

Background: How energy metabolism contributes to the extended life span of calorie-restricted animals remains an enigma.
Results: We identified enhanced fuel oxidation and a preference of fatty acids as the major energy source in calorie-restricted nematodes.
Conclusion: Enhanced fuel utilization rather than total ingested calories contributes to the beneficial effects of calorie restriction.
Significance: Enhanced fuel oxidation to extend life span is a conserved mechanism across phylogeny.
Keywords: Aging, C. elegans, Energy Metabolism, Phosphoenolpyruvate Carboxykinase, Proteomics, Caloric Restriction
Abstract
Caloric restriction (CR) markedly extends life span and improves the health of a broad number of species. Energy metabolism fundamentally contributes to the beneficial effects of CR, but the underlying mechanisms that are responsible for this effect remain enigmatic. A multidisciplinary approach that involves quantitative proteomics, immunochemistry, metabolic quantification, and life span analysis was used to determine how CR, which occurs in the Caenorhabditis elegans eat-2 mutants, modifies energy metabolism of the worm, and whether the observed modifications contribute to the CR-mediated physiological responses. A switch to fatty acid metabolism as an energy source and an enhanced rate of energy metabolism by eat-2 mutant nematodes were detected. Life span analyses validated the important role of these previously unknown alterations of energy metabolism in the CR-mediated longevity of nematodes. As observed in mice, the overexpression of the gene for the nematode analog of the cytosolic form of phosphoenolpyruvate carboxykinase caused a marked extension of the life span in C. elegans, presumably by enhancing energy metabolism via an altered rate of cataplerosis of tricarboxylic acid cycle anions. We conclude that an increase, not a decrease in fuel consumption, via an accelerated oxidation of fuels in the TCA cycle is involved in life span regulation; this mechanism may be conserved across phylogeny.
Introduction
CR5 is the only nongenetic intervention that results in an increase in life span, while maintaining the physiology of much younger animals in a wide range of species, from nematodes to mammals (1–8). In addition, CR delays the onset of various aging-related diseases, such as cancer and diabetes in both humans and animal models (9, 10).
Although the beneficial effects of CR are well established, its molecular and cellular mechanisms remain unclear. Recent studies suggest that CR initiates a regulatory process that monitors the function of proteins and senses changes in nutrients, to elicit appropriate physiological responses to these changes. In Caenorhabditis elegans, for example, genes encoding the energy sensor adenosine monophosphate (AMP)-activated kinase AAK-2 (11), the nutrient sensor target of rapamycin kinase (12), the NAD+-dependent deacetylase SIRT1/SIR-2.1 (13), and transcription factors such as the NF-E2-related factor 2 (Nrf2/SKN-1) (14) and the Foxa subfamily of forkhead box (FOXA/PHA-4) (15) have all been found to mediate the anti-aging effects of CR. At the cellular level, animals that are undergoing CR exhibit many changes that may contribute to the physiological benefits of this regime, such as reduced oxidative damage, suppressed protein synthesis, and a slowed, age-associated decline of DNA repair (7, 9, 10). CR also has been shown to alter the content of hormones and induce metabolic changes, including a decreased concentration of glucose, triglycerides, insulin, and growth factors in the blood (9, 10). Several studies addressing the effect of CR on the metabolome and the transcriptome further suggest a connection between metabolism and the phenotypes of CR animals (17–19).
Despite these achievements, the influence of CR on metabolism at the organismal level and the consequent impact of this regulation on health and life span of animals are still poorly defined. For example, it has been hypothesized that CR slows the metabolism of animals, leading to a reduced production of reactive oxygen species that elicits an anti-aging effect (20). However, CR nematodes demonstrate increased oxygen consumption (14, 21), arguing against this hypothesis. Therefore, it is critical to quantify the metabolism of living animals and examine the physiological outcome of this modification.
The major questions arising from research involving a broad number of species is the mechanism of the effect of CR on energy metabolism as follows. 1) Is there a decreased energy expenditure to match the decreased food intake, and if so, which metabolic processes are altered to accommodate CR? 2) Is there a switch in the utilization of fuels caused by CR? 3) What regulates specific metabolic changes? A major limitation of research in this area has been the lack of a comprehensive assessment of the expression of specific proteins in response to CR and a correlation of these changes with the metabolic function of the organism. In this study, we report a more comprehensive approach using target proteomic analysis that is combined with metabolic flux measurements to answer some of the questions posed above.
We first applied “stable isotope labeling by amino acids in cell culture” (SILAC) to C. elegans. SILAC is a quantitative proteomic technique established in cell culture (22) in Drosophila (23), mouse (24), and recently C. elegans (25, 26) that reliably detects differences in protein abundance among samples. Then our proteomic, bioinformatic, and immunoblotting studies suggested an energy source shift in metabolism from carbohydrates to fatty acids in eat-2 nematodes, a genetic model that manifests chronic CR. The eat-2 gene encodes an acetylcholine receptor subunit that is exclusively found in pharyngeal muscles and functions to regulate pharyngeal pumping (27). The eat-2 mutant nematodes have a pharyngeal swallowing defect that compromises their food intake and thus serve as a genetic model of CR (28). We also determined the rate of TCA cycle flux by utilizing a number of specifically labeled substrates, and we related these values to various times in the life span of eat-2 and control nematodes to directly assess the alterations in energy metabolism that were induced by CR. Based on our findings, we propose that an accelerated rate of energy metabolism is a conserved process that is associated with longevity in nematodes.
MATERIALS AND METHODS
C. elegans Strains, Their Maintenance, and Age Synchronization
WT Bristol N2 strain nematodes were used in this study. The mutants eat-2 (ad465 and ad1116), enol-1 (ok2210), and pck-1 (ok2098) were obtained from the Caenorhabditis Genetics Center. The eat-2 strain was out-crossed three times with N2 nematodes. Nematodes were maintained by standard methods that included culture on NGM plates (0.25% peptone, 51 mm NaCl, 25 mm K3PO4, 5 μg/ml cholesterol, 1 mm CaCl2, 1 mm MgSO4) seeded with OP50 bacteria, cryostorage, and recovery from stocks. The composition of the media and solutions and the detailed protocols for their use were described previously (29). To synchronize their age, gravid nematodes were bleached according to a published protocol (29), and the surviving eggs were hatched as age-synchronized nematodes. In all experiments, the pre-fertile period of adulthood was identified as t = 0.
Generation of a pck-1 Overexpression Line in C. elegans
For the generation of transgenic animals, a plasmid DNA mix was microinjected into the gonad of young adult hermaphrodite animals, using the standard method (30). F1 progeny were selected on the basis of the roller phenotype. Individual F2 progenies were isolated to establish independent lines. For the generation of the EQ239 (pck-1 o.e.) strain, the plasmid DNA mix consisted of 5 ng/μl pAH103 (pck-1p::pck-1 genomic DNA) and 80 ng/μl pRF4 (rol-6). The pAH103 plasmid was constructed by cloning the entire 3.4-kb pck-1 gene and 2.7-kb upstream region from genomic DNA. Wild-type (N2) animals were microinjected to generate this strain. Microinjecting N2 animals with 80 ng/μl pRF4 (rol-6) alone did not affect the mean life span of N2 animals grown on OP50 (data not shown).
Labeling Bacteria with Heavy Lysine
Arginine and lysine auxotrophic Escherichia coli strain AT713 was obtained from the E. coli Genetic Stock Center at Yale University. To label AT713 bacteria with heavy lysine, bacteria were first streaked on a lysogeny broth (LB) plate and cultured overnight at 37 °C. A single bacterial colony was then inoculated into 3 ml of LB and cultured overnight in an incubator shaker (I2500, New Brunswick Scientific, Edison, NY) (set at 200 rpm) at 37 °C. Ten μl of bacterial culture was next inoculated into 5 ml of M9 basal medium (50 mm Na2HPO4, 20 mm KH2PO4, 10 mm NaCl, 20 mm NH4Cl, 2 mm MgSO4, 0.1 mm CaCl2, and 0.2% glucose) supplemented with arginine (100 μg/ml), cysteine (100 μg/ml), and lysine (100 μg/ml, either regular or heavy lysine (Cambridge Isotope Laboratories, Andover, MA)) (M9 with amino acid supplementation) and continuously cultured in an incubator shaker (37 °C, 200 rpm) until the absorbance of the culture at 600 nm (A600) reached 1.0. A600 was measured with a Beckman Du® 640 spectrophotometer (Beckman Coulter, Brea, CA). Two ml of the resulting labeled bacteria were next inoculated into 200 ml of M9 basal medium, with amino acid supplementation, and cultured in an incubator shaker (37 °C, 200 rpm) until A600 reached 1.5. Bacteria were next pelleted by a brief centrifugation (8,000 × g, 10 min, Beckman Avanti J-26 XP, Beckman Coulter) and resuspended in 10 ml of M9 basal medium. Bacteria then were spread onto a peptone-free NGM plate (500 μl for each 100-mm plate and 200 μl for each 60-mm plate) and exposed to 1000 mJ/cm2 of ultraviolet (UV) light (SpectroLinker XL-1500, Spectronics Corp.).
Labeling Nematodes with Heavy Lysine
Six animals from each strain were transferred onto a peptone-free NGM plate that had been seeded with heavy lysine-labeled or unlabeled AT713 bacteria, obtained by the methods described above. Gravid animals from the next generation of these nematodes were bleached to collect their live eggs, according to a protocol described previously (29). Eggs were transferred onto peptone-free NGM plates seeded with heavy lysine-labeled or unlabeled AT713 bacteria. Hatched, age-synchronized animals were next cultured to L4 (day 0) and then transferred to peptone-free NGM plates seeded with heavy lysine-labeled or unlabeled AT713 plates containing 25 mg/liter 5-fluoro-2′-deoxyuridine (Acros Organics). At day 3, nematodes were harvested using 2 ml of M9 basal medium and washed into 1.5-ml low retention tubes (Fisher). AT713 bacteria were then separated from nematodes with 6–8 washes with 1 ml of H2O, combined by centrifugation at 2000 × g for 1 min (accuSpin Mirco17, Fisher). Nematode pellets from the last centrifugation were defined as wet nematodes and subjected to proteomic sample preparation as described below.
Proteomic Sample Preparation
Equal weights (5.5 mg) of eat-2 (heavy lysine-labeled) and WT (unlabeled) worms were pooled in forward labeling proteomic experiments, whereas equal weights (7.7 mg) of WT (heavy lysine-labeled) and eat-2 (unlabeled) animals were pooled in reverse labeling experiments. The pooled worms were suspended in 100 μl of 200 mm ammonium bicarbonate, containing 2% perfluorooctanoic acid (PFOA) (w/v), protease inhibitor mixture (Sigma), and phosphatase inhibitor mixture 3 (Sigma), and proteins were then extracted by ultrasonication (4.5 kHz three times for 9 s with a 3-min pause on ice between the strokes) using a Virsonic 100 ultrasonic cell disrupter (SP Scientific). The resulting protein extract was centrifuged at 15,000 × g for 10 min (Eppendorf Centrifuge 5417 C), and the supernatant was collected. Pellets were then resuspended in 100 μl of the same extraction buffer, sonicated, and centrifuged, and supernatants were saved as described above. The resulting supernatants from the two extractions were next combined, reduced with 10 mm dithiothreitol (DTT) at 37 °C for 1 h, and then S-alkylated by 25 mm iodoacetamide at 25 °C for 1 h in the dark. Proteins were then precipitated by mixing with a 9-fold excess volume of ice-cold acetone and left for 2 h at −20 °C. The precipitated proteins were then centrifuged at 2,400 × g for 10 min at 4 °C (Eppendorf Centrifuge 5417 C), and the pellet was washed twice with ice-cold acetone. Protein pellets were air-dried for 10 min and then redissolved in 50 μl of 200 mm ammonium bicarbonate, containing 2% PFOA (w/v) by sonication in a water bath for 10 min in a Bransonic Ultrasonic 2510R-MT (Danbury, CT). The resulting solution was diluted with 150 μl of 100 mm ammonium bicarbonate to reduce PFOA concentration to 0.5%, and the amount of dissolved protein was determined with a DC protein assay kit (Bio-Rad). A total of 1.736 mg of protein in the forward labeling and 1.736 mg of protein in the reverse labeling experiments was digested by Lys-C (1:50 Lys-C to protein ratio w/w) at 37 °C for 18 h. The protein digests were dried in a speed-vac concentrator (Savant SC210A, Fisher) at 25 °C and then subjected to repeated reconstitution in 100 μl of ethanol/ethyl acetate/water/TFA (0.33:0.33:0.33:0.01, v/v) and evaporation to thoroughly remove PFOA as described previously (31).
Peptide Fractionation by Reverse Phase-Alkaline pH Chromatography
Lys-C digests were dissolved in 400 μl of 30 mm ammonium formate buffer at pH 10. Reverse phase chromatography was performed by using an XTerra MS C18 column (3.5 μm, 4.6 × 150 mm, Waters) and a Thermo-Finnigan Surveyor MS pump Version 2.4 coupled to an Agilent 1100 series diode-array detector, Version 1.4. Peptides were next chromatographed using a linear gradient of 5–35% acetonitrile in aqueous 0.1% formic acid over 20 min at a 400 μl/min flow rate, followed by acetonitrile from 35 to 80% over 5 min. Eluents was directly introduced to the diode-array detector to monitor the peptide elution at 215 nm. Xcalibur software (Version 1.4 SR1, Thermo-Finnigan Inc.) was used for instrument control, data acquisition, and data processing. Column eluents were manually collected at 1-min intervals into 1.5-ml low retention tubes. Twenty peptide fractions were dried in a speed-vac concentrator. The residual salts were removed by reconstitution of each fraction in 100 μl of water, followed by drying in a speed-vac concentrator. Each peptide fraction was dissolved in 12 μl of 0.1% formic acid and subjected to LC-MS/MS.
LC-MS/MS Analysis
LC-MS/MS analyses used an UltiMate 3000 LC systems (Dionex Inc.) interfaced to an LTQ-Orbitrap XL mass spectrometer (Thermo-Finnigan, Bremen, Germany). The platform was operated in the nano-LC mode, using the standard nano-ESI API source fitted with a PicoTip emitter that had an uncoated fitting and 10-μm spray orifice (New Objective, Inc.). The solvent flow rate through the column was maintained at 300 nl/min. The protein digests (typically 5 μl) were injected into a reversed-phase 0.3 × 5 mm C18 PepMap trapping column with a 5-μm particle size (Dionex Inc.) that was equilibrated with 0.1% formic acid, 2% acetonitrile (v/v). The column was washed for 5 min with the equilibration solution at a flow rate of 25 μl/min using an isocratic loading pump operated through an auto sampler. Next, the trapping column was switched in-line with a reversed-phase 0.075 × 150-mm C18 Acclaim PepMap 100 column (Dionex Inc.), and the peptides were chromatographed by using a linear gradient of acetonitrile from 6 to 60% in aqueous 0.1% formic acid over 100 min at a flow rate of 300 nl/min. The eluent was directly introduced to the mass spectrometer. The mass spectrometer was operated in a data-dependent MS to MS/MS switching mode, with five most intense ions in each MS scan subjected to MS/MS analysis. The full MS scan was performed at a resolution of 60,000 in the Orbitrap detector, and the MS/MS scans were performed in the ion trap detector in collision-induced dissociation mode. The threshold intensity for the MS/MS trigger was always set at 1000. The fragmentation was carried out using the collision-induced dissociation mode with normalized collision energy of 35. The data were entirely collected in the profile mode for the full MS scan and the centroid mode for the MS/MS scans. The dynamic exclusion function for previously selected precursor ions applied the following parameters: repeat count of 2, repeat duration of 45 s, exclusion duration of 60 s, and exclusion size list of 150. Xcalibur software (Version 2.0.7, Thermo-Finnigan Inc., San Jose, CA) was used for instrument control, data acquisition, and data processing.
Protein Identification
Proteins were identified by comparing all of the experimental peptide MS/MS spectra to the Wormpep (November 1, 2010) using Mascot database search software (Version 2.1.04, Matrix Science, London, UK). The carbamidomethylation of cysteine was set as a fixed modification, whereas the oxidation of methionine to methionine sulfoxide and the modification of C-terminal lysine to heavy lysine were considered to be variable modifications. The mass tolerance was set to 10 ppm for the precursor ion and to 0.8 Da for the product ion. Strict Lys-C specificity was applied, and missed cleavages were not allowed. Peptides with at least six amino acid residues and with a minimum mascot score of 20 were considered significant. False discovery rate was calculated from N(decoy)·2/N(decoy) + N(target) and set to be at ≤0.01.
Calculation of Ratios of Heavy Lysine-labeled Proteins over Their Unlabeled Counterparts
The proteomics software Tool Suite Version 2.4.1 was used to calculate the labeled/unlabeled intensities of all peptides identified from the same protein. The protein ratios were calculated by use of an in-house software package written in Python. For each replicate, protein ratios were calculated from the observed intensities of labeled/unlabeled peptides. Consider a protein match with n peptide spectral matches. Let Ii,H and Ii,L (i = 1, 2, … , n) denote the observed intensities of ith peptide spectral match of the protein with and without isotope (13C6) labeling. A protein ratio, r, was calculated using linear regression Equation 1,
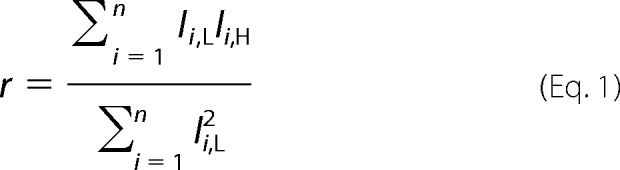 |
The standard error of this ratio is calculated by Equation 2,
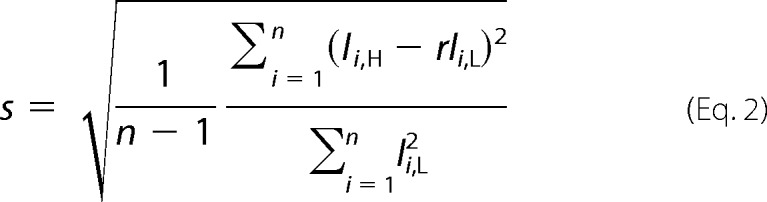 |
To minimize inevitable systematic experimental variation, all protein ratios were normalized by tubulin within each replicate as shown in Equation 3,
 |
 |
The quantitative results from all replicates were merged into a single report. The mean ratio of a protein across m replicates is a weighted mean calculated as shown in Equation 4,
 |
and are the normalized ratio and its standard error of the protein from replicate j (j = 1, 2, … , m).
The standard error for the weighted mean ratio is shown in Equation 5,
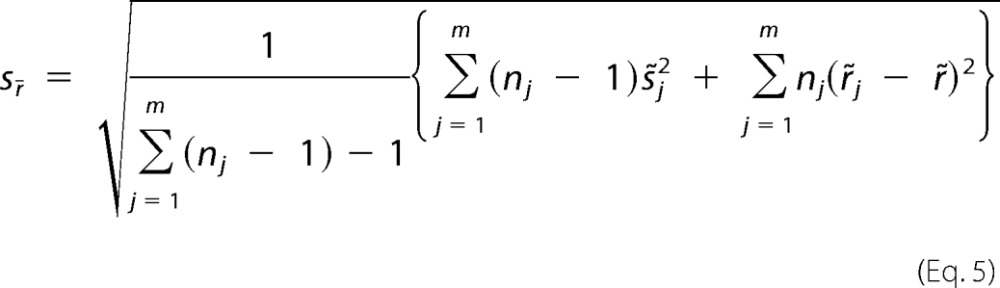 |
where nj is the number of peptide spectral matches for the protein in replicate j, represents the variances within the replicates, and represents the variances between replicates. A hypothesis test was used to test whether the obtained protein ratios are statistically significant by the following Equation 6.
 |
where Ft(v) is the cumulative density function of the t distribution with a degree of freedom shown in Equation 7
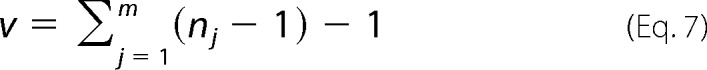 |
Immunoblotting
Nematodes were washed three times with M9 buffer (32) and lysed in buffer A (15 mm HEPES (pH 7.6), 10 mm KCl, 1.5 mm MgCl2, 44 mm glucose, and 1 mm DTT) containing a protease inhibitor and phosphatase inhibitor mixture (Roche Applied Science) followed by brief sonication (33). For human HepG2 cells, 4 × 106 cells were collected and washed with PBS, harvested, and lysed in radio immunoprecipitation assay buffer containing a protease inhibitor and phosphatase inhibitor mixture. Insoluble cell debris was removed by centrifugation (13,000 rpm, 10 min, accuSpin Mirco17 centrifuge, Fisher) at 4 °C, and the resulting supernatants were used for immunoblotting. Lysates of mouse liver were prepared as described previously (34). The antibodies used were as follows: anti-rat PEPCK (35), anti-ENOA_HUMAN (Sigma), and anti-CPT1A_RAT (34, 36, 37). Quantification of signals for immunoblots was done using National Instruments Vision Assistant 8.5 (Austin, TX) to analyze digital pictures of membranes.
Measurement of TCA Cycle Flux
The technique for determining the rate of TCA cycle flux in the worms used in this study is essentially as described by Jomain and Hanson (38). As a general fuel for the worms, 2 ml of AT713 bacteria (∼5 mg) were pelleted and then pretreated with 5 ml of 200 proof ethanol for 30 min. The ethanol was removed by washing several times with 5 ml of H2O and combined by centrifugation (10,000 × g, for 1 min). The concentrated AT713 bacteria were then resuspended in 200 μl of H2O, which contained 1000 or 3000 nematodes; the bacteria were then placed in 300 μl of DMEM/F-12 medium (Mediatech, Inc. Manassas, VA), which contained 1 mm sodium acetate and 0.5 μCi of [2-14C]sodium acetate (PerkinElmer Life Sciences), or 0.1 mm palmitate and 0.5 μCi of [1-14C] palmitate (the palmitate was mixed with 3% defatted bovine serum albumin), or 2.5 mm sodium glutamate and 0.5 μCi of [5-14C]sodium glutamate (PerkinElmer Life Sciences), or 1 mm glucose and 0.5 μCi of [u-14C]glucose (PerkinElmer Life Sciences). Under these conditions, the substrates gave maximal rates of oxidation to CO2. The medium containing the worms was then transferred to siliconized 25-ml Erlenmeyer flasks containing 8 ml of NGM agar medium. The flasks were equipped with a rubber stopper, which served both to seal the flask and to hold a plastic bucket suspended above the medium. At the end of an incubation period of 2 h, 0.3 ml of Hyamine hydroxide was injected through the stopper into the plastic bucket, and 0.5 ml of 1 m sulfuric acid was injected into the flask to complete the liberation of CO2. After 1 h, the bucket was removed and immersed in vials containing 5 ml of liquid scintillation fluid, to remove the Hyamine for the subsequent determination of radioactivity in a liquid scintillation counter (Beckman LS-8000, Beckman Instruments, Inc.). All metabolic experiments were carried out with three independent nematode cultures, at room temperature, using a metabolic shaker set at 30 rpm.
Quantification of PEPCK Activity
Pellets of nematodes were sonicated in 0.25 m sucrose, 1 mm DTT, and 5 mm Tris-HCl (pH 7.4) in an ice bath as described under “Immunoblotting.” Cytosolic fraction of the homogenate was obtained with centrifugation (30,000 × g, 30 min, 4 °C) and then used to determine PEPCK activity at room temperature, as described previously (39).
RNA Interference (RNAi) Clones
The identities of all RNAi clones were verified by sequencing the inserts using the M13-forward primer. All clones were from Julie Ahringer's RNAi library. HT115 bacteria transformed with RNAi vectors expressing dsRNA of the genes of interest were grown at 37 °C in LB with 10 μg/ml tetracycline and 50 μg/ml carbenicillin, and then seeded onto NGM-carbenicillin plates supplemented with 100 μl 0.1 m isopropyl β-d-1-thiogalactopyranoside (IPTG).
Life Span Analysis
Life span analysis was conducted at 20 °C as described previously (40, 41), unless otherwise stated. Animals were grown at 20 °C on NGM plates for at least two generations before the experiments were initiated. RNAi treatments were carried out by adding synchronized eggs to plates containing bacteria with an empty vector (L4440) and then transferring the larva to plates seeded with the RNAi bacteria 3 days later (L4/Young Adult). Nematodes were moved to plates with fresh RNAi bacteria every 2 days until reproduction was completed and then moved to new plates every 5–7 days for the rest of the life span analysis. Viability of the nematodes was scored every 2–3 days. In all experiments, the pre-fertile period of adulthood was used as t = 0 for life span analysis. Stata 12 software (StataCorp) was used for statistical analysis. In all cases, the log-rank (Mantel-Cox) test was used to test the hypothesis that the survival functions among groups were equal.
Statistical Analysis
Statistical significance was analyzed using Statistica software (StatSoft). t tests, analysis of variance with Bonferroni corrections, or Dunnett's post hoc analyses were used for their appropriate applications as indicated in the figure legends.
RESULTS
Global Protein Expression Is Down-regulated in Eat-2 Mutant
Protocols of SILAC were adapted for use with C. elegans to identify potential metabolic alterations that were induced by CR. AT713, an arginine and lysine auxotrophic E. coli variant (42), was cultured with medium containing 99% enriched [13C6]lysine. The resulting [13C6]lysine-labeled bacteria were then fed to the worms as their only food source on peptone-free NGM plates; this generated [13C6]lysine-labeled wild-type (WT) and eat-2 mutant nematodes. Nematodes of the eat-2 mutant lived ∼30% longer than WT nematodes under these culturing conditions (Fig. 1), similar to nematodes of these two strains under standard nematode culturing conditions (27).
FIGURE 1.

Effect of the proteomics culture conditions on eat-2(−) versus N2 life span. Survival curves are shown of WT (N2, triangles) or eat-2(ad1116) mutants (circles). All worms are grown on NGM plates without peptone and fed with AT713 bacteria used in proteomic studies at 20 °C. Statistics detail of the experiment is listed in the table.
Two standard SILAC protocols were used to compare the proteome of WT and eat-2 mutant nematodes as follows: 1) a “forward” labeling, where labeled eat-2 mutant and unlabeled WT nematodes were mixed; and 2) a “reverse” labeling, where labeled WT and unlabeled eat-2 mutant nematodes were mixed. In both comparisons, the same weights, rather than number of animals from each nematode strain, were used, because eat-2 mutant nematodes are noticeably smaller than WT worms, and the comparison of mass spectral peaks with similar intensity provides a more accurate quantification.
Proteins extracted from mixtures were digested with Lys-C, an endoproteinase that specifically hydrolyzes proteins at the carboxyl side of lysine (43), and analyzed by LC-MS/MS. The mass spectrometry data has been deposited to PRIDE database.
The ion intensities of Lys-C peptides from labeled and unlabeled WT nematodes were plotted in Fig. 2A. Linear regression analysis of the plot yielded a slope value of 1.0 and a correlation coefficient (r2) value of 0.9889, demonstrating that the heavy isotope labeling did not influence the proteome of C. elegans. In contrast to 1:1 mixed labeled and unlabeled WT nematodes, the correlation of the intensity of labeled and unlabeled Lys-C peptides of 1:1 mixed eat-2 mutant and WT nematodes in both forward and reverse labeling SILAC samples was scattered and was not fit well in linear regression analysis (Fig. 2, B and C, respectively), indicating that eat-2 mutant worms have a distinctive proteome from that of WT worms. In the forward labeling SILAC samples (Fig. 2B), the slope of the linear regression (slope = 0.60, r2 = 0.79) and the mean (0.75 ± 0.21 (S.D.)) Lys-C peptide ratios (eat-2 over WT) were less than 1. This suggests that the concentration of most of the observed proteins was down-regulated in eat-2 mutant nematodes compared with the same weight of WT worms; this provides evidence for suppressed global protein expression in eat-2 nematodes (44). We obtained a consistent result from the reverse labeling experiment (Fig. 2C), in which the slope value of the linear regression (slope = 1.61, r2 = 0.88) (Fig. 2C) was multiplicatively inversed to that obtained from the forward labeling experiment. Based on this observation, we estimated that the proteome of eat-2 mutant nematodes was ∼61% of the proteome of the same weight of WT nematodes.
FIGURE 2.

Global protein expression is down-regulated in eat-2. A, Lys-C peptides from a 1:1 (by weight) mixture of [13C6]lysine-labeled (WT*) and unlabeled (WT) WT nematodes were analyzed by LC-MS/MS. The observed intensities of labeled and unlabeled peptides were normalized in respect to TBB-1. The normalized intensities of those peptides identified from both WT* and WT were plotted against each other (gray dots). The solid lines are linear regressions of respective peptide ratio data. The linear regression slopes and values of r2 are labeled in each panel. The black dots indicate peptides from TBA-1, TBA-2, TBB-1, ACT-2, ACT-3, ACT-4, and ACT-5. B and C, Lys-C peptides were first identified from a 1:1 (by weight) mixture of forward ([13C6]lysine-labeled eat-2 mutant (eat-2*) and unlabeled WT (WT) nematodes) or reversal ([13C6]lysine-labeled WT mutant (WT*) of unlabeled eat-2 (eat-2) nematodes) of labeling SILAC assays. The un-normalized intensities of those peptides identified from both eat-2 and WT were plotted against each other in B (forward labeling) and C (reverse labeling) (gray dots). Dashed lines indicate the linear regressions with their fitting formula and r2 values labeled below the dashed lines. The black dots indicate peptides from TBA-1, TBA-2, TBB-1, ACT-2, ACT-3, ACT-4, and ACT-5. The solid lines are the linear regressions of these housekeeping proteins. The linear regression slopes and values of r2 are labeled.
Proteomic Analysis Indicates Potentially Altered Energy Metabolism in Eat-2 Mutant Nematodes
The intensity ratios of the Lys-C peptides were then normalized with respect to TBB-1 (a nematode homolog of β-tubulin) (45) to minimize systematic variation, a common practice in quantitative proteomics (46). The intensity ratio of a protein, defined as the linear regression of the intensity ratios of all its identified peptides, was calculated for both forward (supplemental Table S1a) and reverse (supplemental Table S1b) labeling samples. A total of 1758 proteins were identified, of which 1128 (64.2%) were found in both the forward and reverse labeling experiments. The intensity ratios, presented as eat-2 over WT, and the molecular identities of these proteins in the two experiments are summarized in supplemental Table S1c.
We then focused on key proteins in the pathways of carbohydrate, fatty acid, and protein metabolism, important components of energy metabolism in all organisms (Fig. 3). In C. elegans; these pathways are well defined and are conserved in mammals (47). Nearly all of the enzymes in the pathways of glycolysis and gluconeogenesis were identified in our screen, and the majority of them had unaltered abundance in eat-2 mutant nematodes, i.e. changes in protein intensity ratios were less than 20% and/or the forward and reversal labeling experiments did not agree with each other in protein abundance change of direction (supplemental Table S1c). Interestingly, several enzymes that play a key regulatory role in glycolysis and gluconeogenesis were consistently down-regulated in eat-2 mutant nematodes in both forward and reverse labeling samples, although such a regulation was only over 20% in one of these samples (Table 1). Among these enzymes are the following: 1) ENOL-1 (a nematode ortholog of mammalian α-enolase) (48) and PYK-1 and PYK-2 (nematode homologs of mammalian pyruvate kinase) (49) that catalyze the last two steps of glycolysis and control the flow of glucose carbon into the TCA cycle; 2) FBP-1 (a nematode homolog of mammalian fructose-1,6-biphosphatase) (49) and PYC-1 (a nematode ortholog of human pyruvate carboxylase) (50); and 3) the nematode PEPCK-C encoded by pck-1 (49) that is critical for gluconeogenesis and cataplerosis from amino acids. These data suggest that carbohydrate metabolism is reduced in eat-2 mutant nematodes.
FIGURE 3.

Pathways of fuel utilization in C. elegans. The metabolic pathways for the major fuels used by C. elegans, and which were tested in this study, are included in the figure. Of special significance is the transport of long-chain fatty acids across the mitochondrial membrane by CPT-1. Several of the important genes identified in the proteomics study are included. These include F57B10.3, phosphoglycerate mutase; ENOL-1, enolase; PYK-1/PYK-2, pyruvate kinase; ACDH-1, short-chain fatty acid specific acyl-CoA dehydrogenase; ACDH-12, long-chain fatty acid specific acyl-CoA dehydrogenase; ECH-9, enoyl-CoA hydratase; CPT-1, carnitine palmitoyltransferase 1. The proteins that were induced in the eat-2 mutant are indicated with a * notation, and proteins that were decreased at indicted by a # notation.
TABLE 1.
Identified proteins related to carbohydrate, fatty acid, and protein metabolism
| Mammalian metabolic enzymes of following | C. elegans genes | Average ratioa | p valuea | Abundance changeb |
|---|---|---|---|---|
| Carbohydrate | ||||
| Hexokinase | C50D2.7 | 0.83 | NA | |
| Phosphoglucose isomerase | gpi-1 | 0.87 | 0.172 | |
| Phosphofructokinase | Y71H10A.1 | 1.08 | 0.014 | |
| Aldolase | aldo-1 | 0.90 | 0.003 | |
| Aldolase | aldo-2 | 0.79 | 0.036 | − |
| Triose-phosphate isomerase | tpi-1 | 0.85 | 0.081 | |
| Fructose bisphosphatase | fbp-1 | 0.79 | <0.001 | − |
| Glyceraldehyde phosphate dehydrogenase | gpd-1;gpd-4 | 0.88 | <0.001 | |
| Phosphoglycerate kinase | pgk-1 | 0.86 | 0.029 | |
| Phosphoglycerate mutase | F57B10.3 | 0.75 | 0.049 | − |
| Enolase | enol-1 | 0.83 | <0.001 | − |
| Pyruvate kinase | pyk-1; pyk-2 | 0.82 | <0.001 | − |
| Pyruvate dehydrogenase A | T05H10.6 | 0.89 | 0.066 | |
| Pyruvate dehydrogenase B | C04C3.3 | 0.90 | NA | |
| Dihydrolipoamide dehydrogenase | F23B12.5 | 0.94 | 0.059 | |
| PEPCK-C | pck-1 | 0.74 | 0.014 | − |
| Pyruvate carboxylate | pyc-1 | 0.91 | 0.261 | − |
| Fatty acid | ||||
| Acyl-CoA dehydrogenase (short chain) | acdh-1 | 1.25 | 0.239 | + |
| Acyl-CoA dehydrogenase (short chain) | acdh-3 | 0.93 | 0.001 | |
| Dehydrogenase (short chain) | dhs-30 | 1.05 | 0.403 | + |
| Enoyl-CoA hydratase | ech-7 | 1.16 | 0.053 | + |
| 3-l-Hydroxyacyl-CoA dehydrogenase | B0272.3 | 0.88 | 0.040 | |
| β-Ketothiolase | B0303.3;F53A2.7 | 0.86 | <0.001 | |
| Acyl-CoA dehydrogenase (medium chain) | acdh-7 | 0.89 | 0.044 | |
| Acyl-CoA dehydrogenase (medium chain) | acdh-8;acdh-10 | 0.83 | 0.071 | |
| Acyl-CoA dehydrogenase (medium chain) | acdh-9 | 0.79 | <0.001 | − |
| Acyl-CoA dehydrogenase (very long chain) | acdh-11 | 0.80 | 0.094 | − |
| Acyl-CoA dehydrogenase (very long chain) | acdh-12 | 0.77 | <0.001 | − |
| Steroid reductase (very long chain) | art-1 | 0.78 | 0.053 | − |
| Acetoacetyl-CoA synthetase | sur-5 | 0.54 | <0.001 | − |
| Acyl-CoA synthetase (long chain) | acs-5 | 0.68 | 0.073 | − |
| Acyl-CoA synthetase (long chain) | acs-13 | 0.68 | NA | |
| Acyl-CoA synthetase | acs-19 | 0.81 | NA | |
| Acyl-CoA synthetase | acs-14 | 0.76 | NA | |
| Acyl-CoA synthetase | acs-12 | 1.11 | NA | |
| Acyl-CoA synthetase | acs-4 | 0.94 | 0.378 | |
| Acyl-CoA synthetase | acs-11 | 0.91 | NA | |
| Lipase | T21H3.1 | 1.44 | 0.016 | + |
| Acetyl-CoA carboxylase | pod-2 | 0.68 | <0.001 | − |
| Fatty-acid synthase | fasn-1 | 0.88 | 0.032 | |
| β-Ketoacyl-ACP reductase | dhs-25 | 0.41 | 0.044 | − |
| Fatty-acid desaturase | fat-2;fat-6 | 0.60 | 0.052 | − |
| C-18 polyunsaturated fatty acid elongase | elo-1 | 0.88 | <0.001 | |
| Carnitine palmitoyltransferase 1 | cpt-1 | 0.64 | 0.157 | − |
| Carnitine palmitoyltransferase 2 | cpt-2 | 0.76 | NA | |
| Protein | ||||
| Branched chain keto acid dehydrogenase E1 | tag-173 | 0.77 | <0.001 | − |
| Isovaleryl-CoA dehydrogenase | ivd-1 | 0.82 | 0.036 | − |
| Aromatic amino acid hydroxylase | pah-1 | 0.61 | <0.001 | − |
| Fumarylacetoacetate hydrolase | fah-1 | 0.75 | <0.001 | − |
| Glu/Leu/Phe/Val dehydrogenase | ZK829.4 | 0.81 | 0.004 | − |
| 4-Hydroxyphenylpyruvate dioxygenase | hpd-1 | 0.62 | <0.001 | − |
| mino acid transporter glycoprotein subunit | atgp-1 | 1.42 | <0.001 | + |
| Branched chain aminotransferase | bcat-1 | 1.13 | 0.313 | + |
a The procedures to calculate the average ratios and p values are described under “Materials and Methods.”
b The + and − signs indicate that the ratios for those proteins were either larger than 1.2 (+) or smaller than 0.8 (−) in one of the two proteomics experiments, and the ratios in the other experiment were consistently changed to the same direction (>1.0 or <1.0), respectively.
Three enzymes that are involved in fatty acid metabolism, ACDH-1, the nematode homolog of short-chain fatty acid-specific acyl-CoA dehydrogenase, which catalyzes the initial chemical reaction of fatty acid β-oxidation (51); DHS-30, a dehydrogenase that is specific to short-chain fatty acid metabolism (52); and ECH-7, the nematode homolog of mammalian enoyl-CoA hydratase, a key enzyme in β-oxidation that provides energy and acetyl-CoA from fatty acids (49); were significantly up-regulated (Table 1), i.e. the forward and reversal labeling experiments agreed with each other in up-regulation, and the data from one of the experiments indicated at least a 20% change in eat-2 mutant nematodes. The content of the other two enzymes of the β-oxidation pathway, 3-l-hydroxyacyl-CoA dehydrogenase (encoded by B0272.3) and β-ketothiolase (encoded by F53A2.7 and B0303.3 (49)), remained unchanged in eat-2 mutant nematodes, as compared with control worms. In contrast, all identified enzymes that are involved in the metabolism of medium- and long-chain fatty acids were either decreased or unchanged. These included ACDH-9 (the nematode homolog of mammalian medium-chain acyl-CoA dehydrogenase) (53), ACDH-11 and ACDH-12 (the nematode homologs of mammalian long chain acyl-CoA dehydrogenase) (54), ART-1 (a steroid reductase required for elongation of the long chain fatty acids) (55), SUR-5 (the nematode homolog of mammalian acetoacetyl-CoA synthetase) (56), and ACS-5 the nematode homolog of mammalian (the nematode homolog of mammalian long-chain acyl-CoA synthetase) (Table 1) (49). Enzymes that catalyze key chemical reactions in fatty acid synthesis and storage, such as POD-2 (a nematode homolog of acetyl-CoA carboxylase that catalyzes the initial step in the de novo synthesis of fatty acids) (57), FAT-2 (the nematode homolog of mammalian Δ-12 fatty acyl desaturase), and FAT-6 (the nematode homolog of mammalian Δ-9 fatty acid desaturase) (58), were down-regulated. Other identified proteins related to medium- to long-chain fatty acid metabolism or fatty acid storage were not changed in abundance.
Short-chain fatty acids freely translocate between the cytosol and the mitochondria of cells. Consistent with an increased metabolism of short-chain fatty acids as an energy source and down-regulated medium to long-chain fatty acid metabolism in eat-2 mutant nematodes, we found that CPT-1, the nematode ortholog of human carnitine palmitoyltransferase I, which transports medium- or long chain-fatty acid across the mitochondrial membrane for β-oxidation (53), was down-regulated. Moreover, a lipase encoded by T21H3.1 (59) that converts triglycerides into monoglycerides and free fatty acids to produce energy was up-regulated, as was peroxisomal acyl-CoA oxidase, which initiates long-chain fatty acid oxidation in peroxisomes (60). We did not, however, identify any putative nematode peroxisomal acyl-CoA oxidase encoded by F08A8.2, F08A8.3, or F08A8.4 (53).
An analysis of a limited number of the identified proteins involved in amino acid metabolism pathways indicates that the catabolism of amino acids was down-regulated, and re-uptake of amino acids was up-regulated (Table 1). For example, the abundance of IVD-1 and PAH-1, the nematode aromatic amino acid hydroxylase and isovaleryl-CoA dehydrogenase, key enzymes in amino acids metabolism, was down-regulated (61). In contrast, ATGP-1, a nematode amino acid transport glycoprotein subunit that facilitates amino acid uptake (62), and PEPT-1 (63), a low affinity/high capacity oligopeptide transporter required for uptake of intact peptides from the intestine, were all elevated in eat-2 mutant nematodes.
Immunoblotting Identifies Altered ENOL-1 and CPT-1 Abundance in Eat-2
Although proteomic data from our forward and reverse labeling experiments agreed well with each other in two independent experimental duplicates in our analysis, we further confirmed our initial observation of changes in the level of specific proteins by using other methods. We performed immunoblotting with proteins extracted from the eat-2 mutant and WT nematodes raised on regular NGM plates or NGM plates absent of peptone, a protein supplement routinely used to grow nematodes. We initially tested four antibodies against murine long-chain acyl-CoA synthetase, murine PEPCK-C, murine carnitine palmitoyltransferase I, and human ENOLASE 1. This strategy was used after we verified that there was significant sequence homology between epitopes recognized by the antibodies and the sequence of the enzymes in nematodes. The epitopes of these and 13 other antibodies were searched using Blast against the nematode genome; we found that the epitopes of these four antibodies have medium (∼45%) to high (>70%) sequence identity with their nematode homologs (34, 36, 37), namely ACS-5, the nematode homolog of PEPCK-C encoded by PCK-1, ENOL-1, and CPT-1 (data not shown). We identified expected immunoreactive bands against mammalian antibodies for ENOL-1, PEPCK-C, and CPT-1 but not ACS-5.
With nematodes raised without peptone, the content of ENOL-1 and CPT-1 relative to actin was lower in the eat-2 mutant than in WT nematodes (Fig. 4). Specifically, quantification of the immunoreactive bands of ENOL-1 and CPT-1 relative to actin indicated a ∼20% decrease in both ENOL-1 and CPT-1. As a control, the immunoreactive band of ENOL-1 was absent from an enol-1 deletion mutant (Fig. 4).
FIGURE 4.

Immunochemical quantification of ENOL-1 and CPT-1. Immunoblotting of ENOL-1 and CPT-1 in extracts from eat-2 mutant (ad465) and WT (N2) nematodes that were raised on NGM plates, with (+) or without (−) peptone, is shown. A and B, top panel, representative SDS-polyacrylamide gels. Images of immunoreactive bands to actin (Actin) with different exposure times are shown. The molecular markers of 50 and 37 kDa are labeled. Two immunoreactive bands against ENOL-1 were identified. The lower band with a smaller molecular weight is specific to eat-2 nematodes. It is possible that this immune-reactive band represents an ENOL-1 isoform that is specifically expressed in eat-2 nematodes. A and B, bottom panel, and C, quantification of immunoreactive bands of five independent experiments. In these sample gels, the immunoreactive bands to actin with shorter exposure time were used for quantification. Lysates of human HepG2 cells ((h)HepG2) and mouse liver ((m)Liver) were loaded as positive controls for ENOL-1 and CPT-1, respectively. Nematodes with enol-1 deletion were used as a control, but deletion mutant of cpt-1 was not available. *, p < 0.05; **, p < 0.001; ***, p < 0.005; t test. Error bars indicate S.E.
Interestingly, eat-2 mutant worms raised on peptone-containing NGM plates had less ENOL-1 but more CPT-1, as compared with WT worms that were similarly cultured on peptone-free NGM plates. Because peptone is an enriched source of dietary protein, this observation indicates that the metabolism of nematodes is subject to adaptation and to dietary modification even during CR. In support of this contention, the absence of peptone from NGM medium significantly down-regulated CPT-1 but not ENOL-1 in both WT and eat-2 mutant nematodes (Fig. 4).
Increased Oxidation of Glucose, Amino Acids, and Short-chain Fatty Acids in Eat-2 Mutant Nematodes
The metabolism of specific nutrients has not yet been determined in nematodes, although the basal metabolic rate, accessed by oxygen consumption and heat production, was reportedly up-regulated in WT nematodes raised with reduced food and/or in the eat-2 mutant (14, 21, 64). We measured the capacity of eat-2 mutant and WT nematodes to oxidize specific substrates to CO2. The oxidation rates of [2-14C]acetate, [1-14C]palmitate, [5-14C]glutamate, and [U-14C]glucose were used in this study; both acetate, a short chain fatty acid, and glucose enter the TCA cycle as acetyl-CoA and generate 14CO2, whereas glutamate enters the TCA cycle as α-ketoglutarate and is immediately decarboxylated via α-ketoglutarate dehydrogenase, generating 14CO2 (65–67). Palmitate is a long chain fatty acid that is transported into the mitochondria by CPT-1, converted to acetyl-CoA via β-oxidation, and then oxidized to CO2 in the TCA cycle. The rate of 14CO2 production from these labeled compounds can be used to quantify the rate at which nematodes metabolize major fuels. AT713 bacteria, which had been killed by ethanol to minimized background 14CO2 generation, are the major fuel for the nematodes. Ethanol pretreatment eliminated growth and metabolism of the bacteria (data not shown), but it had little or no effect on the feeding behavior of the nematodes (data not shown). The amount of 14CO2 produced from the oxidation of acetate by WT and eat-2 mutant nematodes was dependent on both the incubation time of and the nematode number included in the assay (data not shown). The rate of acetate oxidation was 0.13 ± 0.03 nmol/h/1000 worms for the WT and 0.094 ± 0.01 nmol/h/3 mg nematodes, as compared with 1.77 ± 0.24 nmol/h/1000 and 1.98 ± 0.16 nmol/h/3 mg nematodes for the eat-2 mutant (Fig. 5A), indicating that eat-2 mutant nematodes have 21 times the rate of acetate oxidation to 14CO2 when expressed by weight and 13.4 times when expressed by nematode number. Similarly, the rate of glutamate oxidation was quantified as 2.19 ± 0.22 nmol/h/1000 or 2.44 ± 0.25 nmol/h/3 mg nematodes for eat-2 mutant and 0.64 ± 0.01 nmol/h/1000 or 0.45 ± 0.01 nmol/h/3 mg nematodes for WT nematodes; the rate of glucose oxidation was 0.96 ± 0.23 nmol/h/1000 or 1.07 ± 0.26 nmol/h/3 mg nematodes for eat-2 mutant and 0.21 ± 0.04 nmol/h/1000 or 0.15 ± 0.03 nmol/h/3 mg nematodes for WT nematodes. Thus, the rate of glutamate oxidation in eat-2 mutant nematodes was 5.4 (by weight) and 3.4 (by nematode number) times faster than WT (Fig. 5B), and the rate of glucose oxidation in eat-2 mutant nematodes was 7.3 (by weight) and 4.7 (by nematode number) times faster than WT (Fig. 5C). The relative acetate oxidation rate of eat-2 mutant over WT nematodes was markedly higher than the relative oxidation rates of glutamate and glucose (Fig. 5E). In contrast to the oxidation of acetate, the rate of palmitate oxidation to carbon dioxide was similar in the WT and eat-2 mutant worms (Fig. 5D). This finding suggests that the pathway involved in the conversion of palmitate to acetyl CoA is not induced in the eat-2 mutant worms and agrees with the proteomic and immunoblotting data that are discussed above, which demonstrated that the concentration of the mitochondrial fatty acid transport protein CPT-1 (Fig. 3) was markedly reduced in eat-2 mutant nematodes (Fig. 4).
FIGURE 5.

Oxidation of [2-14C]acetate, [5-14C]glutamate, [U-14C]glucose, and [1-14C]palmitate to 14CO2 by eat-2 and control nematodes. 1000 or 3000 age-synchronized, day 3 eat-2 mutant, and WT nematodes, which were raised on NGM plates seeded with untreated AT713 bacteria, were separated from the bacteria and mixed with AT713 bacteria that had been treated with ethanol, along with [2-14C]acetate (A), [5-14C]glutamate (B), [U-14C]glucose (C), or [1-14C]palmitate (D). The mixture was transferred onto the NGM agar surface at the bottom of 50 ml flask and incubated for 4 h. 14CO2 that was generated by oxidative metabolism was collected and quantified using the methods detailed under “Materials and Methods.” The mean 14C radioactivity of same amount of bacteria was deducted from 14C radioactivity obtained from samples. A–D, oxidation rates of acetate (A), glutamate (B), glucose (C) or palmitate (D) were calculated in respect to 1000 or 3 mg nematodes. p < 0.005; t test. Error bars indicate the S.E. of triplicates. E, relative oxidation rates of acetate, glutamate, glucose, and palmitate by eat-2 mutant as compared with WT nematodes indicates that eat-2 mutant nematodes use more fatty acids as an energy source than do WT nematodes. ***, p < 0.005.
Energy Source Switch and Enhanced Metabolism Contributes to CR-mediated Life Span
We next examined whether the life span extension is mediated, at least in part, by the energy metabolism strategy used by eat-2 mutant nematodes by selecting 21 proteins from Table 1 and supplemental Table S1, the protein ratios of which were consistently altered in both forward and reverse labeling samples. The genes encoding these 21 proteins include the following: 1) a number of genes involved in the above-mentioned metabolic changes, such as acdh-1, acdh-12, enol-1, pod-2, fat-2, pyk-1, fbp-1, and pck-1; 2) several genes that were previously implicated in longevity regulatory pathways other than CR, for example, sbds-1, hpd-1, gei-7, vit-5, sodh-1, and asp-1 (51, 68); and 3) several genes among the top 5% most up-regulated or most down-regulated in eat-2 mutant nematodes (Fig. 6). The expression of these genes was knocked down using RNAi, and the life span of those RNAi-treated animals using the standard method was analyzed (69).
FIGURE 6.

Life span analysis of proteins identified by SILAC. Twenty one out of the 286 proteins identified from SILAC were tested for their effects on WT (N2) or eat-2(ad1116) life span. A, survival curves of WT or eat-2 mutant (eat-2) worms grown on either vector control (blue and green) or sbds-1 RNAi bacteria (red and orange) at 20 °C. The RNAi treatments were initiated at day 1 of adulthood. B, survival curves of WT or eat-2 mutant (eat-2) worms grown on either vector control (blue and green) or hpd-1 RNAi bacteria (red and orange) at 20 °C. C, results of the life span analysis for all 21 CR-responsive proteins were summarized in this table. “+,” RNAi knockdown of the indicated gene significantly extended the life span of WT or eat-2 mutant nematodes, when compared with the vector control. p < 0.05, log rank test. “−,” RNAi knockdown of the indicated gene significantly shortened the life span of WT or eat-2 mutant nematodes, when compared with the vector control. p < 0.05, log rank test. “ns”, the life span of RNAi treated animals did not exhibit a statistically significant life span changes. “*”, the experiment was carried out. Please see supplemental Table S2 for statistical details of each experiment.
Among the 21 selected genes, the expression of seven genes was found to be up-regulated in eat-2 mutant nematodes (supplemental Table S1 and Fig. 6C). It is possible that CR initiates the longevity response by increasing the content of these proteins. To test this idea, we examined whether reducing the content of these proteins suppressed the extended life span of the eat-2 mutant. RNAi knockdown of four of these genes, acdh-1, asp-3, sbds-1, and unc-52, significantly decreased the life span of the eat-2 worms without affecting the life span of WT nematodes (Fig. 6 and supplemental Table S2a). Thus, an increase in the content of proteins produced by these genes is required, at least partially, for the longevity effect of CR.
In addition to the hypothesis that CR elicits an increase in the expression of acdh-1, asp-3, sbds-1, and unc-52 to extend life span, CR also may initiate a decrease in the expression of other genes that are important for longevity. Life span analyses was performed on 14 genes (Fig. 6C), whose protein content was down-regulated in eat-2 mutants, as noted in the SILAC experiments. If the reduction of these proteins was critical for CR-mediated life span extension, one would predict that RNAi knockdown of these genes should mimic the life span extension noted with CR nematodes. Indeed, RNAi knockdown of four of these genes, including hpd-1, gei-7, fat-2, and vit-5, significantly extended the life span of WT animals (Fig. 6 and supplemental Table S2a). We then examined the effects of hpd-1, gei-7, fat-2, or vit-5 knockdowns on eat-2 life span. Interestingly, although knockdown of hpd-1 significantly extended the life span of both WT and eat-2 mutant worms, a knockdown of gei-7, fat-2, and vit-5 did not alter or even shorten the life span of the eat-2 mutants (Fig. 6B and supplemental Table S2a). Because the content of these proteins have already been decreased in eat-2 nematodes, further reducing the expression of these genes may not provide additional beneficial effects or may even be detrimental. Previous dosage-dependent studies of CR in C. elegans suggested that instead of extending life span the extreme CR conditions result in a shorter life span (70). This also may explain why some of the genes we tested, such as pod-2, pyc-1, and acdh-12, significantly decreased eat-2 life span without affecting the life span of the WT worms (Fig. 6B and supplemental Table S2a). It is worth noting that 3 of the 14 genes we examined, enol-1, pyk-1, and pck-1 (which encodes PEPCK-C), significantly shortens the life span of both WT and eat-2 worms. All three genes code for key enzymes of glycolysis or gluconeogenesis/glyceroneogenesis; a dysfunction in these genes in the human is linked to metabolic diseases (71, 72).
Because some of these genes may be essential for proper larval development, the initial life span analyses were done by initiating the RNAi treatment at late L4 larva stage, i.e. adult-only treatment. Indeed, RNAi knockdown of enol-1, pyk-1, and pck-1 from hatching caused larval lethality. Nevertheless, similar results were obtained when we repeated the life span analyses by initiating the RNAi treatment from hatching (i.e. whole life treatment) for those genes that do not interfere with development (supplemental Table S2a).
To further explore whether altered energy metabolism influences life span, we overexpressed pck-1, the gene encoding the nematode homolog for PEPCK-C, a key enzyme involved in the cataplerosis of TCA cycle anions. The overexpression of the gene for PEPCK-C in skeletal muscle of mice markedly increased the activity of the mice, the number of mitochondria in their skeletal muscle, and the utilization of fatty acids during strenuous exercise (73); these mice lived ∼30% longer than control animals.6 Overexpressing the nematode form of PEPCK-C (pck-1 (o.e.)) greatly increased the content of PEPCK-C in the worms (Fig. 7A) and markedly induced enzyme activity, when compared with control worms (Fig. 7B). No significant PEPCK activity was detected in nematodes with pck-1 deletion.
FIGURE 7.

Overexpression of the nematode PEPCK-C homolog, pck-1, extends the life span of C. elegans. A, immunoblotting of PEPCK-C in WT, pck-1 (o.e.), and pck-1 knock-out mutant (pck-1 (KO)) nematodes. B, quantification of PEPCK-C activity in WT, pck-1 (o.e.), rol-6, and pck-1 mutant nematodes. The unit of PEPCK-C activity is defined as 1 μmol of substrate converted to product/min at room temperature. Lysates of mouse liver ((m)Liver) were loaded as a positive control. C, survival curves of WT (N2, squares) or worms that overexpress the gene for the nematode homolog of PEPCK-C (pck-1(o.e.), circles). The worms were grown on NGM plates at 20 °C as described under “Materials and Methods.”
The overexpression of pck-1 significantly extended the life span of transgenic worms by 22%, as compared with their nontransgenic wild-type siblings (Fig. 7 and supplemental Table S2b). Thus, in both the mouse and C. elegans increasing the activity of PEPCK-C increases the life span of the animal. The mechanism that is responsible for this effect is presently under investigation.
DISCUSSION
In this study, we have used a multidisciplinary approach to determine how CR affects energy metabolism and how this regulation contributes to life span. To do so, we first applied SILAC to C. elegans and profiled the proteome of eat-2 mutant nematodes. This proteomic and immunoblotting analyses suggested a switch in fuel utilization from carbohydrates to short chain fatty acids in the eat-2 mutant nematode, a suggestion that was confirmed by metabolic quantification. Surprisingly, we noted that the eat-2 mutant had a markedly increased rate of oxidative metabolism of glucose, glutamate, and acetate but not palmitate as compared with control worms. The metabolism of acetate was 21-fold higher in the eat-2 mutant worms when expressed relative to body weight. It is generally believed that CR increases life span, in part by decreasing fuel utilization and thus oxidative metabolism.
Does this metabolic re-patterning influence the health and life span of nematodes undergoing CR? The genetic knockdown of ACDH-1 specifically shortened the extended life span of eat-2 mutant nematodes but had no effect on the life span of WT nematodes. Ablating the expression of two key genes (enol-1 and pyk-1) of glycolysis and pck-1 encoding PEPCK-C, however, led to a nonspecific shortened life span (during adulthood) or caused lethal effect (throughout life) in both eat-2 mutant and WT nematodes. It is likely that the arbitrary suppression of glycolysis or gluconeogenesis and glyceroneogenesis in the RNAi knockdown experiments cannot totally mimic the physiological impact of CR and can itself elicit detrimental effects on the health of nematodes. Animals undergoing CR clearly require both glycolysis and fatty acid oxidation as an energy source to survive, but the animals use fatty acid oxidation to ensure an adequate generation of energy under fuel-restricted conditions. In this regard, a genetic deficiency in pyruvate kinase, enolase, or PEPCK in humans results in clinically documented diseases (71, 72, 74). Nevertheless, overexpression of PEPCK-C, which presumably enhances fuel oxidation by accelerating the cataplerosis of TCA cycles anions, increased the life span of nematodes. Interestingly, the long-lived PEPCK-Cmus mice ate more than controls and mainly utilized fatty acids as energy sources during strenuous exercise (73). In yeast, CR shunted carbon metabolism from fermentation to the oxidation of glucose via the TCA cycle, a metabolic pathway generating more energy. This helped extend life span (75).
Together, these observations indicate that CR-mediated changes in energy metabolism have a critical but complex relationship with the health and life span of C elegans. This also clearly demonstrates that enhanced oxidation of ingested calories, rather than the number of calories that are consumed, contributes to the enhanced life span caused by CR, a mechanism that is likely conserved across phylogeny.
How does this metabolic re-patterning influence the health and life span of nematodes undergoing CR? The overall flux of substrates such as glucose, glutamate, and acetate through the TCA cycle was markedly induced in the eat-2 mutant worms; however, the rate of oxidation of palmitate was not different in the eat-2 as compared with the same weight of WT worms. This may be a metabolic strategy used by eat-2 worms to respond to CR by adapting to the composition of the dietary lipid in the host bacteria (i.e. there is an abundant source of short chain fatty acids) and by conserving long-chain fatty acids. It is important to note that the relative levels of key enzymes in glycolysis, such as ENOL-1, in eat-2 mutant nematodes are lower than in the same weight of WT nematodes, as indicated by both proteomic and immunoblotting. This comparison was made by using the abundance of actin or tubulin as the denominator. One argument is that the proteome, rather than the abundance of several housekeeping proteins, should be used as the denominator. With our SILAC data, the proteome of eat-2 mutant nematodes was estimated as ∼61% of that of the same weight of WT nematodes. Using this comparison strategy, the protein levels of these key enzymes of glycolysis are not changed or even enhanced in the eat-2 mutants. For example, the average “un-normalized” ENOL-1 protein ratio (eat-2 over WT) is 0.73 (supplemental Table S1), suggesting that there are 19% more ENOL-1 proteins in eat-2 mutant nematodes. Additionally, an enzyme, although an important predictor of substrate flux, is not an absolute indicator of the overall flux of carbon over the pathway where the enzyme functions; most enzymes are present in activities that exceed the rate of flux of the specific metabolic pathways and are primarily regulated by allosteric mechanisms (76). The supply of substrates and their relative rate of removal by subsequent metabolic processes are the critical factors in determining the control of the utilization of major metabolites, such as long chain fatty acids. These findings underline the importance of verifying the metabolic predictions of proteomic studies with actual measurements of substrate flux.
A major question raised by these studies is as follows: Why is fuel oxidation increased so markedly in the eat-2 mutant worms? As noted previously, the mutant worms have an ∼20-fold higher rate of acetate metabolism and an ∼7-fold higher rate of exogenous glutamate and glucose metabolism than WT nematodes. Energy metabolism in all organisms is tightly controlled by coupling respiration to ATP synthesis (respiratory control); this ensures that fuel utilization will occur mainly to satisfy the energy demands of the organism. The eat-2 mutants are considerably smaller and are not more active than controls worms, so the reason for their enhanced rate of oxidative metabolism is not apparent. It is known, however, that both eat-2 mutant worms and CR nematodes have a higher rate of oxygen consumption and heat generation than do the N2 controls (21, 64), which may be due to the uncoupling of mitochondrial respiration from ATP synthesis. The advantage of this strategy for the animal, especially when calories are normally limiting, is not clear. It is likely an adaptation of the mutant to the impaired ability to take in fuel. As a result, when fuel is available, the eat-2 mutant largely oxidizes it to CO2 as determined by enhanced TCA cycle flux, instead of depositing it, as do WT nematodes, due to a defect in respiratory control; this leads to the small and lean bodies that are characteristic of the eat-2 mutants. It is also important to consider the possibility that a mutation of the acetylcholine receptor itself may have effects on energy metabolism that are independent of alterations in dietary intake. Future analysis of mitochondrial function in nematodes with CR may answer these and other fundamental questions regarding how CR improves health and life span.
Supplementary Material
Acknowledgments
We thank Kristopher Kramp for technical support; Drs. Leslie Webster and Charles L. Hoppel for critically reading of the manuscript; Dr. Charles L. Hoppel for rat mitochondrial outer membranes and antibodies; Dr. Hui Jin for providing the human HepG2 cell line; Jasmine W. Feng for data analysis, and Caenorhabditis Genetics Center for nematode strains.
This work was supported, in whole or in part, by National Institutes of Health Grants 1R01 AG028516 (to A. L. H.) and P20-CA-103736 (to R. W. H.). This work was also supported by the Mt. Sinai Foundation (to Z. F.), a pilot award from the Proteomics Center, School of Medicine, Case Western Reserve University (to Z. F. and M. M.), and by Grants AG-SS-2420-10 from the Ellison Foundation (to R. W. H.).

This article contains supplemental Tables 1 and 2.
P. Hakimi, N. Berger, and R. W. Hanson, unpublished results.
- CR
- caloric restriction
- PEPCK
- phosphoenolpyruvate carboxykinase
- TCA
- tricarboxylic acid
- SILAC
- stable isotope labeling by amino acids in cell culture
- PFOA
- perfluorooctanoic acid
- o.e.
- overexpression.
REFERENCES
- 1. Chapman T., Partridge L. (1996) Female fitness in Drosophila melanogaster. An interaction between the effect of nutrition and of encounter rate with males. Proc. Biol. Sci. 263, 755–759 [DOI] [PubMed] [Google Scholar]
- 2. Lane M. A., Mattison J., Ingram D. K., Roth G. S. (2002) Caloric restriction and aging in primates. Relevance to humans and possible CR mimetics. Microsc. Res. Tech. 59, 335–338 [DOI] [PubMed] [Google Scholar]
- 3. Masoro E. J. (2000) Caloric restriction and aging. An update. Exp. Gerontol. 35, 299–305 [DOI] [PubMed] [Google Scholar]
- 4. Jiang J. C., Jaruga E., Repnevskaya M. V., Jazwinski S. M. (2000) An intervention resembling caloric restriction prolongs life span and retards aging in yeast. FASEB J. 14, 2135–2137 [DOI] [PubMed] [Google Scholar]
- 5. Weindruch R. (1996) Caloric restriction and aging. Sci. Am. 274, 46–52 [DOI] [PubMed] [Google Scholar]
- 6. Houthoofd K., Vanfleteren J. R. (2006) The longevity effect of dietary restriction in Caenorhabditis elegans. Exp. Gerontol. 41, 1026–1031 [DOI] [PubMed] [Google Scholar]
- 7. Masoro E. J. (2005) Overview of caloric restriction and ageing. Mech. Ageing Dev. 126, 913–922 [DOI] [PubMed] [Google Scholar]
- 8. Weindruch R., Walford R. L., Fligiel S., Guthrie D. (1986) The retardation of aging in mice by dietary restriction. Longevity, cancer, immunity and lifetime energy intake. J. Nutr. 116, 641–654 [DOI] [PubMed] [Google Scholar]
- 9. Weindruch R., Walford R. L. (1988) The Retardation of Aging and Disease by Dietary Restriction, Charles C. Thomas Publishers, Springfield, IL [Google Scholar]
- 10. Masoro E. J. (2003) Subfield history. Caloric restriction, slowing aging, and extending life. Sci. Aging Knowledge Environ. 2003, RE2. [DOI] [PubMed] [Google Scholar]
- 11. Apfeld J., O'Connor G., McDonagh T., DiStefano P. S., Curtis R. (2004) The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to life span in C. elegans. Genes Dev. 18, 3004–3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jia K., Chen D., Riddle D. L. (2004) The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism, and life span. Development 131, 3897–3906 [DOI] [PubMed] [Google Scholar]
- 13. Wang Y., Oh S. W., Deplancke B., Luo J., Walhout A. J., Tissenbaum H. A. (2006) C. elegans 14-3-3 proteins regulate life span and interact with SIR-2.1 and DAF-16/FOXO. Mech. Ageing Dev. 127, 741–747 [DOI] [PubMed] [Google Scholar]
- 14. Bishop N. A., Guarente L. (2007) Two neurons mediate diet restriction-induced longevity in C. elegans. Nature 447, 545–549 [DOI] [PubMed] [Google Scholar]
- 15. Panowski S. H., Wolff S., Aguilaniu H., Durieux J., Dillin A. (2007) PHA-4/Foxa mediates diet restriction-induced longevity of C. elegans. Nature 447, 550–555 [DOI] [PubMed] [Google Scholar]
- 16.Deleted in proof
- 17. Wang Y., Lawler D., Larson B., Ramadan Z., Kochhar S., Holmes E., Nicholson J. K. (2007) Metabonomic investigations of aging and caloric restriction in a life-long dog study. J. Proteome Res. 6, 1846–1854 [DOI] [PubMed] [Google Scholar]
- 18. Kayo T., Allison D. B., Weindruch R., Prolla T. A. (2001) Influences of aging and caloric restriction on the transcriptional profile of skeletal muscle from rhesus monkeys. Proc. Natl. Acad. Sci. U.S.A. 98, 5093–5098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee C. K., Klopp R. G., Weindruch R., Prolla T. A. (1999) Gene expression profile of aging and its retardation by caloric restriction. Science 285, 1390–1393 [DOI] [PubMed] [Google Scholar]
- 20. Harman D. (1972) The biologic clock. The mitochondria? J. Am. Geriatr. Soc. 20, 145–147 [DOI] [PubMed] [Google Scholar]
- 21. Houthoofd K., Braeckman B. P., Lenaerts I., Brys K., De Vreese A., Van Eygen S., Vanfleteren J. R. (2002) No reduction of metabolic rate in food-restricted Caenorhabditis elegans. Exp. Gerontol. 37, 1359–1369 [DOI] [PubMed] [Google Scholar]
- 22. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 23. Sury M. D., Chen J. X., Selbach M. (2010) The SILAC fly allows for accurate protein quantification in vivo. Mol. Cell. Proteomics 9, 2173–2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krüger M., Moser M., Ussar S., Thievessen I., Luber C. A., Forner F., Schmidt S., Zanivan S., Fässler R., Mann M. (2008) SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell 134, 353–364 [DOI] [PubMed] [Google Scholar]
- 25. Larance M., Bailly A. P., Pourkarimi E., Hay R. T., Buchanan G., Coulthurst S., Xirodimas D. P., Gartner A., Lamond A. I. (2011) Stable-isotope labeling with amino acids in nematodes. Nat. Methods 8, 849–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fredens J., Engholm-Keller K., Giessing A., Pultz D., Larsen M. R., Højrup P., Møller-Jensen J., Færgeman N. J. (2011) Quantitative proteomics by amino acid labeling in C. elegans. Nat. Methods 8, 845–847 [DOI] [PubMed] [Google Scholar]
- 27. Avery L. (1993) The genetics of feeding in Caenorhabditis elegans. Genetics 133, 897–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lakowski B., Hekimi S. (1998) The genetics of caloric restriction in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 95, 13091–13096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stiernagle T. (2006) Maintenance of C. elegans. WormBook, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mello C. C., Kramer J. M., Stinchcomb D., Ambros V. (1991) Efficient gene transfer in C. elegans. Extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kadiyala C. S., Tomechko S. E., Miyagi M. (2010) Perfluorooctanoic acid for shotgun proteomics. PLoS One 5, e15332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brenner S. (1974) The genetics of Caenorhabditis elegans. Genetics 77, 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim D. H., Feinbaum R., Alloing G., Emerson F. E., Garsin D. A., Inoue H., Tanaka-Hino M., Hisamoto N., Matsumoto K., Tan M. W., Ausubel F. M. (2002) A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297, 623–626 [DOI] [PubMed] [Google Scholar]
- 34. Hoppel C., Kerner J., Turkaly P., Minkler P., Tandler B. (2002) Isolation of hepatic mitochondrial contact sites. Previously unrecognized inner membrane components. Anal. Biochem. 302, 60–69 [DOI] [PubMed] [Google Scholar]
- 35. Ballard F. J., Hanson R. W. (1969) Purification of phosphoenolpyruvate carboxykinase from the cytosol fraction of rat liver and the immunochemical demonstration of differences between this enzyme and the mitochondrial phosphoenolpyruvate carboxykinase. J. Biol. Chem. 244, 5625–5630 [PubMed] [Google Scholar]
- 36. Lee K., Kerner J., Hoppel C. L. (2011) Mitochondrial carnitine palmitoyltransferase 1a is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 286, 25655–25662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Distler A. M., Kerner J., Lee K., Hoppel C. L. (2009) Post-translational modifications of mitochondrial outer membrane proteins. Methods Enzymol. 457, 97–115 [DOI] [PubMed] [Google Scholar]
- 38. Jomain M., Hanson R. W. (1969) Dietary protein and the control of fatty acid synthesis in rat adipose tissue. J. Lipid Res. 10, 674–680 [PubMed] [Google Scholar]
- 39. Ballard F. J., Hanson R. W. (1967) Phosphoenolpyruvate carboxykinase and pyruvate carboxylase in developing rat liver. Biochem. J. 104, 866–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hsu A. L., Murphy C. T., Kenyon C. (2003) Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300, 1142–1145 [DOI] [PubMed] [Google Scholar]
- 41. Kenyon C., Chang J., Gensch E., Rudner A., Tabtiang R. (1993) A C. elegans mutant that lives twice as long as wild type. Nature 366, 461–464 [DOI] [PubMed] [Google Scholar]
- 42. Taylor A. L., Trotter C. D. (1967) Revised linkage map of Escherichia coli. Bacteriol. Rev. 31, 332–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jekel P. A., Weijer W. J., Beintema J. J. (1983) Use of endoproteinase Lys-C from Lysobacter enzymogenes in protein sequence analysis. Anal. Biochem. 134, 347–354 [DOI] [PubMed] [Google Scholar]
- 44. Hansen M., Taubert S., Crawford D., Libina N., Lee S. J., Kenyon C. (2007) Life span extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell 6, 95–110 [DOI] [PubMed] [Google Scholar]
- 45. Wright A. J., Hunter C. P. (2003) Mutations in a β-tubulin disrupt spindle orientation and microtubule dynamics in the early Caenorhabditis elegans embryo. Mol. Biol. Cell 14, 4512–4525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dong M. Q., Venable J. D., Au N., Xu T., Park S. K., Cociorva D., Johnson J. R., Dillin A., Yates J. R., 3rd (2007) Quantitative mass spectrometry identifies insulin signaling targets in C. elegans. Science 317, 660–663 [DOI] [PubMed] [Google Scholar]
- 47. Braeckman B. P., Houthoofd K., Vanfleteren J. R. (2009) Intermediary metabolism. WormBook, 1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mendenhall A. R., LaRue B., Padilla P. A. (2006) Glyceraldehyde-3-phosphate dehydrogenase mediates anoxia response and survival in Caenorhabditis elegans. Genetics 174, 1173–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Van Gilst M. R., Hadjivassiliou H., Yamamoto K. R. (2005) A Caenorhabditis elegans nutrient response system partially dependent on nuclear receptor NHR-49. Proc. Natl. Acad. Sci. U.S.A. 102, 13496–13501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liao V. H., Freedman J. H. (2001) Characterization of a cadmium-inducible isoform of pyruvate carboxylase from Caenorhabditis elegans. DNA Seq. 12, 137–145 [DOI] [PubMed] [Google Scholar]
- 51. Murphy C. T., McCarroll S. A., Bargmann C. I., Fraser A., Kamath R. S., Ahringer J., Li H., Kenyon C. (2003) Genes that act downstream of DAF-16 to influence the life span of Caenorhabditis elegans. Nature 424, 277–283 [DOI] [PubMed] [Google Scholar]
- 52. Joo H. J., Yim Y. H., Jeong P. Y., Jin Y. X., Lee J. E., Kim H., Jeong S. K., Chitwood D. J., Paik Y. K. (2009) Caenorhabditis elegans utilizes dauer pheromone biosynthesis to dispose of toxic peroxisomal fatty acids for cellular homoeostasis. Biochem. J. 422, 61–71 [DOI] [PubMed] [Google Scholar]
- 53. Brock T. J., Browse J., Watts J. L. (2007) Fatty acid desaturation and the regulation of adiposity in Caenorhabditis elegans. Genetics 176, 865–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li Z., Zhai Y., Fang J., Zhou Q., Geng Y., Sun F. (2010) Purification, crystallization, and preliminary crystallographic analysis of very long-chain acyl-CoA dehydrogenase from Caenorhabditis elegans. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 426–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kuwabara P. E., Shah S. (1994) Cloning by synteny. Identifying C. briggsae homologs of C. elegans genes. Nucleic Acids Res. 22, 4414–4418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gu T., Orita S., Han M. (1998) Caenorhabditis elegans SUR-5, a novel but conserved protein, negatively regulates LET-60 Ras activity during vulval induction. Mol. Cell. Biol. 18, 4556–4564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tagawa A., Rappleye C. A., Aroian R. V. (2001) Pod-2, along with pod-1, defines a new class of genes required for polarity in the early Caenorhabditis elegans embryo. Dev. Biol. 233, 412–424 [DOI] [PubMed] [Google Scholar]
- 58. Horikawa M., Nomura T., Hashimoto T., Sakamoto K. (2008) Elongation and desaturation of fatty acids are critical in growth, lipid metabolism and ontogeny of Caenorhabditis elegans. J. Biochem. 144, 149–158 [DOI] [PubMed] [Google Scholar]
- 59. McGhee J. D., Sleumer M. C., Bilenky M., Wong K., McKay S. J., Goszczynski B., Tian H., Krich N. D., Khattra J., Holt R. A., Baillie D. L., Kohara Y., Marra M. A., Jones S. J., Moerman D. G., Robertson A. G. (2007) The ELT-2 GATA factor and the global regulation of transcription in the C. elegans intestine. Dev. Biol. 302, 627–645 [DOI] [PubMed] [Google Scholar]
- 60. Coudron P. E., Frerman F. E., Schowalter D. B. (1983) Chemical and catalytic properties of the peroxisomal acyl-coenzyme A oxidase from Candida tropicalis. Arch Biochem. Biophys. 226, 324–336 [DOI] [PubMed] [Google Scholar]
- 61. Calvo A. C., Pey A. L., Ying M., Loer C. M., Martinez A. (2008) Anabolic function of phenylalanine hydroxylase in Caenorhabditis elegans. FASEB J. 22, 3046–3058 [DOI] [PubMed] [Google Scholar]
- 62. Veljkovic E., Stasiuk S., Skelly P. J., Shoemaker C. B., Verrey F. (2004) Functional characterization of Caenorhabditis elegans heteromeric amino acid transporters. J. Biol. Chem. 279, 7655–7662 [DOI] [PubMed] [Google Scholar]
- 63. Fei Y. J., Fujita T., Lapp D. F., Ganapathy V., Leibach F. H. (1998) Two oligopeptide transporters from Caenorhabditis elegans. Molecular cloning and functional expression. Biochem. J. 332, 565–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Braeckman B. P., Houthoofd K., Vanfleteren J. R. (2002) Assessing metabolic activity in aging Caenorhabditis elegans. Concepts and controversies. Aging Cell 1, 82–88 [DOI] [PubMed] [Google Scholar]
- 65. Hanson R. W., Ballard F. J. (1967) The relative significance of acetate and glucose as precursors for lipid synthesis in liver and adipose tissue from ruminants. Biochem. J. 105, 529–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ballard F. J., Hanson R. W. (1967) Changes in lipid synthesis in rat liver during development. Biochem. J. 102, 952–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Baldner G. L., Flatt R. E., Shaw R. N., Beitz D. C. (1985) Fatty acid biosynthesis in liver and adipose tissue from dogs. Comp. Biochem. Physiol. B 82, 153–156 [DOI] [PubMed] [Google Scholar]
- 68. Samuelson A. V., Carr C. E., Ruvkun G. (2007) Gene activities that mediate increased life span of C. elegans insulin-like signaling mutants. Genes Dev. 21, 2976–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hansen M., Hsu A. L., Dillin A., Kenyon C. (2005) New genes tied to endocrine, metabolic, and dietary regulation of life span from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet. 1, 119–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ching T. T., Paal A. B., Mehta A., Zhong L., Hsu A. L. (2010) drr-2 encodes an eIF4H that acts downstream of TOR in diet restriction-induced longevity of C. elegans. Aging Cell 9, 545–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Etiemble J., Picat C., Dhermy D., Buc H. A., Morin M., Boivin P. (1984) Erythrocytic pyruvate kinase deficiency and hemolytic anemia inherited as a dominant trait. Am. J. Hematol. 17, 251–260 [DOI] [PubMed] [Google Scholar]
- 72. Stefanini M. (1972) Chronic hemolytic anemia associated with erythrocyte enolase deficiency exacerbated by ingestion of nitrofurantoin. Am. J. Clin. Pathol. 58, 408–414 [DOI] [PubMed] [Google Scholar]
- 73. Hakimi P., Yang J., Casadesus G., Massillon D., Tolentino-Silva F., Nye C. K., Cabrera M. E., Hagen D. R., Utter C. B., Baghdy Y., Johnson D. H., Wilson D. L., Kirwan J. P., Kalhan S. C., Hanson R. W. (2007) Overexpression of the cytosolic form of phosphoenolpyruvate carboxykinase (GTP) in skeletal muscle repatterns energy metabolism in the mouse. J. Biol. Chem. 282, 32844–32855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Vidnes J., Sovik O. (1976) Gluconeogenesis in infancy and childhood. III. Deficiency of the extramitochondrial form of hepatic phosphoenolpyruvate carboxykinase in a case of persistent neonatal hypoglycemia. Acta Paediatr. Scand. 65, 307–312 [DOI] [PubMed] [Google Scholar]
- 75. Lin S. J., Kaeberlein M., Andalis A. A., Sturtz L. A., Defossez P. A., Culotta V. C., Fink G. R., Guarente L. (2002) Calorie restriction extends Saccharomyces cerevisiae life span by increasing respiration. Nature 418, 344–348 [DOI] [PubMed] [Google Scholar]
- 76. Changeux J. P. (1965) The control of biochemical reactions. Sci. Am. 212, 36–45 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.