGF receptor (i.e. for the formation of inactive pre-dimers), homodimerization of the EGF receptor is actually slightly preferred over heterodimerization with ErbB2. However, once the pre-dimers are formed, EGF binds with ∼7-fold higher affinity to the EGFR/ErbB2 heterodimer than to the first site on the EGF receptor homodimer. Thus, dimerization with ErbB2 allows the EGF receptor to adopt a conformation that is more favorable for ligand binding than dimerization with another EGF receptor.
The structure of the half-occupied Drosophila EGF receptor may represent a good model for the EGFR/ErbB2 heterodimer (38). In that structure, the binding of the Spitz ligand wedges apart subdomains I and III of the extracellular domain and bends the dimerization arm. These changes are readily accommodated in the singly ligated dimer but cannot be reproduced when Spitz binds to the second subunit. As a result, binding of Spitz to the second site of the dEGF receptor is of substantially lower affinity than binding to the first site. We hypothesize that in the EGFR/ErbB2 heterodimer, ErbB2 serves as a “constitutively unoccupied” partner. This would allow the formation of an asymmetric dimer of the EGF receptor and ErbB2 extracellular domains that optimizes the affinity of the lone EGF receptor subunit for EGF. This is consistent with the observations of Liu et al. (39) who noted that the relative orientation of subdomains II and IV in the extracellular domain of ErbB2 allow it to align optimally with a liganded partner and serve as the unliganded subunit in singly ligated ErbB heterodimers.
Based on the parameters derived from our binding studies, we were able to predict the levels of the various EGF receptor and ErbB2 species at equilibrium under several different conditions. Our simulations revealed that in cells expressing only EGF receptors, relatively few EGF receptors exist as dimers even at saturating concentrations of ligand. This is because the negative cooperativity in the homodimer leads to the dissociation of doubly occupied dimers, forming ligand-occupied monomers. By contrast, when ErbB2 is coexpressed with EGF receptors in cells, a much higher level of dimers (specifically, EGFR/ErbB2 heterodimers) is maintained.
The fundamental importance of negative cooperativity in determining the level of receptor dimers is highlighted by the results of the simulation carried out on an EGF receptor that (theoretically) lacks negative cooperativity (i.e. K21 = K22). In this case, the extent of dimer formation is very similar to that seen for the EGFR/ErbB2 heterodimer. Thus, negative cooperativity appears to play a major role in setting the level of EGF receptor homodimers. As the EGFR/ErbB2 heterodimer is not subject to negative cooperativity, ErbB2 appears to be the preferred dimerization partner largely because it does not bind EGF.
Interestingly, much of the effect of ErbB2 on the function of the EGF receptor appears to be independent of the kinase activity of ErbB2. Luciferase complementation assays using EGF receptors and ErbB2 that lacked the entire intracellular domain demonstrated that expression of the truncated ErbB2 still resulted in an increase in the EC50 of EGF for the EGF receptor. Similarly, in luciferase assays using full-length receptors, the kinase-dead ErbB2 interacted with the EGF receptor in the same way as wild type ErbB2. Furthermore, expression of kinase-dead ErbB2 shifted the EGF binding isotherm to the left, enhanced EGF receptor autophosphorylation, and reduced receptor down-regulation. ErbB2 was also phosphorylated normally when co-expressed with wild type EGF receptors. These findings suggest that ErbB2-mediated alterations in EGF receptor function arise, to a significant extent, from structural effects rather than from the introduction of a kinase domain with a different substrate specificity.
We have recently shown that the mechanics of EGFR/ErbB2 heterodimer activation are determined by the activity status of the kinase domain of the EGF receptor, but are independent of the kinase activity of ErbB2 (23). A heterodimer containing wild type EGF receptor and kinase-dead ErbB2 behaves as if both partners were kinase-active. But a heterodimer containing kinase-dead EGF receptors and wild type ErbB2 behaves as if both partners were kinase-dead and neither the EGF receptor nor ErbB2 are phosphorylated in response to EGF. Thus, even when the heterodimer contains one active kinase (ErbB2), no EGF-stimulated phosphorylation is observed in the absence of an active EGF receptor kinase domain. When considered together with the present findings that the ability of ErbB2 to modulate EGF receptor binding affinity and other signaling functions is independent of the catalytic activity of the ErbB2 kinase, it seems warranted to consider the possibility the ErbB2 subunit is simply an allosteric modulator of the EGF receptor and that its kinase is never actually activated within the EGFR/ErbB2 heterodimer.
Although the effects of ErbB2 on EGF receptor function are independent of ErbB2 kinase activity, they clearly require the formation of back-to-back dimers by the extracellular domain. Mutation of Tyr-253 in the dimerization arm of ErbB2 blocked its interaction with the EGF receptor in luciferase complementation assays and abolished the ability of ErbB2 to shift the EGF binding isotherm, enhance EGF receptor autophosphorylation, or limit EGF receptor down-regulation. These findings are consistent with the notion that ErbB2 modulated EGF receptor function through structural mechanisms that involve the extracellular dimerization interface.
This extracellular domain-mediated dimerization appears to be required for phosphorylation of ErbB2 as the Y253D-ErbB2 subunit was not phosphorylated in response to EGF in cells co-expressing wild type EGF. These data suggest that the majority of ErbB2 phosphorylation occurs within the context of an EGFR/ErbB2 heterodimer, or possibly a higher order oligomer assembled from EGFR/ErbB2 heterodimers. Although the possibility of lateral phosphorylation of ErbB2 as an “exogenous” substrate for an EGF receptor homodimer under some circumstances cannot be ruled out, it is clear that at moderate levels of ErbB2 expression this does not represent a major mechanism for the EGF-dependent phosphorylation of ErbB2.
In summary, we have quantitated the effect of ErbB2 on EGF receptor binding affinity and shown that ErbB2 is the preferred dimerization partner for the EGF receptor largely because it alleviates the effect of negative cooperativity on dimer stability. Thus, one purpose for the ligand-less ErbB2 subunit may be to provide flexibility in determining the overall level of signaling active, kinase dimers that can be maintained at any given concentration of growth factor. The data demonstrate that differences in the extracellular domain dimerization interface give rise to changes in the ligand binding properties of the EGF receptor and are consistent with the hypothesis that ErbB2 modifies EGF receptor function through protein-protein interactions rather than through its catalytic activity.
Supplementary Material
Footnotes
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM82824 (to L. J. P.) and P50 CA94056 (to D. P.-W.).

This article contains supplemental Materials.
REFERENCES
- 1. Ullrich A., Coussens L., Hayflick J. S., Dull T. J., Gray A., Tam A. W., Lee J., Yarden Y., Libermann T. A., Schlessinger J., Downward J., Mayes E. L. V., Whittle N., Waterfield M. D., Seeburg P. H. (1984) Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 309, 418–425 [DOI] [PubMed] [Google Scholar]
- 2. Ferguson K. M. (2008) Structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 37, 353–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lemmon M. A., Schlessinger J. (2010) Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lemmon M. A. (2009) Ligand-induced ErbB receptor dimerization. Exp. Cell Res. 315, 638–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wilson K. J., Gilmore J. L., Foley J., Lemmon M. A., Riese D. J., 2nd (2009) Functional selectivity of EGF family peptide growth factors. Implications for cancer. Pharmacol. Ther. 122, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hofman E. G., Bader A. N., Voortman J., van den Heuvel D. J., Sigismund S., Verkleij A. J., Gerritsen H. C., van Bergen en Henegouwen P. M. (2010) Ligand-induced EGF receptor oligomerization is kinase-dependent and enhances internalization. J. Biol. Chem. 285, 39481–39489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu P., Sudhaharan T., Koh R. M., Hwang L. C., Ahmed S., Maruyama I. N., Wohland T. (2007) Investigation of the dimerization of proteins from the epidermal growth factor receptor family by single wavelength fluorescence cross-correlation spectroscopy. Biophys. J. 93, 684–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sako Y., Minoghchi S., Yanagida T. (2000) Single-molecule imaging of EGFR signalling on the surface of living cells. Nat. Cell Biol. 2, 168–172 [DOI] [PubMed] [Google Scholar]
- 9. Tao R. H., Maruyama I. N. (2008) All EGF(ErbB) receptors have preformed homo- and heterodimeric structures in living cells. J. Cell Sci. 121, 3207–3217 [DOI] [PubMed] [Google Scholar]
- 10. Yu X., Sharma K. D., Takahashi T., Iwamoto R., Mekada E. (2002) Ligand-independent dimer formation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Mol. Biol. Cell 13, 2547–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yarden Y., Schlessinger J. (1987) Self-phosphorylation of epidermal growth factor receptor. Evidence for a model of intermolecular allosteric activation. Biochemistry 26, 1434–1442 [DOI] [PubMed] [Google Scholar]
- 12. Graus-Porta D., Beerli R. R., Daly J. M., Hynes N. E. (1997) ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 16, 1647–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tzahar E., Waterman H., Chen X., Levkowitz G., Karunagaran D., Lavi S., Ratzkin B. J., Yarden Y. (1996) A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell. Biol. 16, 5276–5287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang X., Gureasko J., Shen K., Cole P. A., Kuriyan J. (2006) An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149 [DOI] [PubMed] [Google Scholar]
- 15. Ferguson K. M., Berger M. B., Mendrola J. M., Cho H. S., Leahy D. J., Lemmon M. A. (2003) EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell 11, 507–517 [DOI] [PubMed] [Google Scholar]
- 16. Garrett T. P., McKern N. M., Lou M., Elleman T. C., Adams T. E., Lovrecz G. O., Zhu H. J., Walker F., Frenkel M. J., Hoyne P. A., Jorissen R. N., Nice E. C., Burgess A. W., Ward C. W. (2002) Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor α. Cell 110, 763–773 [DOI] [PubMed] [Google Scholar]
- 17. Ogiso H., Ishitani R., Nureki O., Fukai S., Yamanaka M., Kim J. H., Saito K., Sakamoto A., Inoue M., Shirouzu M., Yokoyama S. (2002) Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 110, 775–787 [DOI] [PubMed] [Google Scholar]
- 18. Adak S., DeAndrade D., Pike L. J. (2011) The tethering arm of the EGF receptor is required for negative cooperativity and signal transduction. J. Biol. Chem. 286, 1545–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lu C., Mi L. Z., Grey M. J., Zhu J., Graef E., Yokoyama S., Springer T. A. (2010) Structural evidence for loose linkage between ligand binding and kinase activation in the epidermal growth factor receptor. Mol. Cell. Biol. 30, 5432–5443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Macdonald J. L., Pike L. J. (2008) Heterogeneity in EGF-binding affinities arises from negative cooperativity in an aggregating system. Proc. Natl. Acad. Sci. U.S.A. 105, 112–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Macdonald-Obermann J. L., Pike L. J. (2009) The intracellular juxtamembrane domain of the epidermal growth factor (EGF) receptor is responsible for the allosteric regulation of EGF binding. J. Biol. Chem. 284, 13570–13576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karunagaran D., Tzahar E., Beerli R. R., Chen X., Graus-Porta D., Ratzkin B. J., Seger R., Hynes N. E., Yarden Y. (1996) ErbB-2 is a common auxiliary subunit of NDF and EGF receptors. Implications for breast cancer. EMBO J. 15, 254–264 [PMC free article] [PubMed] [Google Scholar]
- 23. Macdonald-Obermann J. L., Piwnica-Worms D., Pike L. J. (2012) Mechanics of EGF receptor/ErbB2 kinase activation revealed by luciferase fragment complementation imaging. Proc. Natl. Acad. Sci. U.S.A. 109, 137–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Contreras M. A., Bale W. F., Spar I. L. (1983) Iodine monochloride (IC1) iodination techniques. Methods Enzymol. 92, 277–292 [PubMed] [Google Scholar]
- 25. Yang K. S., Macdonald-Obermann J. L., Piwnica-Worms D., Pike L. J. (2010) Asp-960/Glu-961 controls the movement of the C-terminal tail of the epidermal growth factor receptor to regulate asymmetric dimer formation. J. Biol. Chem. 285, 24014–24022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang K. S., Ilagan M. X., Piwnica-Worms D., Pike L. J. (2009) Luciferase fragment complementation imaging of conformational changes in the epidermal growth factor receptor. J. Biol. Chem. 284, 7474–7482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sarup J. C., Johnson R. M., King K. L., Fendly B. M., Lipari M. T., Napier M. A., Ullrich A., Shepard H. M. (1991) Characterization of an anti-p185HER2 monoclonal antibody that stimulates receptor function and inhibits tumor cell growth. Growth Regul. 1, 72–82 [PubMed] [Google Scholar]
- 28. Cho H. S., Mason K., Ramyar K. X., Stanley A. M., Gabelli S. B., Denny D. W., Jr., Leahy D. J. (2003) Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421, 756–760 [DOI] [PubMed] [Google Scholar]
- 29. Garrett T. P., McKern N. M., Lou M., Elleman T. C., Adams T. E., Lovrecz G. O., Kofler M., Jorissen R. N., Nice E. C., Burgess A. W., Ward C. W. (2003) The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol. Cell 11, 495–505 [DOI] [PubMed] [Google Scholar]
- 30. Adak S., Yang K. S., Macdonald-Obermann J., Pike L. J. (2011) The membrane-proximal intracellular domain of the epidermal growth factor receptor underlies negative cooperativity in ligand binding. J. Biol. Chem. 286, 45146–45155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chantry A. (1995) The kinase domain and membrane localization determine intracellular interactions between epidermal growth factor receptors. J. Biol. Chem. 270, 3068–3073 [PubMed] [Google Scholar]
- 32. Clayton A. H., Walker F., Orchard S. G., Henderson C., Fuchs D., Rothacker J., Nice E. C., Burgess A. W. (2005) Ligand-induced dimer-tetramer transition during the activation of the cell surface epidermal growth factor receptor. A multidimensional microscopy analysis. J. Biol. Chem. 280, 30392–30399 [DOI] [PubMed] [Google Scholar]
- 33. Martin-Fernandez M., Clarke D. T., Tobin M. J., Jones S. V., Jones G. R. (2002) Preformed oligomeric epidermal growth factor receptors undergo an ectodomain structure change during signaling. Biophys. J. 82, 2415–2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luker K. E., Smith M. C., Luker G. D., Gammon S. T., Piwnica-Worms H., Piwnica-Worms D. (2004) Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc. Natl. Acad. Sci. U.S.A. 101, 12288–12293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Offterdinger M., Bastiaens P. I. (2008) Prolonged EGFR signaling by ERBB2-mediated sequestration at the plasma membrane. Traffic 9, 147–155 [DOI] [PubMed] [Google Scholar]
- 36. Wang Z., Zhang L., Yeung T. K., Chen X. (1999) Endocytosis deficiency of epidermal growth factor (EGF) receptor-ErbB2 heterodimers in response to EGF stimulation. Mol. Biol. Cell 10, 1621–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wada T., Qian X. L., Greene M. I. (1990) Intermolecular association of the p185neu protein and EGF receptor modulates EGF receptor function. Cell 61, 1339–1347 [DOI] [PubMed] [Google Scholar]
- 38. Alvarado D., Klein D. E., Lemmon M. A. (2010) Structural basis for negative cooperativity in growth factor binding to an EGF receptor. Cell 142, 568–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu P., Cleveland T. E., 4th, Bouyain S., Byrne P. O., Longo P. A., Leahy D. J. (2012) A single ligand is sufficient to activate EGFR dimers. Proc. Natl. Acad. Sci. U.S.A. 109, 10861–10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.