

Background: Current bacterial methods to measure TM homodimerization are limited to only TM domains expressed in an inverted orientation.
Results: We developed an assay (AraTM) to measure homodimerization of both TM and soluble receptor domains expressed in their native orientation.
Conclusion: CYTO domain of RAGE drives homodimerization in multi-domain constructs, including extracellular and TM domain.
Significance: AraTM enables identification of key receptor domains driving homodimerization in cell membranes.
Keywords: Membrane Biophysics, Membrane Proteins, Protein-Protein Interactions, Receptor Regulation, Signal Transduction, Receptor Oligomerization, Transmembrane Helix Sequence-Structure, Transmembrane-Juxtamembrane Interactions
Abstract
Dimerization is a key regulatory mechanism in activation of transmembrane (TM) receptors during signal transduction. This process involves a coordinated interplay between extracellular (EX), TM, and cytoplasmic (CYTO) regions to form a specific interface required for both ligand binding and intracellular signaling to occur. While several transcriptional activator-based methods exist for investigating TM interactions in bacterial membranes, expression of TM chimera in these methods occurs in a reverse orientation, and are limited to only TM domains for proper membrane trafficking and integration. We therefore developed a new, AraC-based transcriptional reporter assay (AraTM) that expresses EX-TM-CYTO chimera in their native orientation, thereby enabling membrane trafficking to occur independent of the TM chimera used as well as permitting analysis of EX-TM-CYTO interactions in biological membranes. Using integrin αIIb TM-CYTO as a model, we observe a large increase in homodimerization for the constitutively active TM mutant L980A relative to wild-type in the TM-CYTO construct (A963-E1008). We also characterized the receptor for advanced glycation endproducts (RAGE), whose homooligomeric state is critical in ligand recognition, and find the specific juxtamembrane region within the CYTO (A375-P394) mediates homodimerization, and is dominant over effects observed when the extracellular C2 domain is included. Furthermore, we find good agreement between our AraTM measurements in bacterial membranes and BRET measurements made on corresponding RAGE constructs expressed in transfected HEK293 cells. Overall, the AraTM assay provides a new approach to identify specific interactions between receptor EX-TM-CYTO domains in biological membranes that are important in regulation of signal transduction.
Introduction
Transmembrane receptors play critical roles in regulating diverse biological processes, including cell-cell communication, adhesion, and proliferation. In particular, receptor dimerization is a key mechanism by which extracellular cues such as ligand binding are communicated across the cell membrane to activate intracellular signaling processes through formation of a multimeric, signaling complex (1). Recent work has illustrated the importance of specific interfaces involving transmembrane (TM)2 regions of single- and multi-pass membrane proteins in stabilizing receptor signaling complexes, as well as cooperative interactions between TM, cytoplasmic (CYTO) and extracellular (EX) regions that regulate ligand-dependent signal transduction across cell membranes (2). One such example are the integrins, which are a family of heterodimeric, cell surface receptors involved in cell adhesion and cell-cell communication (3). For platelet integrin αIIbβ3, heterodimeric complexes stabilized through specific EX, TM and CYTO interactions occur in the inactive form, and both αIIb and β3 undergo homooligomeric clustering into larger focal adhesion complexes upon inside-out activation (3–7). Likewise, basic residues in the juxtamembrane (JM) region of glycoprotein Ib CYTO are important in stabilizing the glycoprotein Ib-IX complex (8, 9). In the case of platelet-derived growth factor β receptor (PDGFβ), while TM domain interactions involving a conserved, GX3G interface drive homodimerization, purified peptide corresponding to a JM-TM-CYTO fragment of PDGFβ forms a obligate dimer, which suggests the CYTO rather than the TM enforces the stoichiometry of the signaling complex (10). Thus, the interplay between TM, CYTO and EX interactions is central to defining both the active and inactive forms of oligomeric receptor complexes during signal transduction.
Transcription factor-based selection systems such as TOXCAT, POSSYCAT, TOXluc and GALLEX are essential tools for examining specific motifs important in TM-mediated dimerization (11–15). In particular, TOXCAT and POSSYCAT are based on TM domain fusions to ToxR, which is a transmembrane transcription factor from V. cholera (16). Upon dimerization, ToxR binds the ctx promoter and activates gene transcription through its N-terminal DNA binding domain (16). Unlike most single-pass TM receptors, which contain an N-terminal signal peptide to direct membrane insertion (type I) as well as a TM domain (17), ToxR is a type II integral membrane protein, where the C-terminal TM domain functions to direct insertion and integration into the cytoplasmic membrane (18). For TOXCAT, TOXluc, and POSSYCAT, the TM domain of ToxR is replaced with a given TM domain of interest and the periplasmic domain is replaced with a truncated form of maltose-binding protein (MBP) that lacks its N-terminal signal peptide sequence (12–14). For each assay, the heterologous TM domain of interest functions as a surrogate signal peptide to direct insertion of the TM-MBP fusion in an inverted orientation in the inner membrane of E. coli (12–14). The orientation of expressed chimera using ToxR is illustrated in Fig. 1. The extent of TM-mediated oligomerization is reflected in the level of chloramphenicol acetyltransferase (CAT), β-galactosidase, or luciferase reporter gene transcription, all of which are under control of the ctx promoter (12–14). Although ToxR-based methods are useful for studying the homodimeric interaction of TM domains, the N-terminal DNA-binding domain and type II orientation of ToxR requires TM constructs to be expressed as C-terminal fusions directly adjacent to ToxR. Thus, addition of EX or CYTO fragments (i.e. EX-TM-CYTO) to ToxR-TM-MBP chimera interfere with the ability of the TM to act as a signal peptide to properly traffic chimera to the bacterial inner membrane. Additionally, the LexA-based GALLEX method has been used with success to investigate TM domain homo- and heterodimerization (11). However, as with ToxR, the N-terminal orientation of the LexA DNA binding domain necessitates expression of TM-MBP fusions in a reverse orientation in order to insert into the bacterial inner membrane (Fig. 1), thereby permitting only TM domains for analysis (11). Furthermore, wild-type LexA is a soluble, cytosolic protein, so the heterologous TM domain of interest fused to LexA acts as a surrogate signal peptide sequence to the MBP fragment to promote insertion of LexA-TM-MBP chimera into the bacterial inner membrane in an inverted orientation. Therefore, two limitations of current LexA- and ToxR-based methods are that they can only be applied to TM domains, and they place TM domains in an inverted orientation in the bacterial inner membrane.
FIGURE 1.

Organization of AraC and ToxR Fusions Used in TM Interaction Assays. In ToxR-based assays (12–14), constructs are configured with ToxR as an N-terminal fusion to a TM domain of interest, with mature MBP lacking its signal peptide (SP) fused at the C terminus. ToxR is a type II integral membrane protein (18), and therefore the TM domain of interest acts both as a signal peptide to direct membrane trafficking as well as membrane integration with the TM domain in a reverse orientation. In the AraTM assay, the C-terminal orientation of the DNA-binding domain within AraC enables constructs to be expressed with a N-terminal MBP fusion, which includes its native signal peptide to direct membrane integration. Thus, AraTM constructs are expressed in their native orientation, and membrane integration is decoupled from the specific sequence fused to MBP and AraC.
To address these limitations, we developed a new approach based on the Escherichia coli AraC transcription factor to investigate EX-TM-CYTO receptor domain homodimerization (AraTM). Unlike ToxR or LexA, AraC contains a C-terminal DNA binding domain, and is therefore compatible with expressing EX-TM-CYTO fusions in their native orientation as an N-terminal fusion to full-length MBP that includes its native signal peptide (Fig. 1). The native signal peptide present in MBP promotes membrane trafficking, eliminating the need for the TM domain to act as a surrogate signal peptide (19). To quantifying dimerization, we developed an eGFP reporter under control of the AraC-regulated PBAD promoter that enables measurements on whole cells directly from culture. We used integrin αIIb TM-CYTO as a model system to benchmark the sensitivity of our assay to specific TM domain mutations known to affect homodimerization, and extended this approach to identify domains involved in the homodimerization of the receptor for advanced glycation endproducts (RAGE). Our results demonstrate the sensitivity of the AraTM method to specific point mutations or domain deletions within EX-TM-CYTO constructs, and illustrates the utility of AraTM as a complementary approach to other mammalian cell-based assays to investigate the key domains and motifs responsible for mediating receptor dimerization.
EXPERIMENTAL PROCEDURES
Subcloning
Unless otherwise stated, standard molecular biology techniques were used. All constructs used were verified by DNA sequencing (Department of Genetics Core Facility, University of Pennsylvania School of Medicine). A detailed list of primers is provided in supplemental Table S1.
Plasmid pTrcRSF was generated by amplifying the tac promoter region and multiple cloning site from plasmid pTrc99a as a SpeI fragment and RSF origin of replication and kanamycin resistance marker from plasmid pRSF1b as a SpeI fragment, and ligating these two fragments after SpeI digestion and dephosphorylation of the RSF fragment using CIP. AraC was amplified from plasmid pDS439 and subcloned into pTrcRSF as a KpnI/HindIII fragment (20). Full-length MBP, including the N-terminal signal peptide sequence for direct trafficking to the periplasmic membrane, was amplified from pMAL-p2e (NEB) with the reverse primer containing an HA epitope sequence (YPYDVPDYA) and subcloned into pTrcRSF as a NcoI/SacI fragment. A fragment of the MCS from pET28a with additional, unique restriction enzyme sites was subcloned between SacI and KpnI sites (Sequence S1). The resultant plasmid (pAraTM) was used for all subsequent experiments (Sequence S2), with RAGE and integrin αIIb domains cloned in-frame as fusions with MBP and AraC (amino acids 168–293) as SacI/KpnI fragments. Integrin αIIb inserts were generated using overlap-extension PCR with synthetic oligonucleotides corresponding to the TM and JM regions) and RAGE inserts were amplified from plasmid (OriGene) containing the full-length receptor.
The negative control plasmid pAraCY used for the maltose complementation test, which lacks both a TM domain and N-terminal signal peptide sequence, was generated by amplifying the MBP fragment lacking N-terminal signal peptide from plasmid pToxR1, and ligating after NcoI/SacI digestion into plasmid pTrcRSF containing AraC as described above. The negative control plasmid used for all other experiments was pTrcRSF.
The reporter plasmid pAraGFP (Sequence S3) was derived from plasmid pDS439 by amplifying a fragment containing the PBAD promoter, MCS, ampicillin resistance marker and pBR322 origin of replication. This fragment also contains GFP under control of the PBAD promoter, and removes the AraC transcriptional activator present on plasmid pDS439. The reporter plasmid pAraGFP was generated by digestion with SpeI and self-ligation with T4 DNA ligase.
For BRET, full-length human RAGE receptor and RAGE receptor fragments were amplified from plasmid (OriGene) and cloned as fragments into plasmids pGFP2-N3 as NheI/SacI fragments and pRluc-N2 as BglII/KpnI fragments (BioSignal Packard).
AraTM Dimerization Assay
Plasmids pAraTM and pAraGFP were co-transformed into the AraC-deficient E. coli strain SB1676 (The E. coli Genetic Stock Center at Yale University) and streaked on selective LB plates (100 μg/ml ampicillin, 50 μg/ml kanamycin). 8 colonies were picked for each construct and grown in selective LB media for 16 h at 37 °C. After 16 h, 1 μl of saturated culture was added to 1 ml of fresh, selective LB media and grown for an additional 16 h. 400 μl of cultures from each sample was transferred to a black 96-well, clear bottom plate (Greiner) and a series of 2-fold serial dilutions was prepared using selective LB media. A600 measurements and GFP fluorescence emissions spectra (excitation maximum at 485 nm and emissions maximum at 530 nm) were collected using a M200 Infinity plate reader (Tecan). Results are reported as the ratio of fluorescence emission at 530 nm to absorbance at 560 nm.
For FACS measurements, 1 ml of each AraTM construct was grown in selective LB (100 μg/ml ampicillin, 50 μg/ml kanamycin) for 16 h at 37 °C and 200 rpm overnight. After 16 h, 1 μl of saturated culture was added to 1 ml of selective LB media and grown for an additional 16 h. Afterward, cultures were analyzed by flow cytometry (BC FACSCanto II) using 515–545 nm emissions filter for GFP. 100,000 events were collected for each construct and the gate was set so that 0% of the negative control cells were GFP-positive.
Maltose Complementation Test
Plasmid pAraTM containing TM inserts were transformed into the MBP-deficient E. coli strain MM39, streaked onto selective LB plates (50 μg/ml kanamycin) and incubated at 37 °C. The following day, colonies from each plate were grown in selective LB media (50 μg/ml kanamycin) at 37 °C and 200 rpm overnight. 5 μl of saturated culture from each construct was streaked on selective M9 minimal media plates (50 μg/ml kanamycin) containing 0.4% (w/v) maltose and incubated at 37 °C for 2 days to assess growth (12).
Spheroplast Protection Assay and Immunoblotting
For spheroplast immunoblotting, pAraTM plasmids were transformed into MBP-deficient MM39 cells and cultures grown to A600 of 0.6 at 37 °C and 200 rpm before collecting cells. For immunoblotting to detect AraC-chimera expression from whole-cell extracts, plasmid pAraTM containing TM inserts and pAraGFP were transformed into SB1676 cells and cultures were grown for 16 h at 37 °C and 200 rpm. The following morning, cell cultures were diluted back down to A600 of 0.6 before processing cell lysates.
20 μl of culture was mixed with 5 μl of 5× Lammeli sample buffer, heated briefly at 90 °C, then loaded onto a 12% acrylamide gel. Samples were run for 1 h at 200 V using Lammeli running buffer, then transferred to a nitrocellulose membrane (Amersham Biosciences Hybond ECL) for 90 min, blocked for 1 h at room temperature using 5% milk in TBST, then incubated with 1:10,000 dilution of HRP-conjugated anti-MBP monoclonal antibody (NEB) for 1 h at room temperature. Membranes were developed using a chemiluminescent substrate (GE) and imaged using a Typhoon imager.
We used a previously described spheroplast protection assay to assess membrane insertion and integration of MBP-containing chimeras (12). Briefly, cultures were processed using osmotic lysis (Epicenter Biotechnologies) to remove the outer membrane and isolate periplasts and spheroplasts. Spheroplasts were separated from periplasts by centrifugation (12,000 × g) for 5 min, and treated with proteinase K (to determine chimera orientation in theperiplasmic membrane) and Nonidet P-40 (to dissolve spheroplasts and release cytosolic proteins). Chimera expression levels in periplasts, spheroplasts, and cytosolic fractions were detected by immunoblotting as described above.
Bioluminescence Resonance Energy Transfer (BRET)
8 μg of RAGE-GFP and -Rluc fusions were transfected into HEK293 cells by electroporation (GenePulser, Bio-Rad) using the HEK293 preset protocol in HEBS buffer (pH 7.05). Immediately after electroporation, cells were transferred to a white, round-bottom 96 well plate (100 μl of cells per well) in DMEM containing 10% FBS with l-glutamine/VitaMax supplements and 1% Penn/Strep, and incubated at 37 °C and 5% CO2 for 48 h. Media was removed, cells were washed once with 100 μl of PBS per well before 100 μl of BRET buffer (0.1 g/liter CaCl2, 0.1 g/liter MgCl2, and 1 g/liter d-glucose in PBS) was added to each well. 5 μl of Deep Blue C (GoldBio) was added to each sample well, and luminescence measurements (Filter 1: Green and Filter 2: Magenta) were collected using a M200 Infinity plate reader (Tecan) over the course of the 10 s. The energy transfer efficiency is calculated by dividing the intensity of the signal for the green channel by the intensity of the signal for the magenta channel (21) in Equation 1.
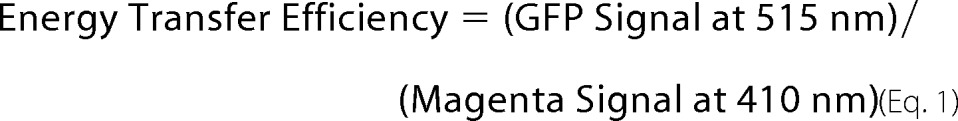 |
The expression level of each eGFP2-fused and Rluc-fused RAGE truncation was evaluated through Western blotting against anti-GFP and anti-Rluc antibody, respectively. Loading control was evaluated through Western blotting against anti-tubulin antibody and HEK293 cells expressing empty eGFP2 and Rluc vectors were used as negative control for BRET.
RESULTS
Design of AraTM Assay
The AraTM assay is based on transcriptional activity of the E. coli AraC transcription factor, which is active at the PBAD promoter as a homodimer (22). Wild-type AraC includes an N-terminal dimerization and ligand-binding domain (amino acids 1–87), which undergoes a conformational change upon l-arabinose binding to bind the I1 and I2 half-sites in the PBAD promoter, thereby driving gene expression (19). Genetic interaction studies indicate that replacement of the wild-type AraC dimerization domain with homo- and heterodimeric coiled-coils such as GCN4 and Fos-Jun causes constitutive activation at the PBAD promoter, leading to the view that AraC is a modular transcriptional activator whose activity can be modified through controlling N-terminal domain dimerization (23, 24). We therefore modified wild-type AraC by replacing the N-terminal dimerization domain with receptor EX-TM-CYTO domains, and utilize GFP expression under control of the PBAD promoter as a measure of receptor domain dimerization. To ensure that EX-TM-CYTO fusions are properly integrated into the bacterial inner membrane, we generated chimeras that also contain an N-terminal fusion to full-length maltose-binding protein (MBP), including its native signal peptide sequence. MBP is a soluble, periplasmic protein responsible for high affinity binding and transport of maltose across the periplasmic membrane (25). Thus, full-length MBP directs the correct expression and integration of receptor EX-TM-CYTO fusions into the periplasmic membrane through its native signal peptide sequence, thereby eliminating the need for the TM domain to act as a surrogate signal peptide. Proper integration of MBP-EX-TM-CYTO-AraC chimera can be confirmed using both complementation experiments on growth media containing maltose as the sole carbon source as well as immunoblotting with anti-MBP antibodies (12). An overview of the AraTM assay is given in Fig. 2, and nucleic acid sequences for plasmids and primers used to develop the AraTM assay are provided in supplemental materials.
FIGURE 2.

Overview of AraTM assay. Chimeric proteins containing N-terminal MBP and C-terminal AraC domains fused with an in-frame receptor fragment are expressed by the regulator plasmid (pAraTM; kanamycin resistant). Once expressed, MBP directs expression of the chimera to the inner membrane of E. coli. Homodimerization brings AraC transcriptional factors in close proximity, enabling binding to the PBAD promoter on the reporter plasmid (pAraGFP; ampicillin resistance) and activating transcription of the reporter gene GFP.
Homodimerization of Integrin αIIb TM-CYTO Domains
To confirm the AraTM assay is sensitive to specific residues known to be responsible for driving TM-CYTO domain dimerization, we used the TM-CYTO of human integrin αIIb as a model system (A963-E1008; Fig. 3A). Integrin αIIbβ3 is the major platelet receptor for fibrinogen, and is comprised of 2 type I integral membrane proteins that form a non-covalent heterodimer in the inactive state stabilized by both specific TM and CYTO interactions (3). Specifically, the integrin αIIb TM domain contains a conserved, GX3G motif that is important for homodimerization, which has led to a proposed push-pull mechanism for regulation of the inactive, active and clustered states in which the interplay between TM and CYTO interactions are important (4, 26, 27). For the wild-type integrin αIIb TM-CYTO (Figs. 3 and 4), significant homodimer formation occurs, with a GFP signal more than twice that of the negative control. Furthermore, when we introduce the specific TM mutation L980A into the TM-CYTO (A963-E1008), which was shown to substantially increase homooligomer formation measured using TOXCAT on a truncated TM construct (W968-K989) as well as by analytical ultracentrifugation and gel-shift assays on a purified TM-CYTO construct (A958-E1008), we observe a 1.5-fold increase in GFP signal relative to wild-type integrin αIIb TM-CYTO (Fig. 3, B and C) (6, 26). To correct for variation in homodimerization signal with cell density, results are reported as the slope of fluorescent intensity for GFP emission versus cell density for a series of cell dilutions. Both wild-type and L980A integrin αIIb TM-CYTO gives consistent results across several independent replicates at varying cell densities, with the majority of individual measurements falling within the estimated 95% confidence interval for the entire dataset (Fig. 3B). We also quantified homodimerization of integrin αIIb TM-CYTO constructs in individual cells using flow cytometry (Fig. 4). Compared with the wild-type integrin αIIb TM-CYTO, the constitutively active L980A mutant has a nearly 2-fold increase in GFP-positive cell population (Fig. 4, A and B), with a clear shift in the overall cell population to higher GFP expression levels rather than skew caused by a small subpopulation of highly fluorescent cells. Thus, the increase in dimerization observed for the L980A mutant reflects a true population average throughout the entire cell suspension. Furthermore, the magnitude of the increase in GFP fluorescence for wt versus L980A is similar for measurements using a microplate reader (Fig. 3) and flow cytometer (Fig. 4), which indicates the robustness of the AraTM method to multiple, independent methods for quantitation.
FIGURE 3.

AraTM assay results for wild-type integrin αIIb TM-CYTO and its mutations. A, integrin αIIb TM-CYTO sequence subcloned into pAraTM. The L980A mutation is also highlighted. B, fluorescence intensity at 530 nm of wild-type integrin αIIb TM-CYTO and mutant L980A from serial dilutions of bacterial cultures plotted against the corresponding cell density (OD600). Solid lines represent the best-fit line through the experimental data, and the dotted lines represent the upper and lower bound of the 95% confidence interval for each data set. C, average slopes from fluorescence intensity versus OD600 for each construct are compared from eight independent replicates, with error bars representing standard error.
FIGURE 4.

Population distribution of GFP-positive cells for various integrin αIIb AraTM constructs by flow cytometry. A, compared with wild-type, the mutant L980A shows an increase in the % of total cell population (100,000 events) that is GFP-positive as well as a shift in the overall cell population measured in terms of FSC to a higher average GFP signal, which is in good agreement with whole-cell fluorescence measurements from cell suspension (Fig. 3). B, integrin αIIb L980A mutant shows a 2-fold increase in the GFP-positive population as compared with wild-type.
Integrin αIIb wild-type and L980A TM-CYTO were both properly integrated in the E. coli inner membrane, as evidenced by growth on maltose M9 minimal plates (Fig. 5A), and expressed at similar levels, as confirmed by immunoblotting from whole-cell lysates (Fig. 5B). For integrin αIIb TM-CYTO mutant L980A, we also confirmed proper membrane integration and orientation using a modified spheroplast digestion assay described previously (12, 26). AraTM constructs were detected using an HRP-conjugated anti-MBP antibody. In particular, we observe intact chimera in the spheroplast fraction, but not the periplasmic or extracellular space, consistent with an inner membrane-integrated TM-CYTO chimera (Fig. 5C) rather than a secreted, soluble periplasmic TM-CYTO chimera. Furthermore, degradation of MBP by proteinase K treatment occurs in the spheroplast fraction in the absence of Nonidet P-40 (Fig. 5C), which indicates the correct, N-terminal orientation of MBP in the periplasmic space accessible to proteinase K rather than accumulation of improperly trafficked chimera in the cytoplasmic space. If the MBP-TM-CYTO-AraC chimera was unable to integrate into the bacterial inner membrane, the membrane would act as a barrier to prevent degradation by proteinase K, resulting in detection of full-length chimera after proteinase K treatment of intact spheroplasts. Whereas in TOXCAT, the spheroplast prevents complete cleavage of MBP by proteinase K for properly integrated chimera (26), the addition of the flexible linker, including an HA epitope, between the N-terminal MBP and integrin αIIb TM-CYTO domain in AraTM increases proteinase K accessibility, which explains the loss of detectable chimera observed in intact spheroplasts by anti-MBP immunoblotting. In addition, for integrin αIIb TM-CYTO L980A mutant, chimera expression begins after 3 h and remaining constant overnight as detected by immunoblotting from whole-cell lysates with anti-MBP antibody (Fig. 6). Thus, the AraTM assay is able to capture relative changes in homodimerization between wild-type integrin αIIb TM-CYTO (A963-E1008) and the key mutant L980A that has been show previously to cause constitutive activation in the full-length receptor (26). Furthermore, the AraTM assay is capable of properly integrating larger TM-CYTO fragments (A963-E1008; Fig. 3) into the bacterial inner membrane, in contrast to truncated TM domain fragments (W968-K989) used previously in TOXCAT (26).
FIGURE 5.

The integrin αIIb TM-CYTO chimera express at similar levels and are properly integrated into the inner membrane of E. coli. A, AraTM chimeras containing wild-type and mutant integrin αIIb TM-CYTO expressed in MalE-deficient MM39 cells were streaked on a 0.4% maltose M9 plate and incubated for 48 h at 37 °C. Each construct is properly integrated into the inner membrane of E. coli, as indicated by robust growth on the 0.4% maltose M9 plates similar to the positive control (pTrcRSF containing MBP-AraC chimera). As expected, no growth is observed on the negative control (AraCY). B, wild-type and mutant integrin αIIb TM-CYTO chimera were expressed at equal levels as determined by immunoblotting with HRP-conjugated anti-MBP antibody, and the observed chimera MWs were consistent with the expected MWs. C, periplasts and spheroplasts were prepared for mutant integrin αIIb L980A TM-CYTO, treated with and without Nonidet P-40 (1% v/v) and proteinase K (50 μg/ml), and blotted against anti-MBP antibody (WC: whole cell, P: periplast, SP: spheroplast, SN: supernatant, PK: proteinase K, and Nonidet P-40: Detergent Nonidet P-40). No chimera is detected in intact spheroplasts treated with proteinase K (SP + PK) nor in the periplasmic fraction of the cell, consistent with the expected periplasmic orientation and membrane integration of the MBP fusion.
FIGURE 6.
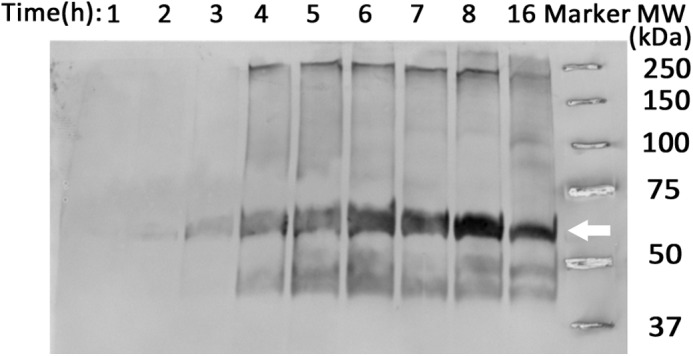
Monitoring expression of AraTM chimera in cell culture. Cells expressing integrin αIIb L980A chimera were collected hourly to monitor expression level and degradation products by immunoblotting with an anti-MBP antibody. Chimera expression began 3 h post-induction at 37 °C and remained constant overnight.
Identifying Domains Critical for Homodimerization of RAGE
RAGE is a 43 kDa type I TM receptor that belongs to the immunoglobulin superfamily (28). RAGE is expressed in a wide range of tissues, including brain, liver, and heart, where it activates pro-inflammatory signaling as part of the innate immune system in response to external stress (29). RAGE is capable of binding several ligands such as S100-family proteins, amyloid-β peptide and advanced glycation endproducts (AGE), and the binding of each ligand as well as subsequent signaling depends on the oligomeric state of the receptor (28, 30). RAGE exists in 2 major forms (Fig. 7A): a full-length, TM receptor (RAGE) as well as a soluble, extracellular domain (sRAGE) that acts as a dominant-negative to inhibit ligand-dependent signaling (28). Most work has focused on determining the structural basis for dimerization of sRAGE, which includes the V-domain (Fig. 7) that is responsible for binding AGE, as well as the V-C1-C2 domain fragment (Fig. 7) that is responsible for binding multimeric S100A-family ligands (31, 32). However, much less is known about specific TM-CYTO interactions that occur in oligomeric states of RAGE.
FIGURE 7.

RAGE. A, graphic illustration of domain structure in the full-length receptor, including the TM and CYTO regions. B, annotation of amino acid sequences in full-length RAGE corresponding to specific domains within the receptor. Sequences for the TM and CYTO region of RAGE are given, including positions (A375, P394) for specific truncations in the CYTO region (SP: signal peptide, V: V domain, C1: C1 domain, C2: C2 domain, PR: proximal domain).
We generated a series of RAGE domain deletions, including deletions of extracellular C2 and CYTO domains, and quantified their homodimerization using the AraTM assay. Domains and numbering used to delineate the specific domains with RAGE used are given in Fig. 7B. Full-length RAGE (V-C1-C2-PR-TM-CYTOfull; A23-P404) and RAGE chimera containing C1-domains (C1-C2-PR-TM-CYTOfull; P124-P404) were unable to complement grow on maltose minimal media, and were therefore excluded from further analysis (Fig. 8A). However, all other RAGE constructs were properly integrated into the bacterial inner membrane, as indicated by robust growth on maltose M9 minimal plates (Fig. 8A), and each was expressed at similar levels as indicated by whole-cell immunoblotting with anti-MBP (Fig. 8B). Isolation of spheroplasts from cells expressing RAGE PR-TM-CYTOfull (R314-P404) indicated they were properly oriented into the inner membrane (Fig. 8C), as degradation of MBP in intact spheroplasts occurred only after proteinase K treatment in the absence of Nonidet P-40 (12).
FIGURE 8.

Expression of RAGE constructs in AraTM assay. A, RAGE C2-PR-TM-CYTOfull AraTM chimera as well as additional truncations of RAGE C2-PR-TM-CYTOfull are able to complement growth on maltose M9 minimal plates. B, cells expressing RAGE chimera were expressed at similar levels and the chimera MWs are consistent with the expected MWs as determined by immunoblotting from whole-cell lysates with anti-MBP antibody. C, periplasts and spheroplasts of the RAGE PR-TM-CYTOfull chimera were prepared, treated with/without Nonidet P-40 (1% v/v) and proteinase K (50 μg/ml), and blotted against anti-MBP antibody (WC: whole cell, P: periplast, SP: spheroplast, SN: supernatant, PK: proteinase K, and Nonidet P-40: detergent Nonidet P-40). No chimera is detected in intact spheroplasts treated with proteinase K (SP + PK) nor in the periplasmic fraction of the cell, consistent with the expected periplasmic orientation and membrane integration of the MBP-RAGE-AraC fusion.
We find that deletion of the C2 domain has little impact on homodimerization in constructs containing the CYTO domain (C2-PR-TM-CYTOfull; P224-P404 and PR-TM-CYTOfull; R314-P404), whereas deletion of the CYTO domain significantly reduces homodimerization largely independent of the C2 domain (C2-PR-TM; P224-R365 and PR-TM; R314-R365) (Fig. 9). We made additional deletions within RAGE CYTO to identify which regions were most important for homodimerization. We find that removal of the C-terminal distal region (PR-TM-CYTOP394; R314-P394) has no measurable impact on homodimerization, whereas deletion of the glutamic acid-rich region of the CYTO (PR-TM-CYTOA375; R314-A375) causes a modest decrease in homodimerization. However, deletion of the entire CYTO domain (PR-TM; R314-R365) causes a greater than 2-fold decrease in dimerization (Fig. 9), which indicates the juxtamembrane region (R365-A375) present within the CYTO is key for dimerization of TM RAGE constructs. Thus, we conclude that the RAGE CYTO exhibits strong homodimerization in the context of C2-TM-CYTO domains expressed in cell membranes, particularly the juxtamembrane region (R365-A375) present within the CYTO.
FIGURE 9.

Cytoplasmic truncations of RAGE reduce homodimerization in cell membranes. Removing the last 10 amino acids in the cytosolic domain of RAGE (PR-TM-CTYOP394) had minimal affect on RAGE dimerization, whereas removal of the central domain (PR-TM-CYTOA375) reduced dimerization, but not to background levels observed in the cytoplasmic domain deletion construct. Results shown are from three independent replicates and the error bars represent standard error.
Homodimerization of RAGE Receptor Expressed in Mammalian Membranes
To confirm that our results from the AraTM assay are relevant in the context of full-length RAGE, we generated full-length RAGE and RAGE CYTO deletions as fusions with both eGFP2 and Rluc, co-expressed both eGFP2 and Rluc fusions in HEK293 cells, and measured the apparent interaction between each CYTO domain deletion using BRET (33, 34). The extent of energy transfer was calculated by dividing the green signal (515 nm) by the magenta signal (410 nm) as described previously (33). Expression levels for each construct as eGFP2 and Rluc fusions were similar among different truncations as determined by immunoblotting with specific antibodies (supplemental Fig. S1). Consistent with previous FRET measurements in transfected HEK293 cells (35), we find that full-length RAGE receptor exhibits a strong homodimeric signal in the absence of ligand (Fig. 10). Consistent with our AraTM results, removal of the C-terminal distal region (V-C1-C2-PR-TM-CYTOP394; A23-P394) has no impact on homodimerization, but removal of the glutamic acid-rich CYTO region (V-C1-C2-PR-TM-CYTOA375; A23-A375) causes a significant decrease of nearly 25% in the measured BRET signal relative to wild-type RAGE (Fig. 10). It should be noted that the decrease in observed homodimerization for the V-C1-C2-PR-TM-CYTOA375 construct measured using BRET (Fig. 10) is greater than the intermediate reduction in homodimerization signal measured using AraTM (Fig. 9), which indicates a significant, but intermediate effect on dimerization. Deletion of the CYTO reduces the measured BRET signal by 50% to near background levels. Thus, the trend in terms of reduction in homodimerization for the PR-TM-CYTO deletions observed in AraTM (Fig. 9) are consistent with RAGE truncations expressed in HEK293 cells (Fig. 10), and the unliganded receptor is stabilized through interactions involving the juxtamembrane region of RAGE (R365-A375) present in the CYTO.
FIGURE 10.

RAGE domain interactions in mammalian membranes correlate with AraTM results, and highlight the importance of cytoplasmic domain interactions in ligand-independent dimerization. Significant dimerization of RAGE is observed in the absence of the ligand. Removal of the distal C-terminal region (P394) has no impact on homodimerization, but removal of the central CYTO region (A375) causes a significant decrease in homodimerization. Experiments were repeated three times in triplicate, and error bars represent stand error of the mean (Full: full-length, P394: CYTO truncation at P394, A375: CYTO truncation at A375, ΔCYTO: CYTO truncation, and Negative: negative control).
DISCUSSION
The AraTM method provides several advantages in investigating the importance of extracellular (EX), transmembrane (TM), and cytoplasmic (CYTO) domains in defining the dimeric state of TM receptors. In particular, the C-terminal orientation of the DNA-binding domain in AraC enables expression of chimera, including both soluble and transmembrane domains, as a fusion to full-length MBP, including its native signal peptide, to direct membrane insertion. As mentioned previously, the ToxR transcription factor used in TOXCAT, POSSYCAT and TOXluc assays is a type II integral membrane protein containing an N-terminal DNA-binding domain, in which the ToxR TM domain functions as a signal peptide to direct trafficking to the bacterial inner membrane as well as membrane integration of ToxR (17, 18). Thus, modifications to ToxR-TM-MBP chimera that include both soluble EX or CYTO domains fused to TM domains, or specific mutations within a given TM domain, can interfere with membrane trafficking and lead to large differences in chimera expression levels between different constructs (36). In contrast, the C-terminal orientation of the DNA-binding domain in AraC enables expression of constructs in their native orientation, and utilizes the native signal peptide of MBP to direct insertion into the bacterial inner membrane (Figs. 1 and 2). Thus, the AraTM method decouples the dependence of a given TM construct on membrane trafficking from its dimerization in the bacterial inner membrane. This is particularly important given the large impact on extent of dimerization that can occur with cell-to-cell and construct-to-construct variability in ToxR-TM-MBP expression observed using TOXCAT, as well as the need to perform serial N- and C-terminal deletions of specific amino acids within a given TM domain to maximize membrane integration of expressed ToxR-TM-MBP chimera (12, 36). As illustrated for both integrin αIIb (Fig. 5) and RAGE (Fig. 8), the AraTM system is robust to expression and proper membrane integration of a wide range of EX, TM, and CYTO constructs, including RAGE C2-PR-TM-CYTOfull (P224-P404, 21.3 kDa) and integrin αIIb TM-CYTO (A963-E1008; 5.2 kDa), versus truncated TM domains such as that used in TOXCAT for integrin αIIb TM (W968-K989; 2.5 kDa) (26). Thus, the AraTM method (Fig. 2) enables analysis of dimerization in cell membranes of multi-domain receptor chimera that include EX, TM and CYTO, which is of particular use in investigating juxtamembrane interactions important in defining active and inactive states during signal transduction (6, 8–10).
The use of eGFP as a reporter gene in AraTM also allows for assays to be performed directly from cell culture using a microplate reader or flow cytometer, without a need for cell lysis, sample preparation or addition of exogenous substrates required for other common reporters such as CAT or luciferase. As mentioned before, cell-to-cell variability in TOXCAT chimera expression levels has a major impact on the observed dimerization signal (36), which we can measure directly from whole cells using flow cytometry to determine average dimerization per cell for a statistically significant sample size (> 106 cells; Fig. 4). As illustrated by our measurements using integrin αIIb TM-CYTO (A693-E1008), the AraTM method is sensitive to specific point mutations in the context of multi-domain constructs, with a significant increase in homodimerization observed in the case of the TM L980A mutant relative to wt TM-CYTO (Fig. 4) (4, 26). Furthermore, the quantitative agreement between the relative increase in dimerization for wt integrin αIIb TM-CYTO and L980A mutant measured using a microplate fluorescence plate reader (Fig. 3) and flow cytometer (Fig. 4), as well as the shift in total population mean observed between wt and L980A (Fig. 4), indicate the increase in observed GFP signal for L980A is due to a uniform increase in dimerization across the entire cell population rather than skew in the distribution caused by a specific cell subpopulation. Thus, the AraTM method is able to capture effects of specific mutations on homodimerization in the context of TM-CYTO receptor fragments that are consistent with results from previously described in vitro and cell-based assays for integrin αIIb.
In addition, our results point to a direct role for the RAGE CYTO in stabilizing the homooligomeric, unliganded form of the receptor. Previous studies have focused on the soluble, extracellular form of RAGE (sRAGE) containing the V-, C1-, and C2-domains as the primary driving force for homodimerization (31, 32). Purified sRAGE and C1-C2 domains assemble to form stable tetramers in vitro, and upon hexameric calgranulin binding, sRAGE undergoes significant conformational rearrangement in the ectodomain to form higher-order oligomers in the soluble, liganded state (32). Overexpression of full-length RAGE in transfected mammalian cells as mCFP and mYFP fusions result in significant co-localization and FRET transfer efficiency at the surface of transfected HEK293 cells, which supports the idea of RAGE exists in a homooligomeric state in the absence of ligand (35). Using AraTM, we find that the C2 domain does not play a key role in EX-TM-CYTO homodimerization, whereas the CYTO domain plays a key role in stabilizing the unliganded, homodimeric state of RAGE (Fig. 9). Specifically, deletion of the distal C-terminal region (PR-TM-CYTOP394; R314-P394) has no impact on dimerization, whereas deletion of the central cytoplasmic domain (PR-TM-CYTOA375; R314-A375) significantly reduces RAGE homodimerization, and the cytoplasmic domain deletion eliminated homodimerization. Previous work has indicated the extracellular V- and C1-domains as critical for dimerization of sRAGE in the absence of ligand (31, 32), whereas our BRET results for full-length RAGE indicate the strong self-association propensity of the CYTO domain in stabilizing the resting state of the receptor, specifically the juxtamembrane region (A375-P394) within the CYTO important for stabilization of the unliganded, homodimeric state (Fig. 10). Recent NMR data investigating RAGE CYTO binding to the cytosolic signaling protein mDia1 suggests that the TM-proximal region is important for protein-protein interactions involving cytosolic signaling molecules, with the R365-P376 region of the CYTO forming a stable α-turn in solution (37). Our AraTM (Fig. 9) and BRET (Fig. 10) results indicate this region may also play a key role in homodimerization in the unliganded state. Interestingly, there are multiple, conserved families of primarily soluble proteins containing glutamic acid rich regions, in which the glutamic acid-rich region is found to have important roles in protein structure and complex formation. Specifically, in the case of the soluble protein SH3BGR, the C-terminal, glutamic acid-rich region is predicted to adopt coiled-coil like structures capable of mediating protein-protein interactions (38). Other studies have suggested glutamic acid-rich regions may act as low-affinity, calcium-binding domains important in regulating local calcium concentrations (39). Thus, the disordered, glutamic acid region of the CYTO observed previously by NMR may reflect one of several conformations for the RAGE CYTO during signal transduction (37). Furthermore, the overall correlation between the magnitude of homooligomerization measured using AraTM (Fig. 9) and BRET (Fig. 10) emphasizes the utility of the AraTM method to investigate the role of receptor dimerization in the biologically relevant context of cell membranes.
Overall, given the good agreement between our results for integrin αIIb and previously published results investigating homodimerization in vivo and in vitro, as well as the correlation between our results investigating RAGE domain homodimerization in bacterial membranes and BRET signal in transfected mammalian cells, the AraTM provides a useful, complementary method to other mammalian cell-based measurement techniques such as BRET and bacterial 2-hybrid assays such as BACTH to rapidly assess the importance of specific interfaces and domains within transmembrane receptors in defining their dimeric states (33, 40).
Supplementary Material
Acknowledgments
We thank Dr. Joel Bennett (University of Pennsylvania School of Medicine) for feedback and helpful suggestions regarding integrin αIIb.
This work was supported by Lehigh University, including funds provided through a Faculty Research Grant (FRG).

This article contains supplemental Table S1 and Fig. S1.
- TM
- transmembrane
- AraTM
- AraC-based transcriptional reporter assay
- RAGE
- receptor for advanced glycation endproducts
- BRET
- bioluminescence resonance energy transfer
- CYTO
- cytoplasmic domain
- CAT
- chloramphenicol acetyltransferase
- JM
- juxtamembrane
- MBP
- maltose-binding protein.
REFERENCES
- 1. Klemm J. D., Schreiber S. L., Crabtree G. R. (1998) Dimerization as a regulatory mechanism in signal transduction. Annu. Rev. Immunol. 16, 569–592 [DOI] [PubMed] [Google Scholar]
- 2. Moore D. T., Berger B. W., DeGrado W. F. (2008) Protein-protein interactions in the membrane: sequence, structural, and biological motifs. Structure 16, 991–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bennett J. S. (2005) Structure and function of the platelet integrin αIIbβ3. J. Clin. Investig. 115, 3363–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li W., Metcalf D. G., Gorelik R., Li R., Mitra N., Nanda V., Law P. B., Lear J. D., Degrado W. F., Bennett J. S. (2005) A push-pull mechanism for regulating integrin function. Proc. Natl. Acad. Sci. U.S.A. 102, 1424–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Metcalf D. G., Kulp D. W., Bennett J. S., DeGrado W. F. (2009) Multiple approaches converge on the structure of the integrin αIIb/β3 transmembrane heterodimer. J. Mol. Biol. 392, 1087–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li R., Babu C. R., Lear J. D., Wand A. J., Bennett J. S., DeGrado W. F. (2001) Oligomerization of the integrin αIIbβ3: roles of the transmembrane and cytoplasmic domains. Proc. Natl. Acad. Sci. U.S.A. 98, 12462–12467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berger B. W., Kulp D. W., Span L. M., DeGrado J. L., Billings P. C., Senes A., Bennett J. S., DeGrado W. F. (2010) Consensus motif for integrin transmembrane helix association. Proc. Natl. Acad. Sci. U.S.A. 107, 703–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mo X., Luo S. Z., López J. A., Li R. (2008) Juxtamembrane basic residues in glycoprotein Ibβ cytoplasmic domain are required for assembly and surface expression of glycoprotein Ib-IX complex. FEBS Lett. 582, 3270–3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mo X., Luo S. Z., Munday A. D., Sun W., Berndt M. C., López J. A., Dong J. F., Li R. (2008) The membrane-proximal intermolecular disulfide bonds in glycoprotein Ib influence receptor binding to von Willebrand factor. J. Thrombosis Haemostasis 6, 1789–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oates J., King G., Dixon A. M. (2010) Strong oligomerization behavior of PDGFβ receptor transmembrane domain and its regulation by the juxtamembrane regions. Biochim. Biophys. Acta 1798, 605–615 [DOI] [PubMed] [Google Scholar]
- 11. Schneider D., Engelman D. M. (2003) GALLEX, a measurement of heterologous association of transmembrane helices in a biological membrane. J. Biol. Chem. 278, 3105–3111 [DOI] [PubMed] [Google Scholar]
- 12. Russ W. P., Engelman D. M. (1999) TOXCAT: a measure of transmembrane helix association in a biological membrane. Proc. Natl. Acad. Sci. U.S.A. 96, 863–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gurezka R., Langosch D. (2001) In vitro selection of membrane-spanning leucine zipper protein-protein interaction motifs using POSSYCCAT. J. Biol. Chem. 276, 45580–45587 [DOI] [PubMed] [Google Scholar]
- 14. Roth L., Nasarre C., Dirrig-Grosch S., Aunis D., Crémel G., Hubert P., Bagnard D. (2008) Transmembrane domain interactions control biological functions of neuropilin-1. Mol. Biol. Cell 19, 646–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ladant D., Karimova G. (2000) Genetic systems for analyzing protein-protein interactions in bacteria. Res. Microbiol. 151, 711–720 [DOI] [PubMed] [Google Scholar]
- 16. DiRita V. J. (1992) Co-ordinate expression of virulence genes by ToxR in V. cholerae. Mol. Microbiol. 6, 451–458 [DOI] [PubMed] [Google Scholar]
- 17. Ott C. M., Lingappa V. R. (2002) Integral membrane protein biosynthesis: why topology is hard to predict. J. Cell Sci. 115, 2003–2009 [DOI] [PubMed] [Google Scholar]
- 18. Kolmar H., Hennecke F., Götze K., Janzer B., Vogt B., Mayer F., Fritz H. J. (1995) Membrane insertion of the bacterial signal transduction protein ToxR and requirements of transcription activation studied by modular replacement of different protein substructures. EMBO J. 14, 3895–3904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bustos S. A., Schleif R. F. (1993) Functional domains of the AraC protein. Proc. Natl. Acad. Sci. U.S.A. 90, 5638–5642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Siegele D. A., Hu J. C. (1997) Gene expression from plasmids containing the araBAD promoter at subsaturating inducer concentrations represents mixed populations. Proc. Natl. Acad. Sci. U.S.A. 94, 8168–8172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bonde M. M., Hansen J. T., Sanni S. J., Hauns S., Gammeltoft S., Lyngs C., Hansen J. L. (2010) Biased signaling of the angiotensin II type 1 receptor can be mediated through distinct mechanisms. PLoS ONE 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schleif R. (2003) AraC protein: a love-hate relationship. BioEssays 25, 274–282 [DOI] [PubMed] [Google Scholar]
- 23. Weldon J. E., Schleif R. F. (2006) Specific interactions by the N-terminal arm inhibit self-association of the AraC dimerization domain. Protein Science 15, 2828–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eustance R., Schleif R. (1996) In vivo association of protein fragments giving active AraC. Proteins-Structure Function Bioinformatics 25, 501–505 [DOI] [PubMed] [Google Scholar]
- 25. Nikaido H. (1994) Maltose transport system of E. coli: an ABC-type transporter. FEBS Lett. 346, 55–58 [DOI] [PubMed] [Google Scholar]
- 26. Li R., Gorelik R., Nanda V., Law P. B., Lear J. D., DeGrado W. F., Bennett J. S. (2004) Dimerization of the transmembrane domain of Integrin αIIb subunit in cell membranes. J. Biol. Chem. 279, 26666–26673 [DOI] [PubMed] [Google Scholar]
- 27. Schneider D., Engelman D. (2004) Involvement of transmembrane domain interactions in signal transduction by α/β integrins. J. Biol. Chem. 279, 9840–9846 [DOI] [PubMed] [Google Scholar]
- 28. Schmidt A. M., Yan S. D., Yan S. F., Stern D. M. (2001) The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Investig. 108, 949–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hofmann M. A., Drury S., Fu C., Qu W., Taguchi A., Lu Y., Avila C., Kambham N., Bierhaus A., Nawroth P., Neurath M. F., Slattery T., Beach D., McClary J., Nagashima M., Morser J., Stern D., Schmidt A. M. (1999) RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97, 889–901 [DOI] [PubMed] [Google Scholar]
- 30. Ostendorp T., Leclerc E., Galichet A., Koch M., Demling N., Weigle B., Heizmann C. W., Kroneck P. M., Fritz G. (2007) Structural and functional insights into RAGE activation by multimeric S100B. EMBO J. 26, 3868–3878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zong H., Madden A., Ward M., Mooney M. H., Elliott C. T., Stitt A. W. (2010) Homodimerization is essential for the receptor for advanced glycation end products (RAGE)-mediated signal transduction. J. Biol. Chem. 285, 23137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xie J., Burz D. S., He W., Bronstein I. B., Lednev I., Shekhtman A. (2007) Hexameric calgranulin C (S100A12) binds to the receptor for advanced glycated end products (RAGE) using symmetric hydrophobic target-binding patches. J. Biol. Chem. 282, 4218–4231 [DOI] [PubMed] [Google Scholar]
- 33. Ramsay D., Kellett E., McVey M., Rees S., Milligan G. (2002) Homo- and hetero-oligomeric interactions between G-protein-coupled receptors in living cells monitored by two variants of bioluminescence resonance energy transfer (BRET): hetero-oligomers between receptor subtypes form more efficiently than between less closely related sequences. Biochem. J. 365, 429–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Buensuceso C. (2003) Detection of integrin αIIbβ3 clustering in living cells. J Biol. Chem. 278, 15217–15224 [DOI] [PubMed] [Google Scholar]
- 35. Xie J., Reverdatto S., Frolov A., Hoffmann R., Burz D. S., Shekhtman A. (2008) Structural basis for pattern recognition by the receptor for advanced glycation end products (RAGE). J. Biol. Chem. 283, 27255–27269 [DOI] [PubMed] [Google Scholar]
- 36. Duong M. T., Jaszewski T. M., Fleming K. G., MacKenzie K. R. (2007) Changes in apparent free energy of helix-helix dimerization in a biological membrane due to point mutations. J. Mol. Biol. 371, 422–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rai V., Maldonado A. Y., Burz D. S., Reverdatto S., Yan S. F., Schmidt A. M., Shekhtman A. (2012) Signal transduction in receptor for advanced glycation end products (RAGE): solution structure of C-terminal rage (ctRAGE) and its binding to mDia1. J. Biol. Chem. 287, 5133–5144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Scartezzini P., Egeo A., Colella S., Fumagalli P., Arrigo P., Nizetic D., Taramelli R., Rasore-Quartino A. (1997) Cloning a new human gene from chromosome 21q22.3 encoding a glutamic acid-rich protein expressed in heart and skeletal muscle. Human Genetics 99, 387–392 [DOI] [PubMed] [Google Scholar]
- 39. Haber-Pohlmeier S., Abarca-Heidemann K., Körschen H. G., Dhiman H. K., Heberle J., Schwalbe H., Klein-Seetharaman J., Kaupp U. B., Pohlmeier A. (2007) Binding of Ca2+ to glutamic acid-rich polypeptides from the rod outer segment. Biophys. J. 92, 3207–3214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karimova G., Pidoux J., Ullmann A., Ladant D. (1998) A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. U.S.A. 95, 5752–5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.