

Background: Human UDP-xylose synthase (hUXS1) is responsible for conversion of UDP-glucuronic acid to UDP-xylose.
Results: Crystal structure, molecular dynamics simulations, and reaction course analysis give conclusive insight into the enzymatic mechanism in three catalytic steps.
Conclusion: Distortion of sugar pyranose ring in bound substrate facilitates enzymatic reaction.
Significance: A detailed mechanism for catalysis by hUXS1 is proposed.
Keywords: Carbohydrate Biosynthesis, Decarboxylase, Dehydrogenase, Enzyme Mechanisms, Enzyme Structure, Glycosaminoglycan, Enzyme Reaction Coordinate, Short-chain Dehydrogenase/Reductase, Substrate Distortion/Destabilization, UDP-glucuronic Acid
Abstract
UDP-xylose synthase (UXS) catalyzes decarboxylation of UDP-d-glucuronic acid to UDP-xylose. In mammals, UDP-xylose serves to initiate glycosaminoglycan synthesis on the protein core of extracellular matrix proteoglycans. Lack of UXS activity leads to a defective extracellular matrix, resulting in strong interference with cell signaling pathways. We present comprehensive structural and mechanistic characterization of the human form of UXS. The 1.26-Å crystal structure of the enzyme bound with NAD+ and UDP reveals a homodimeric short-chain dehydrogenase/reductase (SDR), belonging to the NDP-sugar epimerases/dehydratases subclass. We show that enzymatic reaction proceeds in three chemical steps via UDP-4-keto-d-glucuronic acid and UDP-4-keto-pentose intermediates. Molecular dynamics simulations reveal that the d-glucuronyl ring accommodated by UXS features a marked 4C1 chair to BO,3 boat distortion that facilitates catalysis in two different ways. It promotes oxidation at C4 (step 1) by aligning the enzymatic base Tyr147 with the reactive substrate hydroxyl and it brings the carboxylate group at C5 into an almost fully axial position, ideal for decarboxylation of UDP-4-keto-d-glucuronic acid in the second chemical step. The protonated side chain of Tyr147 stabilizes the enolate of decarboxylated C4 keto species (2H1 half-chair) that is then protonated from the Si face at C5, involving water coordinated by Glu120. Arg277, which is positioned by a salt-link interaction with Glu120, closes up the catalytic site and prevents release of the UDP-4-keto-pentose and NADH intermediates. Hydrogenation of the C4 keto group by NADH, assisted by Tyr147 as catalytic proton donor, yields UDP-xylose adopting the relaxed 4C1 chair conformation (step 3).
Introduction
Proteoglycans are complex assemblies between proteins and linear glycosaminoglycan polysaccharides such as heparan sulfate and chondroitin sulfate (1). They are found in all connective tissues of vertebrates, and on the surfaces of many cell types as components of the extracellular matrix or the glycocalix (1–3). Due to hydration of their glycosaminoglycan chains, proteoglycans impart hydrostatic and elastic properties to the extracellular matrix. As receptors, proteoglycans are critically involved in cell signaling, with consequent effects on tissue development in health or disease (e.g. cancer) (1–6). Association between glycosaminoglycan molecules and the protein core of the proteoglycan usually involves enzymatic transfer of a xylosyl residue from UDP-xylose to a protein serine (7). Covalent attachment of the xylosyl group is the first step in generating a functional tetrasaccharide linker on which glycosaminoglycan chain elongation takes place. The UDP-xylose precursor is derived from UDP-glucuronic acid through complex oxidoreductive decarboxylation catalyzed by UDP-xylose synthase (UXS; EC 4.1.1.35; other names: UDP-glucuronic acid decarboxylase; UDP-glucuronic acid carboxylyase) (8, 9). Adequate supply of UDP-xylose was shown in zebrafish to be essential for functional deposition of proteoglycans in the extracellular matrix (10). Defects in production and organization of the extracellular matrix resulting from insufficient UXS3 activity were correlated with alterations in cell signaling pathways, mirrored in the impaired morphogenesis of various tissues, including the bone. The central and universal biological importance of UXS in vertebrates, including mammals, therefore raises considerable interest in structural and functional properties of the enzyme. In this work, we present a comprehensive characterization of the human form of UXS (hUXS1) that involved determination of a crystal structure at 1.26-Å resolution.
Phylogenetic analysis reveals that UXS is one of the most strongly conserved nonmitochondrial proteins in nature, with about 57% sequence identity shared between hUXS1 and various bacterial enzyme forms (10). The physiological involvement of UXS differs across the three domains of life, and besides the above described synthesis of protein-linked glycosaminoglycans (7), it also includes the formation of xylose-containing polysaccharides in plants (11) and microorganisms (12–15). Even though UXS activity has been reported in various organisms and cell types, there have only been a few studies of the isolated enzyme. Working with UXS from wheat germ and the fungus Cryptococcus laurentii, Feingold and colleagues (16) have developed an ingenious chemical proposal that is shown in Scheme 1. The reaction catalyzed by UXS starts with oxidation of the C4 alcohol in UDP-glucuronic acid by enzyme-bound NAD+. The resulting UDP-4-keto-glucuronic acid undergoes decarboxylation, followed by protonation to yield UDP-4-keto-pentose. Keto group reduction by the NADH still bound to the enzyme occurs with retention of the original stereochemistry at C4, giving UDP-xylose as product. A recent NMR study of a bacterial UXS ortholog appears consistent with the proposed reaction course (17). These insights into the enzymatic transformation notwithstanding, the catalytic mechanism of UXS is fundamentally unknown. The mechanism merits a detailed investigation as it is both biologically very important and chemically intriguing (18–19).
SCHEME 1.
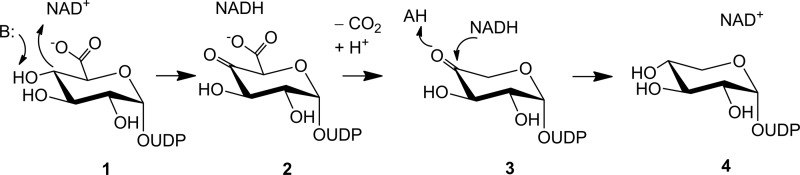
Proposed course of the UXS-catalyzed reaction. See text for further explanations.
Based on similarity at the primary structure level, hUXS1 is classified in the short-chain dehydrogenase/reductase (SDR) superfamily of proteins (20). hUXS1 is most closely related to the divergent group of so-called “extended SDRs” that include nucleotide-sugar epimerization and dehydration as their main activities (for reviews on enzyme mechanisms, see Refs. 18, 19, 21, and 22). Sequence alignment (supplemental Fig. S1) suggests that hUXS1 is equipped with the canonical SDR active site in which a highly conserved triad of residues (Thr118, Tyr147, Lys151) is probably involved in catalytic oxidoreduction. However, there are also variable features (e.g. Glu120, Arg277) that could have a decisive role in the enzymatic conversion. Herein we report on results of structure determination, molecular dynamics (MD) simulation, and reaction course analysis for wild-type and mutated forms of hUXS1, which in combination give conclusive evidence on the enzyme mechanism, delineating how individual active site residues assist the reaction in each of its three catalytic steps. A significant feature of the mechanism is that the catalytic reaction course is underpinned by promoting conformational changes in the pyranose ring structure of the UDP-sugar substrate/intermediate. We have identified the protein residues responsible for the sugar ring distortion and clarified their role in catalysis.
EXPERIMENTAL PROCEDURES
Materials
Unless stated otherwise, all materials were of highest purity available from Roth (Karlsruhe, Germany). UDP-glucuronic acid (>98% purity) was obtained from Sigma and UDP-xylose (>95% purity) from Carbosynth (Compton, UK). D2O (99.8% D) was purchased from ARMAR Chemicals (Döttingen, Switzerland).
Protein Preparation
We used a truncated form of hUXS1 (residues 85–402) that was optimized for crystallization. The construct was designed to encompass the entire Rossmann-fold and ordered regions at the C terminus. Regions predicted to encode a signal peptide and transmembrane helix were deleted from the N terminus, whereas a predicted random coil was deleted from the C terminus. The final construct contained a 23-amino acid long N-terminal peptide comprising the His6 tag. We refer to this construct throughout when hUXS1 is mentioned. Experimental procedures used for recombinant protein preparation are described in full detail in the supplemental “Methods”. Briefly, the enzyme was overexpressed in Escherichia coli Rosetta 2(DE3) using the pET-derived expression vector p11 (wild-type) or the pNIC28-Bsa4 plasmid (mutants). After high-pressure cell disruption (1500 psi), the enzyme was isolated from the crude extract using a Cu2+-loaded IMAC-Sepharose High Performance column (GE Healthcare). Stepwise elution with imidazole was employed to yield highly pure hUXS1 (checked by SDS-PAGE). Imidazole was removed by buffer exchange to 50 mm Tris/HCl (pH 7.5) containing 5% glycerol and 1 mm DTT using a NAP-25 desalting column (GE Healthcare) or Vivaspin-20 centrifugal concentrator (Sartorius Stedim, Göttingen, Germany). Preparations of hUXS1 were stored at −70 °C. For crystallization experiments, SeMet-labeled enzyme was produced in E. coli B834(DE3) grown on SelenoMethionine Medium Base plus Nutrient Mix containing SeMet at 40 mg/liter (Molecular Dimensions, Newmarket, UK). Site-directed mutagenesis was performed using standard protocols. Mutants purified similarly as the wild-type enzyme.
Crystallization, Data Collection, and Refinement
SeMet-labeled hUXS1 containing 5 mm NAD+ and 5 mm UDP was concentrated to 12 mg/ml (∼0.3 mm). Crystals were obtained by vapor diffusion at 20 °C in 150-nl sitting drops, equilibrated against mother liquor containing 20% PEG 6000, 10% ethylene glycol, and 0.2 m NaCl in 0.1 m HEPES (pH 7). Diffraction data to 1.2 Å were collected at 100 K at the Swiss Light Source station X10SA using a MARMOSAIC 225 mm CCD detector at a single wavelength near the selenium absorption edge (λ = 0.9795 Å). Data were indexed and integrated in MOSFLM (23) and scaled in SCALA (23). Merging of the data and analysis of systematic absences identified the space group as P121, with the following cell dimensions: a = 46 Å, b = 45 Å, c = 85 Å; α = 90°, β = 97°, γ = 90°. The program SHELXD (24) located 11 SeMet residues in hUXS1, indicating complete labeling of the protein. SHELXE was used for phasing and automated chain tracing was done with ARP/wARP. The hUXS1 structure was manually rebuilt in COOT (25), and restrained refinement was performed using REFMAC5 (26). When electron density showed the presence of NAD+ and UDP in the active site as well as an additional UDP forming a crystal contact, the ligands were fitted to the density and included in the refinement. Although the full range of data to 1.2 Å was used in refinement, analysis of data completeness and signal to noise suggests that 1.26 Å is a more reasonable estimate of the resolution of the model. Data collection and refinement statistics are summarized in Table 1.
TABLE 1.
Crystallographic data collection and refinement statistics
| Enzyme complex | hUXS1-UDP-NAD+ |
|---|---|
| PDB accession code | 2B69 |
| Synchrotron beamline | SLS, X10SA |
| Wavelength (Å) | 0.9795 |
| Space group | P121 |
| Unit cell dimensions | a = 46, b = 45, c = 85 Å |
| α = γ = 90.0° | |
| β = 97° | |
| Resolution rangea (Å) | 42–1.20 (1.26–1.20) |
| No. unique reflectionsa | 93023 (8576) |
| Completenessa (%) | 91.3 (58.1) |
| Mean I/sdI 〈I/σ(I)〉a | 25.2 (1.3) |
| Rmergea (%) | 6.5 (53.7) |
| Redundancya | 2.1 (1.3) |
| Refinement | |
| No. atoms P/L/Ob | 2439/94/353 |
| Rwork/Rfree (%) | 13.4/16.2 |
| R.m.s. deviation bond lengthc (Å) | 0.014 |
| R.m.s. deviation bond angle (°) | 1.506 |
| Bmean P/L/Ob (Å2) | 14/28/32 |
a Values in parentheses correspond to data in highest resolution shell.
b P/L/O refer to protein, ligand, and solvent atoms.
c R.m.s. deviation is root mean square deviation.
Molecular Dynamics Simulation
All MD simulations were performed using Gromacs (27). For the protein and water a variation of the Amber99sb (28) and the TIP3P (29) force fields was used. For NAD, substrate, intermediates, and product General Amber Force Field (GAFF) parameters (30) and Gromacs input files were generated using the Amber Antechamber suite of programs (31) together with the perl script amb2gmx (32). As starting structure for MD simulations the crystal structure of UXS (PDB code 2B69) was used, approximate pKa values of ionizable residues were established using PROPKA (33), and ionizable residues were protonated accordingly. For the MD simulations the protein with substrate, product, or intermediates was positioned in the center of a cubic box with 6.7-nm side length and solvated in ∼8,000 water molecules. The appropriate number of water molecules was replaced by sodium ions to neutralize the system. A leapfrog algorithm with a time step of 2 fs was used for integrating the equations of motion. The temperature was kept constant at 300 K using a Nosé-Hoover thermostat (34). Electrostatic long range interactions were accounted for using a particle mesh Ewald algorithm (35). A cut-off of 10 Å was used for the real space part of the Ewald summation and the van der Waals interactions. A constant pressure of 1 atm was maintained using a Berendsen thermostat (36). All bonds including hydrogens were constrained using a LINCS algorithm (37). Conformations were saved every 2 ps. The MD simulations covered a time between 4 and 6 ns. In each case the trajectories covering the last 2 ns were used to calculate hydrogen bonding and average structures. For visual inspection of trajectories and conformations Chimera was used. The trajectories were analyzed with various Gromacs tools and in-house scripts.
Analysis of the Enzymatic Reaction Course
Reactions were performed at 25 °C in 50 mm Tris/HCl buffer (pH 7.5). Substrate and enzyme concentrations as well as reaction times differed depending on the specific experimental setup and are therefore mentioned individually. Reactions were stopped by addition of acetonitrile in a 1:1 volume ratio to buffer. Precipitated protein was removed by centrifugation (10 min, 4 °C, 16,000 × g). Samples were analyzed with a reversed phase HPLC (see supplemental “Methods”). It was shown that consumption of UDP-GlcUA proceeded linearly in dependence of reaction time, so that measurements were suitable for calculation of initial rates. Data for kinetic analysis were obtained under conditions in which the concentration of NAD+ or UDP-GlcUA was varied, whereas the concentration of the respective other substrate was constant (wild-type, 0.5 mm NAD+ and 2 mm UDP-GlcUA; mutants, 10 mm NAD+ and 10 mm UDP-GlcUA). Vmax values were derived from nonlinear fits of the Michaelis-Menten equation. The molarity of enzyme active sites (E) was calculated from the protein concentration, assuming a molecular mass of 38,595 Da for the protein subunit. It was used to determine the turnover number, using the relationship kcat = Vmax/E. Experiments were performed in triplicate (S.D. ≤ 15%).
Transient Kinetic Studies
Experiments were performed at 25 °C with an Applied Photophysics (Leatherhead, UK) model SX.18 MV Stopped-flow Spectrometer. Enzyme was used in a limiting concentration (≤25 μm). A 50 mm Tris/HCl buffer (pH 7.5) was used. Enzyme solution was mixed in 1:1 ratio with substrate solution, yielding concentrations of 0.5 mm NAD+ and 10 mm UDP-GlcUA, unless stated otherwise. The absorbance of enzyme-bound NADH was measured at 340 nm and converted to concentration using an extinction coefficient of 6,220 m−1 cm−1. The stopped-flow instrument had a dead-time of 1.5 ms, and data were recorded up to 1,000 s. Slow reactions catalyzed by hUXS1 mutants were followed additionally in a conventional UV/visible spectrophotometer (Beckman Coulter DU 800), measuring the change in absorbance at 340 nm.
Solvent Kinetic Isotope Effect
For examination of the solvent kinetic isotope effect, a 20 mm potassium phosphate buffer (pH/pD 7.5) in H2O or D2O was prepared. The pD was determined as pH meter reading plus 0.4 (38). Concentrations of 10 mm UDP-GlcUA, 0.5 (wild type) or 1 (E120A) mm NAD+, and 15 (wild type) or 36 (E120A) μm enzyme were used. Enzymes were gel-filtered three times employing deuterated phosphate buffer and equilibrated with solvent prior to the experiments in D2O. Reactions were stopped after 8 min (wild-type) or 50 min (E120A) by addition of acetonitrile in 1:1 ratio and samples were analyzed on the HPLC. Solvent isotope effects were calculated from the ratio of kcat values measured in H2O and D2O.
In Situ NMR Analysis
Enzymatic reactions were performed directly in a 5-mm high precision NMR sample tube (Promochem, Wesel, Germany) and monitored in situ over 12 h in the magnet, by recording up to 64 1H NMR spectra in regular intervals. For further measurements, samples were kept in a temperature-controlled water bath (30 °C) and measured in unsteady intervals over ∼48 h. All spectra were obtained with a Bruker DRX-600 AVANCE spectrometer (Bruker, Rheinstetten, Germany) at 600.13 MHz (1H) using the Bruker Topspin 1.3 software. Chemical shifts were referenced to external acetone (δH 2.225 ppm). Further details are provided under supplemental “Methods”.
Other Methods
Force field tests, nondenaturing anionic PAGE, and HPLC analysis are described in supplemental “Methods” and Fig. S5.
RESULTS AND DISCUSSION
High-resolution Crystal Structure of hUXS1
We solved the crystal structure of hUXS1 in complex with NAD+ and UDP to a resolution of 1.26 Å (PDB code 2B69; Table 1). The experimental structure consists of a single polypeptide chain of 312 amino acids and is composed of 12 α-helices (37%) and 17 β-strands (Fig. 1A, supplemental Fig. S1).4 The N-terminal His tag in addition to 3 N-terminal residues and 5 C-terminal residues were not visible in electron density maps and were not modeled. Although the asymmetric unit contains a single hUXS1 subunit, analysis of the crystal packing by the program PISA (39) suggests the protein is dimeric. Similar to other dimeric SDRs, the dimer interface is composed of helices α5 and α7, forming a helical bundle as shown in Fig. 1A. The monomeric form of hUXS1 has a predicted molecular mass of 38.6 kDa, which was confirmed by SDS-PAGE (supplemental Fig. S2A). In nondenaturing anionic PAGE, the enzyme showed a migration behavior consistent with its presence as a homodimer (supplemental Fig. S2B). Furthermore, we used gel filtration to estimate an apparent molecular mass of about 70–75 kDa for hUXS1 in solution (data not shown). The structural and biochemical data therefore support the notion that the enzyme is a functional homodimer (calculated mass 77.2 kDa). Mutants of hUXS1 migrated similarly in nondenaturing PAGE, giving evidence that they are also present as dimers in solution (data not shown).
FIGURE 1.

X-ray structure of hUXS1 bound with NAD+ and UDP. A, α-carbon trace of the hUXS1 crystallographic dimer. The single molecule in the asymmetric unit is colored blue with the UDP-GlcUA binding domain shown lighter blue and the helices at the dimeric interface darker blue. A symmetry-related molecule is shown in gray. B, surface representation of the hUXS1 active site. The NAD+ is buried with C4 14 Å from the surface at the interface between the two domains. Conserved residues are labeled. C and D, electron density (2Fo − Fc contoured at 1σ) and hydrogen-bonding interactions ≤3.2 Å for bound ligands NAD+ (C) and UDP (D). A water molecule coordinated to Thr118 is shown as a red sphere.
Functionally, hUXS1 can be split into two domains: a large NAD+-binding domain (residues 4–181, 216–242, and 279–294) and a considerably smaller UDP-GlcUA binding domain (residues 182–215, 243–278, and 295–315). The large domain of hUXS1 is built of a seven-stranded parallel β-sheet sandwiched between two arrays of parallel helices with a small helix capping one end of the sheet, resulting in a modified version of the classic Rossmann fold. The UDP-GlcUA binding domain is composed of two small 2-stranded β-sheets (one parallel and one anti-parallel) and a 3-helix bundle. The active site of the enzyme is located in a cavity formed between the two domains (Fig. 1B). Fig. 1 shows how NAD+ (panel C) and UDP (panel D) are accommodated in their respective binding sites on the enzyme. The NAD+ is buried deeply in the protein structure, suggesting that coenzyme is bound tightly by the enzyme. This is consistent with biochemical data indicating that hUXS1 displays about 60% of its maximum activity under assay conditions where no external NAD+ is added (supplemental Fig. S2C).
The Active Site of hUXS1
There are 6 highly conserved residues that make up the active site of hUXS1, as shown in Fig. 2. Thr118, Tyr147, and Lys151 form the characteristic signature of a typical SDR catalytic center. Ser119, Glu120, and Arg277 are distinct features of the UXS group of enzymes (Fig. 1B, supplemental Fig. S1). We analyzed binding of the substrate by carrying out an energy-minimized docking of UDP-GlcUA followed by MD simulation of the resulting ternary hUXS1·NAD+·substrate complex. The protein-ligand structure readily converged into a stable conformation that was characterized by a root mean square deviation of ≤0.15 nm as compared with the experimental structure (supplemental Fig. S3A). Fig. 2 shows that in the modeled structure the d-glucuronyl pyranose ring of UDP-GlcUA was distorted from the expected low-energy 4C1 chair conformation to BO,3 boat. Sugar ring distortion is required for Tyr147 to become hydrogen bonded with the C4 hydroxyl, thus positioning the reactive substrate group for general base catalysis by the tyrosine. The C4 hydrogen was within plausible reaction distance (3.6 Å) to the NAD+ nicotinamide C4, suggesting that the modeled complex would be catalytically competent. Hydride transfer would therefore take place with pro-S stereospecificity, fully consistent with expectations for a member of the SDR superfamily. Our model further suggests that Thr118 contributes to the sugar ring distortion by forming a strong hydrogen bond with the carboxylate group at C5. In the experimental structure of hUXS1 bound with UDP, Thr118 is bonded to a water molecule (Fig. 1C). Tyr147 and Lys151 were hydrogen bonded to the hydroxyls at C2 and C3 of the nicotinamide ribose (Fig. 1C), respectively, thus facilitating precise relative positioning of reactive parts of substrate and coenzyme at the enzyme active site, as also seen in classical SDR enzymes (40).
FIGURE 2.

Active site of hUXS1 with the d-glucuronyl ring of UDP-GlcUA accommodated in a distorted BO,3 conformation. Substrate binding mode was obtained by MD simulation. Hydrogen bonds are indicated by blue lines. The interaction between Ala79 and sugar C4 oxygen only occurred in <60% of structures during simulation (supplemental Fig. S3B) and is therefore shown transparently.
Fig. 2 also shows that Ser119 is strongly hydrogen bonded with the substrate carboxylate group. The interaction between Glu120 and Arg277 fastens together two long loops (residues 119–144 and 268–277), thus closing up the active site (Fig. 2, supplemental Fig. S4, C and D). This loop closing is conducive to catalysis because it brings both Thr118 and Ser119 into their reactive positions. In the closed conformation of hUXS1, the glucuronyl moiety of the substrate is buried deeply inside the protein structure, whereas the UDP portion is partially solvent exposed (Fig. 1B, supplemental Fig. S4, A and B).
One of the closest structural neighbors of hUXS1 is the decarboxylase domain of ArnA, a bifunctional enzyme from E. coli that catalyzes conversion of UDP-GlcUA into UDP-4-amino-4-deoxy-l-arabinose (41–43) (supplemental Fig. S4E). Reactions of hUXS1 and ArnA probably proceed on identical paths up to the UDP-4-keto-pentose intermediate, with the exception that ArnA releases the NADH produced. The transformylase domain of ArnA then utilizes UDP-4-keto-pentose in a stereospecific transaminase reaction to give UDP-4-amino-4-deoxy-l-arabinose as the product. A structure of a nonproductive ternary complex of full-length ArnA that has UDP-GlcUA and ATP (as NAD+ surrogate) bound to the decarboxylase domain was solved at 3.0-Å resolution (PDB code 1Z7E). The active sites of hUXS1 and the ArnA decarboxylase domain are similar and the binding sites for substrate and coenzyme appear to be also largely conserved. Interestingly, therefore, the d-glucuronyl ring of substrate bound to ArnA was in the undistorted 4C1 chair conformation (42) (supplemental Fig. S4F). This may be due to the lack of a reactive nicotinamide moiety on ATP (compared with NAD+), preventing the sugar ring distortion from taking place. In ArnA, Thr432 formed a hydrogen bond with the hydroxyl at C4, whereas Tyr463 adopted a position about 5 Å away from the reactive site of the substrate, inconsistent with a direct participation of the tyrosine in catalysis (see later). Our docking experiments reveal that it is possible to accommodate UDP-GlcUA in an “ArnA-like binding mode” in hUXS1. However, under molecular dynamics conditions the enzyme complex structure relaxed immediately to the time-averaged conformation depicted in Fig. 2. Mutagenesis experiments described below strongly support the BO,3 boat model, which therefore constitutes a highly probable representation of the productive hUXS1 ternary complex.
Reaction Course for Wild-type and Mutated Forms of hUXS1
The course of conversion of UDP-GlcUA by wild-type hUXS1 was recorded using HPLC and in situ proton NMR (Fig. 3). The absorbance of NADH was also measured, serving as an on-enzyme spectroscopic probe of the reaction at steady state and under conditions of rapid mixing in a stopped-flow analyzer. Table 2 summarizes the results. UDP-xylose was the sole product of the enzymatic reaction. Traces of UDP-4-keto-pentose were detectable by NMR. Because NADH was also formed in roughly corresponding amounts (≤0.001-fold the molar equivalent of substrate converted) and the relevant NMR signals were relatively broad, it is probable that the enzyme-bound form of the UDP-4-keto-pentose intermediate was observed. Looking at the time domain from milliseconds to about 10 s, in which according to its kcat value the wild-type enzyme would have undergone one full turnover, there was no accumulation of NADH above the steady-state level (Fig. 4A). These results immediately imply that in native hUXS1 the decarboxylation of UDP-4-keto-GlcUA and the reduction of UDP-4-keto-pentose are both substantially faster than the initial oxidation of UDP-GlcUA, which is therefore identified as the rate-determining chemistry in the overall reaction. The NMR data also reveal that during reaction in D2O, the UDP-xylose became deuterium-labeled at the C5 and the incorporation was stereospecific. The relative small 3JH-H coupling constant (∼5.7 Hz) between the remaining proton at C5 and the proton at C4 of the pyranose ring indicate the chiral center at C5 to be in S-configuration and suggest that the deuterium was most probably incorporated at the axial position (Fig. 3, traces A and B).
FIGURE 3.

Results from in situ NMR experiments with hUXS1. Trace A shows some signals of the substrate UDP-GlcUA at the beginning of the transformations. In trace B, additional signals of the final product UDP-xylose are visible. This product is formed during the wild-type catalyzed reaction and contains one deuterium atom in position H-5ax. A further tiny amount of the UDP-4-keto-pentose is detectable, which also carries a deuterium atom in position H-5ax. Transformations catalyzed by the E120A mutant lead to comparable results, as shown in trace C. Here also mainly UDP-xylose is accumulated, whereas UDP-4-keto-pentose is undetectable or present only in very tiny concentrations, respectively. Transformations catalyzed by the Y147F mutant exclusively accumulate the intermediate UDP-4-keto-pentose with one deuterium atom in position H-5ax (trace D). Transformations with the R277Q mutant (trace E) also lead to the same intermediate, which, however, is deuterated twice in position 5. In none of the transformations have any hints for accumulation of the UDP-4-keto-GlcUA been detected. An impurity (TRIS) causes the additional singlet at 3.605 ppm, which is indicated with an asterisk.
TABLE 2.
Kinetic characterization of wild-type and mutated forms of hUXS1
| Enzyme | kcat/limiting reactiona | Km UDP-GlcUA | Reaction productb | NADH formationc | Deuterium incorporation in product at C5d |
|---|---|---|---|---|---|
| s−1 | mm | ||||
| Wild-type | 0.2/oxidatione | 5.1 | 4, traces of 3 | ≤0.001 | Axialf |
| E120A | 1.7 × 10−3/reductione | 0.7 | 4, traces of 3g | ∼0.05g | Axialf |
| (3 and 4) | (∼0.5) | ||||
| Y147F | 1.3 × 10−4/NA | >30h | 3 | ∼1 | Axiali |
| R277Q | 1.5 × 10−4/NA | 0.1 | 3, traces of 4 | ∼1 | Double deuteration |
a Determined from the rate of conversion of UDP-GlcUA.
b Notation: 3, UDP-4-keto-pentose; 4, UDP-xylose.
c Molar ratio of NADH formed and UDP-GlcUA converted; data have S.D. of about 15%.
d Determined by 1H NMR.
e Determined by comparing the rate of NADH formation under conditions of rapid mixing in a stopped-flow apparatus to the steady-state rate of formation of UDP-xylose. Note that reduction is meant here to involve all reaction steps after formation of UDP-4-keto-GlcUA (2), including the decarboxylation. NA, not applicable.
f Stereochemical assignment is made for 4. It follows from the proposed enzymatic reaction that 3 will have deuterium incorporated at C5 with the same stereochemistry as 4.
g The molar ratio of 4 and 3 decreased in response to increase of UDP-GlcUA (1) concentration, from the shown value at 10 mm of 1 to the value in parentheses at 50 mm of 1.
h No exact determination possible, as no substrate saturation could be reached.
i Inferred from equivalence of 1H spectra of 3 formed by Y147F mutant and wild-type enzyme.
FIGURE 4.

Stopped-flow progress curves of NADH formation in reactions catalyzed by wild-type hUXS1 (A) and E120A mutant (B). Reactions were performed using 0.5 mm NAD+ and 10 mm UDP-GlcUA. A, reaction with 25 μm wild-type enzyme. There was no accumulation of NADH above 0.055 mol enzyme equivalents (1.4 μm; dashed line). B, the E120A mutant (15 μm) accumulated up to 0.58 mol enzyme equivalents (8.7 μm; dashed line) of NADH in the pre-steady state, indicating a shift in the rate-determining step from NADH formation to NADH consumption.
Fig. 2 suggests that Glu120, Tyr147, and Arg277 could be catalytically important, and we examined their roles through characterization of E120A, Y147F, and R277Q mutants. All mutants were less active in terms of kcat than the wild-type enzyme (Table 2). Y147F had completely lost the ability to form UDP-xylose as a product of UDP-GlcUA conversion; R277Q produced only traces of UDP-xylose. Therefore, these two mutants accumulated NADH in amounts corresponding to the molar equivalent of UDP-GlcUA used (Table 2). Reactions of the Y147F and R277Q mutants became stalled at the level of UDP-4-keto-pentose. The product made by Y147F in D2O contained a single deuterium at C5, whereas the corresponding product of the reaction of R277Q was doubly deuterated at this position (Table 2 and Fig. 3, traces D and E). NMR data show that deuterium incorporation by Y147F occurred with the same stereoselectivity as in the wild-type enzyme.
We determined that there was no spontaneous H/D exchange at the C5 of UDP-GlcUA, UDP-xylose, and UDP-4-keto-pentose in the time span of our experiments. Therefore, a double incorporation of deuterium label into the UDP-4-keto-pentose product necessitates that in the course of the reaction catalyzed by R277Q, the proton initially present at C5 must have become sufficiently acidic to undergo complete exchange with bulk solvent. Based on chemical considerations, the C5 proton of UDP-4-keto-GlcUA would be a strong candidate. Considering that the R277Q mutant did not produce UDP-4-keto-GlcUA in detectable amounts (Fig. 3), we conclude that the H/D exchange must have taken place at the active site of the mutant. Occurrence of this exchange was not primarily determined by intermediate lifetime, as we could show that the formation of NADH did not proceed ahead of production of UDP-4-keto-pentose during the first turnover. This would be the case if decarboxylation of UDP-4-keto-GlcUA had become the slowest step of the overall reaction of the mutant. Enhanced readiness of R277Q-bound UDP-4-keto-GlcUA to undergo H/D exchange must therefore be ascribed to a direct effect of the mutation on the solvent accessibility of the intermediate, reinforced by results of MD simulations to be described below.
Product distribution of the E120A-catalyzed reaction was dependent on the UDP-GlcUA concentration used, whereupon UDP-4-keto-pentose formation was promoted at high substrate levels. Unlike wild-type enzyme, E120A accumulated substantial amounts of NADH (up to 0.58 molar enzyme equivalents) in the pre-steady state of their reactions (Fig. 4B). Therefore, rate limitation was shifted from NADH formation (substrate oxidation) in wild-type enzyme to NADH consumption (substrate reduction including decarboxylation) in the mutant. Interestingly, the kcat for E120A displayed a solvent kinetic isotope effect of 4.4 (= kcat(H2O)/kcat(D2O)), whereas that of wild-type enzyme was just 1.4.5
Sugar Ring Distortions at the Active Site and Their Role in Promoting Catalysis
Fig. 2 suggests that substrate binding involves considerable strain on the d-glucuronyl pyranose ring. We therefore performed MD simulations on all complexes of the proposed enzymatic reaction (Scheme 1). Results indicate a trajectory of the pyranose ring pucker that moves from the relaxed 4C1 chair in free substrate via BO,3 boat in Michaelis complex (Fig. 5A) to a 2SO skew-boat conformation in the UDP-4-keto-GlcUA intermediate (Fig. 5B). This trajectory, which is plausible as it contains only conformations accessible to the d-glucuronyl ring in solution (44, 45), appears to be highly conducive to UXS catalysis in each step. The 4C1 → BO,3 transition serves to align the catalytic groups for NAD+-dependent oxidation at C4, however, already in expectation of the subsequent decarboxylation because the carboxylate group at C5 is brought into an axial position that makes it an excellent leaving group. The BO,3 → 2SO transition, which accompanies substrate oxidation, additionally involves a slight tilt of the pyranose ring at the active site (cf. Fig. 5, A and B), resulting in distinctly altered hydrogen bonding of the 4-keto-GlcUA moiety with the enzyme as compared with substrate. In particular, contact between Thr118 and the carboxylate group is disrupted and the threonine now interacts with the 4-keto group of the intermediate. This new arrangement of hydrogen bonds (involving Thr118 and Ser119) would provide excellent stabilization for β-keto acid decarboxylation (46). Generating the E120A mutant in silico, we find that due to loss of hydrogen bonding capability in the mutant as compared with wild-type enzyme, substrate distortion in the Michaelis complex is decreased substantially and equatorial-to-axial reorientation of the carboxylate group of the substrate does not occur (Fig. 5E). Therefore, these findings could explain the dual effect of the Glu120 → Ala substitution that reaction is slowed already at the substrate oxidation step, whereas at the same time rate limitation is shifted to the following steps of the catalytic cycle.
FIGURE 5.

Trajectory of sugar ring conformation along the hUXS1 reaction coordinate determined by MD simulation. Numbering of substrate/intermediate corresponds to the designation shown in Schemes 1 and 2. Blue lines indicate hydrogen bonds. Amino acid residues (except mutations) are not labeled here for better clarity (refer to Fig. 2). A, substrate UDP-GlcUA, 1, is in BO,3 boat conformation. B, UDP-4-keto-GlcUA intermediate, 2, in 2SO skew-boat conformation. C, UDP-4-keto-pentose, enol form, in 2H1 half-chair conformation. The water that protonates C5 is shown in white/red. D, UDP-4-keto-pentose intermediate, 3, in 4C1 chair conformation. E, mutant complex E120A·NAD+·UDP-GlcUA. The substrate, 1, is in the 4C1 chair conformation. F, mutant complex R277Q·NAD+·UDP-GlcUA. The substrate, 1, is in the BO,3 boat conformation. G, mutant complex R277Q·NADH·UDP-4-keto-GlcUA. The 4-keto-d-glucuronyl ring of UDP-4-keto-GlcUA, 2, is in a 1C4 chair conformation. MD simulation revealed a water molecule possibly involved in hydrogen/deuterium exchange at C5 of UDP-4-keto-GlcUA in the R277Q mutant. The water is shown in white/red. Note that the modeled structure of the corresponding NADH·UDP-4-keto-GlcUA complex of the wild-type enzyme (panel B) lacked water in a position suitable for hydrogen/deuterium exchange, consistent with only a single deuterium being incorporated during reaction in D2O.
MD simulation of the substrate complex of the R277Q mutant revealed an even larger pyranoside ring distortion than in wild-type enzyme, involving a different way of accommodating the carboxylate at C5 in which Thr118 has no role (Fig. 5F). Reactive carbons on substrate and NAD+ appear to be better aligned for hydride transfer in the wild-type complex as compared with the R277Q complex, providing a tentative explanation for the low reactivity of the mutant in the initial oxidation step of the catalytic reaction. The modeled UDP-4-keto-GlcUA complex of R277Q (Fig. 5G) differs from the corresponding wild-type complex (Fig. 5B) in that the pyranose ring of the 4-keto-GlcUA moiety adopts a 1C4 (rather than 2SO) conformation. However, the carboxylate group of the substrate is still oriented axial, consistent with experimental observation that R277Q forms decarboxylated product exclusively. Interestingly, the mutant complex shows a water molecule in the immediate proximity to the C5 proton of UDP-4-keto-GlcUA, suggesting how H/D exchange could occur at this point (Fig. 5G). Space restriction due to the Arg277 side chain and a different local network of hydrogen bonds presumably exclude water in the corresponding wild-type complex (Fig. 5B).
Proceeding with analysis of the wild-type enzyme, we find that the enol/enolate produced by decarboxylation adopts a 2H1 half-chair conformation that is strained by the presence of the endocyclic double bond (Fig. 5C). We carefully examined the simulated structures for candidate groups involved in stereospecific protonation of the enolate at C5. There are no protein residues close enough to adopt this role. However, a water molecule bonded to Glu120 is found in all structures in a position optimally suited for stereospecific protonation from below the sugar ring through a Si side attack on C5 (Fig. 5C). Solvent deuterium would thus be incorporated in axial position at C5 of UDP-xylose, in agreement with our NMR data and consistent with stereochemical analysis of the UXS reaction by Schutzbach and Feingold (16) using C5 tritium-labeled UDP-GlcUA as substrate. The large solvent isotope effect on kcat for E120A would be consistent with the proposed role for Glu120. The resulting UDP-4-keto-pentose adopts a flattened 4C1 chair (Fig. 5D). The reactive 4-keto group is placed suitably for stereospecific hydride transfer from NADH under general acid catalytic assistance from Tyr147. The UDP-xylose product is in a relaxed 4C1 conformation (data not shown), similar as in Fig. 5D.
The Catalytic Mechanism of hUXS1 and Its Biological Implications
A detailed proposal for the catalytic mechanism of hUXS1 is supported (Scheme 2). Having captured the relevant (stable) intermediates of the enzymatic reaction, we present here the first direct observation that conversion of UDP-GlcUA by UXS proceeds in three discrete catalytic steps. We further assign catalytic functions to the individual active site groups in each reaction step, thus revealing how the classical SDR catalytic center is expanded in UXS to accommodate the specific task of oxidative decarboxylation. Tyr147 is the Brønsted acid-base for catalytic oxidation and reduction. Mutation of the tyrosine to the incompetent phenylalanine was strongly disruptive for activity in substrate dehydrogenation and, not unexpectedly, it completely eliminated reductase activity toward the unactivated keto group in UDP-4-keto-pentose. Thr118 has a key role in positioning the substrate for oxidative decarboxylation by forming a strong hydrogen bond with the carboxylate group at C5. This role differs from the canonical catalytic function of homologous threonine/serine residues in SDR active sites that is in the orientation of the reactive alcohol group on the substrate (20, 40). Arg277 indirectly contributes to catalysis by bonding with Glu120 from the neighboring loop in the structure. Thus, the binding pocket for the carboxylate group of the substrate is created, and the active site is closed up. This prevents escape of UDP-4-keto-pentose during reaction. Glu120 is furthermore important for stereospecific protonation of the enolate formed in the decarboxylation step. A particular component of significance of the hUXS1 mechanism is that the sugar ring pucker in the substrate/intermediates changes along the reaction coordinate to optimally underpin the chemical steps. Glu120 is important for the binding site to exercise strain on the enzyme-bound substrate. Destabilization of substrate is an important general mechanism of enzyme catalysis and hUXS1 appears to have found an ingenious way to apply it. Our findings furthermore provide an explanation for the interesting pattern of residue conservation in extended SDRs that Glu120 and Arg277 of hUXS1 are invariant within the UXS group of enzymes, whereas the same residues are not present in “counterpart” epimerases that prevent decarboxylation at C5 in a highly similar transformation of UDP-GlcUA via oxidation and reduction at C4 (11, 47) (supplemental Fig. S1). Of note, Glu120 and Arg277 of UXS are replaced, respectively, by a serine and a threonine in the epimerases. We predict that in epimerases, the pyranose ring pucker for enzyme-bound substrate and oxidized intermediate will not be the same as in UXS, thus preventing the decarboxylation. Enzymes utilizing UDP-4-keto-pentose as a key intermediate for making products other than UDP-xylose are expected to employ a mechanism of oxidative decarboxylation analogous to that of hUXS1. Molecular strategies for diversification at the level of UDP-4-keto-pentose involve “early” release of NADH as in ArnA (41–43) or catalytic pyranoside ring contraction as in UDP-α-d-apiose synthase (48, 49). The high-resolution structure of hUXS1 provides a valuable basis to further explore these alternative enzymatic reaction mechanisms. Considering the essential role of the enzyme in human extracellular matrix glycobiology, the hUXS1 structure will furthermore be of great interest for exploitation in the design of small-molecule effectors or inhibitors of the enzyme. Evidence from our MD simulations provides useful guidance to such efforts.
SCHEME 2.

Proposed catalytic mechanism of hUXS1.
Supplementary Material
This work was supported by the Austrian Science Fund DK Molecular Enzymology Grant W901-B05 and the SGC. The SGC is a registered charity (number 1097737) that receives funds from multiple sources.

This article contains supplemental “Methods” and Figs. S1–S5.
Amino acid numbering for hUXS1 within the manuscript corresponds to PDB structure 2B69. There is an offset of −85 compared with the gene-derived sequence.
Reaction conditions were used where E120A mutant forms UDP-xylose and only trace amounts of UDP-4-keto-pentose upon conversion of UDP-GlcUA.
- UXS
- UDP-xylose synthase
- UDP-GlcUA
- UDP-glucuronic acid
- hUXS1
- human form of UXS
- SeMet
- selenomethionine
- SDR
- short-chain dehydrogenase/reductase
- MD
- molecular dynamics.
REFERENCES
- 1. Theocharis A. D., Skandalis S. S., Tzanakakis G. N., Karamanos N. K. (2010) Proteoglycans in health and disease. Novel roles for proteoglycans in malignancy and their pharmacological targeting. FEBS J. 277, 3904–3923 [DOI] [PubMed] [Google Scholar]
- 2. Bernfield M., Götte M., Park P. W., Reizes O., Fitzgerald M. L., Lincecum J., Zako M. (1999) Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 68, 729–777 [DOI] [PubMed] [Google Scholar]
- 3. Bishop J. R., Schuksz M., Esko J. D. (2007) Heparan sulfate proteoglycans fine-tune mammalian physiology. Nature 446, 1030–1037 [DOI] [PubMed] [Google Scholar]
- 4. Iozzo R. V. (1998) Matrix proteoglycans. From molecular design to cellular function. Annu. Rev. Biochem. 67, 609–652 [DOI] [PubMed] [Google Scholar]
- 5. Lander A. D., Selleck S. B. (2000) The elusive functions of proteoglycans. In vivo veritas. J. Cell Biol. 148, 227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schaefer L., Schaefer R. M. (2010) Proteoglycans. From structural compounds to signaling molecules. Cell Tissue Res. 339, 237–246 [DOI] [PubMed] [Google Scholar]
- 7. Prydz K., Dalen K. T. (2000) Synthesis and sorting of proteoglycans. Commentary. J. Cell Sci. 113, 193–205 [DOI] [PubMed] [Google Scholar]
- 8. Kearns A. E., Vertel B. M., Schwartz N. B. (1993) Topography of glycosylation and UDP-xylose production. J. Biol. Chem. 268, 11097–11104 [PubMed] [Google Scholar]
- 9. Moriarity J. L., Hurt K. J., Resnick A. C., Storm P. B., Laroy W., Schnaar R. L., Snyder S. H. (2002) UDP-glucuronate decarboxylase, a key enzyme in proteoglycan synthesis. Cloning, characterization, and localization. J. Biol. Chem. 277, 16968–16975 [DOI] [PubMed] [Google Scholar]
- 10. Eames B. F., Singer A., Smith G. A., Wood Z. A., Yan Y. L., He X., Polizzi S. J., Catchen J. M., Rodriguez-Mari A., Linbo T., Raible D. W., Postlethwait J. H. (2010) UDP xylose synthase 1 is required for morphogenesis and histogenesis of the craniofacial skeleton. Dev. Biol. 341, 400–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bar-Peled M., O'Neill M. A. (2011) Plant nucleotide sugar formation, interconversion, and salvage by sugar recycling. Annu. Rev. Plant Biol. 62, 127–155 [DOI] [PubMed] [Google Scholar]
- 12. Bar-Peled M., Griffith C. L., Doering T. L. (2001) Functional cloning and characterization of a UDP-glucuronic acid decarboxylase. The pathogenic fungus Cryptococcus neoformans elucidates UDP-xylose synthesis. Proc. Natl. Acad. Sci. U.S.A. 98, 12003–12008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coyne M. J., Fletcher C. M., Reinap B., Comstock L. E. (2011) UDP-glucuronic acid decarboxylases of Bacteroides fragilis and their prevalence in bacteria. J. Bacteriol. 193, 5252–5259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gu X., Lee S. G., Bar-Peled M. (2011) Biosynthesis of UDP-xylose and UDP-arabinose in Sinorhizobium meliloti 1021. First characterization of a bacterial UDP-xylose synthase, and UDP-xylose 4-epimerase. Microbiology 157, 260–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rosenberger A. F., Hangelmann L., Hofinger A., Wilson I. B. (2012) UDP-xylose and UDP-galactose synthesis in Trichomonas vaginalis. Mol. Biochem. Parasitol. 181, 53–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schutzbach J. S., Feingold D. S. (1970) Biosynthesis of uridine diphosphate d-xylose IV. Mechanism of action of uridine diphosphoglucuronate carboxylyase. J. Biol. Chem. 245, 2476–2482 [PubMed] [Google Scholar]
- 17. Gu X., Glushka J., Yin Y., Xu Y., Denny T., Smith J., Jiang Y., Bar-Peled M. (2010) Identification of a bifunctional UDP-4-keto-pentose/UDP-xylose synthase in the plant pathogenic bacterium Ralstonia solanacearum strain GMI1000, a distinct member of the 4,6-dehydratase and decarboxylase family. J. Biol. Chem. 285, 9030–9040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He X. M., Liu H. W. (2002) Formation of unusual sugars. Mechanistic studies and biosynthetic applications. Annu. Rev. Biochem. 71, 701–754 [DOI] [PubMed] [Google Scholar]
- 19. Tanner M. E. (2008) Transient oxidation as a mechanistic strategy in enzymatic catalysis. Curr. Opin. Chem. Biol. 12, 532–538 [DOI] [PubMed] [Google Scholar]
- 20. Kavanagh K. L., Jörnvall H., Persson B., Oppermann U. (2008) The SDR superfamily. Functional and structural diversity within a family of metabolic and regulatory enzymes. Cell Mol. Life Sci. 65, 3895–3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Frey P. A. (1996) The Leloir pathway. A mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J. 10, 461–470 [PubMed] [Google Scholar]
- 22. Thibodeaux C. J., Melançon C. E., Liu H. W. (2007) Unusual sugar biosynthesis and natural product glycodiversification. Nature 446, 1008–1016 [DOI] [PubMed] [Google Scholar]
- 23. Collaborative Computational Project (1994) The CCP4 Suite. Programs for protein crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 24. Sheldrick G. M., Schneider T. R. (1997) SHELXL. High-resolution refinement. Methods Enzymol. 277, 319–343 [PubMed] [Google Scholar]
- 25. Emsley P., Cowtan K. (2004) COOT. Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 26. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 27. Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008) GROMACS 4. Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 28. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C. (2006) Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926 [Google Scholar]
- 30. Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., Case D. A. (2004) Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 [DOI] [PubMed] [Google Scholar]
- 31. Wang J., Wang W., Kollman P. A., Case D. A. (2006) Automatic atom-type and bond-type perception in molecular mechanical calculations. J. Mol. Graph. Model. 25, 247–260 [DOI] [PubMed] [Google Scholar]
- 32. Mobley D. L., Chodera J. D., Dill K. A. (2006) On the use of orientational restraints and symmetry corrections in alchemical free energy calculations. J. Chem. Phys. 125, 084902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Olsson M. H. M., Søndergaard C. R., Rostkowski M., Jensen J. H. (2011) PROPKA3. Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537 [DOI] [PubMed] [Google Scholar]
- 34. Hoover W. G. (1985) Canonical dynamics. Equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 [DOI] [PubMed] [Google Scholar]
- 35. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. G. (1995) A smooth particle mesh Ewald potential. J. Chem. Phys. 103, 8577–8592 [Google Scholar]
- 36. Berendsen H. J. C., Postma J. P. M., Van Gunsteren W. F., DiNola A., Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684 [Google Scholar]
- 37. Hess B., Bekker H., Berendsen H. J., Fraaije J. G. (1997) LINCS. A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 [Google Scholar]
- 38. Krezel A., Bal W. (2004) A formula for correlating pKa values determined in D2O and H2O. J. Inorg. Biochem. 98, 161–166 [DOI] [PubMed] [Google Scholar]
- 39. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 40. Filling C., Berndt K. D., Benach J., Knapp S., Prozorovski T., Nordling E., Ladenstein R., Jörnvall H., Oppermann U. (2002) Critical residues for structure and catalysis in short-chain dehydrogenases/reductases. J. Biol. Chem. 277, 25677–25684 [DOI] [PubMed] [Google Scholar]
- 41. Gatzeva-Topalova P. Z., May A. P., Sousa M. C. (2004) Crystal structure of Escherichia coli ArnA (PmrI) decarboxylase domain. A key enzyme for lipid a modification with 4-amino-4-deoxy-l-arabinose and polymyxin resistance. Biochemistry 43, 13370–13379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gatzeva-Topalova P. Z., May A. P., Sousa M. C. (2005) Structure and mechanism of ArnA. Conformational change implies ordered dehydrogenase mechanism in key enzyme for polymyxin resistance. Structure 13, 929–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Williams G. J., Breazeale S. D., Raetz C. R., Naismith J. H. (2005) Structure and function of both domains of ArnA, a dual function decarboxylase and a formyltransferase, involved in 4-amino-4-deoxy-l-arabinose biosynthesis. J. Biol. Chem. 280, 23000–23008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mikhailov D., Mayo K. H., Vlahov I. R., Toida T., Pervin A., Linhardt R. J. (1996) NMR solution conformation of heparin-derived tetrasaccharide. Biochem. J. 318, 93–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ochsenbein P., Bonin M., Schenk-Joss K., El-Hajji M. (2011) The 2SO skew-boat conformation in l-iduronic acid. Angew. Chem. Int. Ed. 50, 11637–11639 [DOI] [PubMed] [Google Scholar]
- 46. Pan Y., Kee C. W., Jiang Z., Ma T., Zhao Y., Yang Y., Xue H., Tan C. H. (2011) Expanding the utility of bronsted base catalysis. Biomimetic enantioselective decarboxylative reactions. Chem. Eur. J. 17, 8363–8370 [DOI] [PubMed] [Google Scholar]
- 47. Broach B., Gu X., Bar-Peled M. (2012) Biosynthesis of UDP-glucuronic acid and UDP-galacturonic acid in Bacillus cereus subsp. cytotoxis NVH 391–98. FEBS J. 279, 100–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Choi S. H., Ruszczycky M. W., Zhang H., Liu H. W. (2011) A fluoro analogue of UDP-α-d-glucuronic acid is an inhibitor of UDP-α-d-apiose/UDP-α-d-xylose synthase. Chem. Commun. 47, 10130–10132 [DOI] [PubMed] [Google Scholar]
- 49. Gebb C., Baron D., Grisebach H. (1975) Spectroscopic evidence for formation of a 4-keto intermediate in UDP-apiose UDP-xylose synthase reaction. Eur. J. Biochem. 54, 493–498 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.