

Abstract
Cell–cell recognition often requires the formation of a highly organized pattern of receptor proteins (a synapse) in the intercellular junction. Recent experiments [e.g., Monks, C. R. F., Freiberg, B. A., Kupfer, H., Sciaky, N. & Kupfer, A. (1998) Nature (London) 395, 82–86; Grakoui, A., Bromley, S. K., Sumen, C., Davis, M. M., Shaw, A. S., Allen, P. M. & Dustin, M. L. (1999) Science 285, 221–227; and Davis, D. M., Chiu, I., Fassett, M., Cohen, G. B., Mandelboim, O. & Strominger, J. L. (1999) Proc. Natl. Acad. Sci. USA 96, 15062–15067] vividly demonstrate a complex evolution of cell shape and spatial receptor–ligand patterns (several microns in size) in the intercellular junction during immunological synapse formation. The current view is that this dynamic rearrangement of proteins into organized supramolecular activation clusters is driven primarily by active cytoskeletal processes [e.g., Dustin, M. L. & Cooper, J. A. (2000) Nat. Immunol. 1, 23–29; and Wulfing, C. & Davis, M. M. (1998) Science 282, 2266–2269]. Here, aided by a quantitative analysis of the relevant physico-chemical processes, we demonstrate that the essential characteristics of synaptic patterns observed in living cells can result from spontaneous self-assembly processes. Active cellular interventions are superimposed on these self-organizing tendencies and may also serve to regulate the spontaneous processes. We find that the protein binding/dissociation characteristics, protein mobilities, and membrane constraints measured in the cellular environment are delicately balanced such that the length and time scales of spontaneously evolving patterns are in near-quantitative agreement with observations for synapse formation between T cells and supported membranes [Grakoui, A., Bromley, S. K., Sumen, C., Davis, M. M., Shaw, A. S., Allen, P. M. & Dustin, M. L. (1999) Science 285, 221–227]. The model we present provides a common way of analyzing immunological synapse formation in disparate systems (e.g., T cell/antigen-presenting cell junctions with different MHC-peptides, natural killer cells, etc.).
Important biological events, such as cell fate during development and initiation of various immune responses, are determined by binary cell–cell interactions. Much attention has been directed toward understanding the exquisitely specific molecular scale receptor–ligand binding that is essential for intercellular recognition and signaling. In many instances, successful engagement of a receptor with a complementary ligand on the apposing cell is, by itself, insufficient for sustained intracellular signaling and the concomitant triggering of a specific biological function. For example, the definitive event governing a mature immune response when T lymphocyte or natural killer (NK) cells interact with target cells is the formation of an immunological synapse (e.g., refs. 1–10). Concerted reorganization of the spatial pattern of membrane proteins and cell shape occurs during this recognition process (1–3). The resulting synapse is a clearly organized pattern of protein complexes, several microns in diameter, that forms at the junction between the membranes of the two cells. The mechanisms that drive the formation of synaptic patterns in intercellular junctions are not as well understood as the individual receptor–ligand binding events.
Here, we examine whether the observed characteristics of synaptic patterns (1–3) can emerge spontaneously during cooperative interaction between populations of receptors and ligands in two apposed membranes. We consider a fluctuating two-dimensional membrane containing proteins that can interact with a flat two-dimensional membrane containing complementary protein ligands (Fig. 1). An analogous configuration has been used (e.g., refs. 2, 7, and 10) to visualize synapse formation between T cells and supported lipid membranes. We formulate and study a model for this situation that explicitly incorporates protein binding and dissociation, lateral motion of proteins in the cell membranes, membrane mechanics (bending rigidity and tension), and protein down regulation. The results show that natural coupling of the forces due to these phenomena can lead to spatio-temporal evolution of protein patterns and cell shape resembling that observed during synapse formation in living cells. In this paper, we focus primarily on applying this general model to study the specialized junction between T cells and antigen-presenting cells (APC).
Figure 1.

Schematic depiction of a membrane that can undergo shape changes interacting with a planar membrane. Both membranes contain two proteins (red and green) that are mobile within the membrane. The red (green) proteins are complementary to the red (green) proteins on the apposing membrane and can bind to them selectively if they are sufficiently close. The complexes can dissociate. The red and green proteins are of different size. The proteins in the two membranes are initially not complexed. This cartoon depicts a situation observed in experiments (1, 2) during the early stages of T cell synapse formation.
The formation of functional immunological synapses between T cells and supported bilayers reconstituted to mimic APCs has recently been observed (2). A combination of interference reflection microscopy, fluorescence imaging, and immunoradiometric assays has provided quantitative information on the spatial pattern of membrane protein concentrations and membrane topography during synapse formation. Detailed accounts of these measurements and those with T cells and APCs (which exhibit the same phenomenology) are available (1, 2). Fig. 2 A and B are experimental results taken from Grakoui et al. (2) that show the dynamic reorganization of the membrane proteins, MHC-peptide, and intercellular adhesion molecule-1 (ICAM-1), and cell shape changes in the contact area during the formation of the T cell immunological synapse. When the physico-chemical parameters in the theoretical model developed by us in this paper are set to be those measured in the cellular environment and for the specific proteins involved in the formation of the T cell immunological synapse [MHC-peptide, T cell antigen receptor (TCR), ICAM-1, and lymphocyte function-associated antigen-1 (LFA-1)], calculated results are in near-quantitative agreement with these experimental observations. We have also studied the effects of changing MHC-peptide/TCR binding kinetics over a wide range (antagonist to super agonist). The model predictions are consistent with a large body of experimental observations (11) regarding the correlation between TCR/MHC-peptide dissociation kinetics and biological outcome. A possible mechanism for this correlation is suggested. Our results also shed light on why the immunological synapse for NK cells exhibits a pattern that is inverted (3) compared with that observed for T cells. The effects of altering parameters that characterize membrane mechanics and receptor protein mobility on synapse formation are also briefly explored using our model.
Figure 2.

(A) Interference reflection microscopy (IRM) images from experiments (2) with living T cells. The darker shading represents regions with closer apposition of membranes. (B) Experimental fluorescence microscopy images (taken from ref. 2) showing the accumulated concentration of MHC-peptide (green) and ICAM-1 (red). The experimental errors in the concentration measurements are ±5% (M. L. Dustin, personal communication). (C) Model predictions (our calculations) for local distances between the two apposing membranes. The darker shading represents regions with closer apposition of membranes. (D) Model predictions (our calculations) for the accumulated concentration of MHC-peptide (green). The darker green color represents regions where the concentration is greater than 50 molecules/μm2, and the lighter green corresponds to concentrations between 30 and 50 molecules/μm2. The initial uniform MHC-peptide concentration is 20 molecules/μm2. (E) Model predictions (our calculations) for the accumulated concentration of ICAM-1 (red). The darker red color represents regions where the concentration is greater than 120 molecules/μm2, and the lighter red corresponds to concentrations between 40 and 120 molecules/μm2. The initial uniform ICAM-1 concentration is 40 molecules/μm2. The parameters used are detailed in Methods. These particular data correspond to koff = 1 s−1 and DTCR = DLFA-1 = 1 μm2/s. We have carried out calculations for a range of values of the diffusion coefficients for TCR and LFA-1 in the T cell membrane (0.01–1 μm2/s). The basic phenomenology remains the same across this range of values; reduction in the diffusion coefficient by an order of magnitude lowers the protein concentrations at a given time by about 20%. Thus, the specific values of TCR and LFA-1 mobility, while important, are not critical for synaptic pattern formation.
The results reported in this paper suggest that the cell membrane environment modulates receptor–ligand binding in a way that balances forces because of membrane constraints, protein binding and dissociation, and mobilities, such that complex synaptic patterns can emerge spontaneously. Active cellular interventions (e.g., directed cytoskeletal rearrangements) can amplify or regulate these spontaneous processes.
Model Description
In the system we study (Fig. 1), spatio-temporal evolution of membrane proteins is driven by coupling between receptor–ligand binding/dissociation, protein mobility, and membrane shape changes. The concentration gradients driving transport of uncomplexed membrane proteins are largely determined by the rates of receptor protein binding/dissociation with complementary ligands on the apposing membrane. The ability of the proteins to react and form complexes depends on their size, elasticity of tethering to the membrane, and local separation between the membranes. This coupling is an illustration of the ways in which receptor–ligand binding, membrane shape changes and elasticity, and flux of membrane proteins are inextricably linked. The topographical difference between different types of receptor–ligand complexes (e.g., 42 nm for the ICAM-1/LFA-1 complex and 15 nm for the TCR/MHC complex (12, 13)) provides another source of coupling between receptor–ligand binding and membrane shape. The average separation between the two membranes in a particular region is influenced by the local concentrations of different receptor–ligand complexes. Membrane shape changes that lead to deviations in the average intermembrane separation in a local region from the value expected from a consideration of the natural lengths of the receptor–ligand complexes incur energy penalties. The intrinsic membrane shape changes depend on the surface energy (14), which is represented by phenomenological parameters that penalize the creation of new area (tension) and high curvature surfaces (bending rigidity). The model also incorporates a mechanism to account for TCR down regulation (15, 16).
The physical phenomena described above can be represented mathematically by Smoluchowski (or reaction-diffusion) equations (17) coupled to a time dependent Landau-Ginzburg model (18). These equations, which describe the time evolution of the spatial pattern of membrane protein concentrations in the intercellular junction and membrane shape, are:
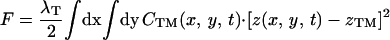 |
1 |
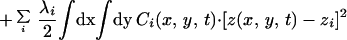 |
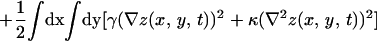 |
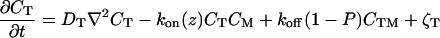 |
2 |
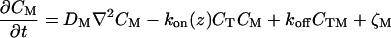 |
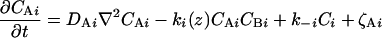 |
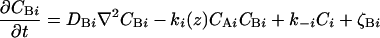 |
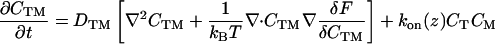 |
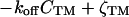 |
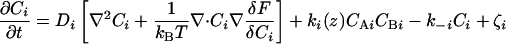 |
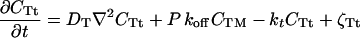 |
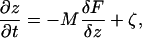 |
where the symbols are defined in Table 1. Although the membrane can undergo significant topographical shape changes during synapse formation, the topology (or genus) of the surface remains unchanged; thus, we ignore the Gaussian curvature term in writing Eq. 1. As has been noted many times (e.g., ref. 19 and references therein), because of elastic tethering of membrane proteins, the two-dimensional receptor–ligand binding constant (Kd) depends on the membrane environment. Factors such as local membrane curvature and orientational flexibility of membrane proteins play a role in determining the two-dimensional Kd (19). A simple approach used commonly to analyze experimental data is to lump these effects and consider the on rate to depend only on intermembrane spacing (z). Consistent with this approach, in Eqs. 2, we take the on rates for receptor–ligand binding to be Gaussian functions of z to reflect the effects of orientational flexibility, elastic tethering, etc., in a coarse-grained way (details in Methods).
Table 1.
| Symbol | Quantity |
|---|---|
| F | Free energy |
| CT | TCR concentration in T cell membrane |
| CM | MHC-peptide concentration in supported membrane |
| CTM | Concentration of TCR/MHC-peptide complex |
| CTt | Concentration of triggered TCRs |
| CAi | Concentration of ith adhesion molecule on the cell (LFA-1 for T cell immunological synapse) |
| CBi | Concentration of the ith complementary ligand on the supported membrane (ICAM-1 for T cell immunological synapse) |
| Ci | Concentration of the complex formed between Ai and Bi |
| kon | On rate for TCR/MHC-peptide binding |
| koff | Off rate for TCR/MHC-peptide binding |
| ki | On rate for the complexation of Ai and Bi |
| k−i | Off rate for the complexation of Ai and Bi |
| Dj | Diffusion coefficient of the jth membrane protein in the appropriate membrane |
| z | Local intermembrane separation |
| zj | Natural length of jth protein complex |
| t | Time |
| γ | Interfacial tension of cell membrane |
| κ | Bending rigidity of cell membrane |
| ζ, kBT | Thermal noise, thermal energy at temperature T |
| M | Phenomenological constant for membrane response to free energy changes |
| λj | Curvature of binding energy well for jth complex |
Methods
The model equations were solved numerically by using a straightforward finite difference scheme. Initially, the membrane proteins were distributed uniformly on the membranes, and no flux boundary conditions were imposed far away from the edge of the contact region. Initially, the upper membrane was given a parabolic shape, and the intermembrane distance was held constant far from the edge of the contact region (see Fig. 4B). The results are insensitive to ±5-nm changes in the initial intermembrane spacing.
Figure 4.

(A) Detailed concentration profile of bound TCR/MHC-peptide complex along a cross section of the contact region at various times during synapse formation. The concentration is zero initially because we are plotting the concentration of the TCR/MHC-peptide complex. The parameters are the same as those used to obtain the calculated results in Fig. 2. (B) Concomitant evolution of cell shape as measured by the coarse-grained average intermembrane spacing.
Parameters Used in the Calculations.
For TCR/MHC-peptide, Kd = koff/kon = 10 molecules/μm2 (2); as noted in the text and in refs. 11 and 20, known values of koff for TCR/MHC-peptide binding range from 0.06 s−1 (strong agonist) to 5.1 s−1 (antagonist); we have carried out calculations for koff = 0.01, 0.05, 0.5, 0.7, 1, 5, and 10 s−1. For LFA-1/ICAM-1, we use the measured values Kd = k−1/k1 = 0.3 μm2/molecule⋅s, and k−1 = 0.1 s−1 (21). We use measured values of the tension and bending rigidity for Dictyostelium discoideum cells (22, 23): γ = 3.1 ± 1.4 μN/m (used 3.1) and κ = 391 ± 156 kBT (used 400). The natural lengths of complexes are: zTCR/MHC-peptide = 15 nm (12, 13); zLFA-1/ICAM-1 = 42 nm (12). For DICAM-1 (or CD54) in the bilayer, we use the measured value for a similarly glycosylphosphatidylinositol (GPI)-linked CD58 = 0.59 μm2/s (10). Typical diffusion coefficients of GPI-linked proteins, including MHC, in supported bilayers are of order 1 μm2/s (24, 25); this value for DMHC-peptide was used. No measurements for DLFA-1 in T cell membranes were found; there is some evidence that DTCR may be as low as 0.01 μm2/s (26); however, TCR is known to translocate to glycolipid-enriched membrane domains during activation (27, 28), which raises the possibility that DTCR may transiently attain higher values; diffusion coefficients for mobile proteins on the cell surface can range from 0.1 to 1 μm2/s (29); to accommodate these possibilities, calculations have been carried out for a range of values of DTCR and DLFA-1 (0.01, 0.1, and 1.0 μm2/s). τ is estimated to be 2–5 s (16); we have used 5 s. For the curvature of the binding wells, λi, we have used 50 kBT/μm2 and M = 10−5 μm2/s; changing these values by factors of 2 or 3 does not affect qualitative results. Two kinds of experiments, micropipet experiments (30) and fluorescence methods (2), have been used to obtain the dependence of two-dimensional Kd on z. The results are presented in terms of a confinement thickness, and the two kinds of experiments yield very different results (confinement thickness ranging from 5 nm to 1 cm). We use a Gaussian distribution centered around z = zj for kon (z); calculations have been carried out with the widths of the distribution equal to 5 nm and 13 nm. The values of the parameters make clear that the thermal noise terms in Eqs. 2 can be ignored at room temperature.
Results
Phenomenology of the T Cell Immunological Synapse.
To apply the general model described above to the T cell immunological synapse, we include TCR down-regulation in Eqs. 2. TCRs that have been engaged with the MHC-peptide for a sufficiently long time (τ) are triggered and ultimately internalized by the cell (i.e., down-regulated). This phenomenon is treated in the simplest possible way after the serial triggering model of Lanzavecchia and coworkers (15, 16). TCR/MHC-peptide complexes dissociate into two possible sets of products: MHC-peptide and triggered TCRs (with kinetic constant P⋅koff; P = exp [−koffτ]) or MHC-peptide and untriggered TCRs [with kinetic constant (1 − P)⋅koff]. The triggered TCRs are down-regulated, and the others return to the pool of available TCRs.
The reaction rate coefficients, diffusion coefficients, membrane tension, and membrane bending rigidity that describe the processes included in the theoretical model have been measured for living cells and the proteins involved in T cell synapse formation (detailed in Methods). The measured values are not intrinsic properties of isolated phospholipid membranes and the relevant proteins; they depend on the cellular environment. For example, experiments have shown that the values of membrane tension and bending rigidity for cells that do not express talin (a protein connecting the lipid bilayer to the cytoskeleton) are 75% lower than that measured for wild-type cells (22). In this way, the cytoskeleton (and possibly other cellular structures) exert a passive influence on global membrane properties and, correspondingly, on synaptic pattern formation. The calculations described here incorporate such passive effects by using parameters measured in the cellular environment. The cytoskeleton is also known to play an active role in the formation of the T cell immunological synapse (4, 5, 31). These active effects of the cytoskeleton and other cellular factors are superimposed on the spontaneous pattern formation considered here.
Fig. 2 shows a comparison of spontaneous temporal evolution of membrane shape and the spatial distribution of MHC-peptide and ICAM-1 with experimental observations (2) of synaptogenesis in T cells. The self-organizing patterns predicted by the model are strikingly similar to the experimental observations. ICAM-1 engages first in the central region of the contact area with a ring of MHC-peptide enclosing it. The MHC-peptide is then transported inward, and ultimately the pattern inverts with MHC-peptide in the central region surrounded by ICAM-1. The cell shape also evolves, with the periphery of the contact region being in closest apposition in the early stages of synapse formation whereas the central region is closest for the mature synapse. The length and time scales characterizing different stages of the evolving patterns and cell shape are in rough quantitative agreement with experiments. Minor differences may reflect active modulation by other factors in the cell or differences in the nature of boundary conditions between our model and the cell. One such difference is apparent at the synapse periphery during early stages of synapse formation (between 0.5 and 3 min): in addition to the MHC-peptide ring, which encloses ICAM-1 in the central region, engagement of ICAM-1 at the periphery of the synapse is predicted by the model, a feature not observed in the cellular system.
In Fig. 2, we make a quantitative comparison between results of our model and observations of synapse formation between a T cell and a supported membrane (2). Because the qualitative features observed in experiments with T cells and APCs are similar (1), our model is also consistent with the phenomenology observed in these experiments. The similarity between the spontaneously evolving patterns predicted by our model and experimental observations with living cells suggests that the essential features of synaptic patterns can emerge because of self-organization processes. The self-organization processes result from the way in which the membrane environment modulates and coordinates receptor–ligand binding (see below).
Effect of TCR/MHC-Peptide Binding Kinetics.
An essential function of the TCR is to distinguish between different MHC-bound peptides. Cellular behavior is known to be strongly correlated with the dissociation rate (koff) for the TCR/MHC-peptide complex (11, 20, 32). For example, the agonist peptide MCC 88–103/Ek is characterized by koff = 0.06 s−1, the weak agonist peptide MCC 102N/Ek exhibits koff = 0.44 s−1, and the antagonist peptide MCC 102G/Ek has a value of koff = 5.1 s−1 (11). We have carried out calculations by using our model for values of koff spanning four orders of magnitude (0.01 s−1 to 100 s−1), thus encompassing a wide range of MHC-peptides. T cell proliferation, cytokine production, and other requisites for a mature immune response do not occur if the accumulated MHC-peptide in the intercellular junction is below a threshold value (≈60 molecules/μm2 in ref. 2). Using this observation along with the spatial pattern of proteins as criteria for effective synapse formation, our calculations show (Fig. 3) that only MHC-peptide complexes that bind TCR with koff in a narrow range (spanning one order of magnitude) lead to spontaneous pattern formation with synaptic characteristics. This finding is consistent with experimental observations that the off-rates of TCR binding to MHC-peptide molecules fall in a similar narrow range (11, 32).
Figure 3.

The maximum value of MHC-peptide accumulation at the center of the junction is plotted for values of koff ranging from 0.01–100 s−1 (t1/2 ranging from ≈70–0.007 s). If 55 molecules/μm2 is used as a criterion for efficient synapse formation, the active range for koff falls in a narrow range (0.5–5 s−1). Examples of the spatial distribution of MHC-peptides when the maximum value for MHC-peptide accumulation is reached are also shown. It is evident that the synaptic pattern does not form for koff = 0.05 and 10 s−1. MHC-peptide is concentrated in the central region, with an outer ring of ICAM-1 for values of koff that lead to effective synapse formation.
Results of our calculations (see Fig. 3) show that differences in koff are amplified by the cell membrane environment to affect biological outcome (i.e., synapse formation). This finding is consistent with experimental observations of the strong correlation between values of koff and biological activity (11, 20). Although we comment briefly on the origin of this behavior later, dissection of the amplification mechanism requires more exhaustive experimental and theoretical studies.
Keeping all other parameters the same, we have carried out calculations with a value of kon that is 500 times larger than that for TCR/MHC-peptide binding. In this case, the synaptic pattern fails to invert; i.e., a pattern with accumulated MHC-peptide in an outer ring and ICAM-1 in the middle is sustained for over 15 min. Intriguingly, this type of synaptic pattern has recently been observed for NK cells (3) where kon for killer cell Ig-like receptor-1 (KIR1)/MHC-peptide binding is known to be 300 times larger (33) than that for TCR. It is worth remarking that synaptic patterns in NK cells are known to form without cytoskeletal motion (3), thus underscoring our hypothesis that synaptic patterns can form spontaneously.
Self-Organization Mechanisms During Synaptic Pattern Formation.
Junction formation begins by the engagement of LFA-1 and ICAM-1 in the center because that is the region of closest approach and this complex has a natural length that is longer than the TCR–MHC-peptide complex. Occupation of the central region by the longer ICAM-1/LFA-1 complex leads to a pivot effect (2) that can bring the membranes into sufficiently close apposition at the periphery of the contact region to allow MHC-peptide binding to TCR. These effects cause TCR/MHC-peptide complexes to form in a ring outside the central region in the early stages of synapse formation. This first stage of junction formation is complete in about 1 min in both our calculated results and the experiments. Fig. 4A shows the formation of a ring of TCR/MHC-peptide complex within 1 min.
Binding provides a concentration gradient for the transport of uncomplexed proteins to the intercellular junction. Transport toward the center of the junction occurs because of the spatial asymmetry in the concentration gradient of the free MHC-peptide molecules. Fig. 4A shows the MHC-peptide transport process by plotting the concentration profile of bound TCR/MHC-peptide complexes along a cross section at various times during the transport process. As a result of the relatively large value of koff for TCR/MHC-peptide binding, and the higher mobility of free proteins in a single membrane compared with a bound complex, the TCR/MHC-peptide complex effectively moves faster than LFA-1 and ICAM-1 toward the center of the T cell/APC junction. The importance of koff in MHC-peptide transport is underscored by the results shown in Fig. 3. If koff is too small, then MHC-peptide transport to the center of the synaptic junction does not occur (e.g., see MHC-peptide concentration pattern in Fig. 3 for koff < 0.1). If koff is too large, MHC-peptide is spread over a very large region with a low concentration (e.g., see MHC-peptide concentration pattern in Fig. 3 for koff > 10).
At first glance, there appear to be two important time scales in the problem: the time associated with protein mobility and that associated with receptor–ligand binding kinetics. However, it is important to understand that the time associated with receptor–ligand binding is inextricably linked to membrane shape changes and fluctuations. Apposed TCR and MHC-peptide molecules in the two membranes can bind only if the intermembrane spacing, membrane fluctuations, and membrane elasticity allow the receptor–ligand pairs to be sufficiently close. Thus, it is more useful to think in terms of an effective reaction time that depends strongly on the membrane characteristics. Eqs. 1 and 2 embody the complex dependence of the effective reaction time on the membrane characteristics and the actual reaction time. In our model, the dependence of the effective reaction time on membrane characteristics provides the mechanism for amplification of effects due to changes in koff on synapse formation. The strong dependence of synaptic pattern formation on membrane characteristics is also made clear by the following results. Experiments have shown that cells that do not express talin (a protein that links the cytoskeleton to the lipid bilayer) have values for the tension and the bending rigidity that are 75% less than those for wild-type cells (22). Disruption of the cytoskeleton by using cytochalisin D is also known to disrupt synapse formation (2). We have carried out calculations with our model with values for the tension and bending rigidity that are 10% of those used to obtain the results in Fig. 2. We find that functional synaptic patterns do not form. In particular, multiple rings with low concentrations of MHC-peptide and ICAM-1 are observed; these multi-ring patterns are not sustained for long.
As is clear from both experimental results (2) and our calculations, the evolution of the TCR/MHC-peptide concentration shown in Fig. 4A and Fig. 2D is coupled with membrane shape changes. Smaller values of the average intermembrane separation are favored in regions where TCR/MHC-peptide exists because of its shorter natural length (≈15 nm). When a sufficient number of MHC-peptide complexes have migrated to the central region, the average intermembrane separation no longer favors the LFA-1/ICAM-1 complex (≈42 nm). This driving force, along with the important fact that high surface energy shapes of the cell membrane are unfavorable, leads to the outward migration of LFA-1/ICAM-1. Fig. 4B plots the evolution of the average intermembrane separation that occurs concomitantly with MHC-peptide transport.
After MHC transport is completed, the region with a high concentration of TCR/MHC-peptide begins to shrink. This results from TCR down-regulation and membrane retraction because of the thermodynamic force driving the cell membrane to acquire its spontaneous curvature.
Concluding Remarks
Our results suggest that evolution has provided an environment for cell–cell recognition wherein an intricate coupling and delicate balance between forces due to binding of more than one type of receptor–ligand pair, membrane shape changes, and protein transport lead to spontaneous evolution of synaptic patterns. This inherent ability to form self-organizing synaptic patterns in the intercellular junction is further faceted by active feedback mechanisms (e.g., directed cytoskeletal motion). The model we have described helps provide an understanding of how the amplification, suppression, or absence of certain key forces can regulate synapse formation. For example, it shows how the TCR/MHC-peptide dissociation rate affects the ability to form synapses. Our results also illustrate how dynamic interactions between just a few proteins, when mediated by the cell membrane environment, can generate functional complexity far exceeding the capabilities of individual receptor–ligand binding.
Synapse organization is rapidly emerging as a broadly significant feature of molecular recognition between cells. Our understanding of how the membrane environment modulates receptor–ligand binding to create synaptic patterns is still immature. We believe that further work with sophisticated descendants of the model described in this paper and synergistic experiments with living cells and reconstituted systems will prove useful in developing a better mechanistic understanding of the key factors that modulate synapse formation and resulting cellular behavior. Such an understanding of the immunological synapse would have important implications for developing modalities to control the immune response and its aberrant regulation. This understanding can also be used to design synthetic systems capable of mimicking recognition processes in biology.
Acknowledgments
We thank Professor C. Bertozzi, Professor M. Dustin, S. J. Lee, and Dr. J. Rabinowitz for discussions. Financial support was provided by the U.S. Department of Energy (Basic Energy Sciences), the Office of Naval Research (MDI2), the Culpeper Biomedical Pilot Initiative, a Burroughs Wellcome Career Award in Biomedical Sciences (to J.T.G.), and Laboratory Directed Research and Development funds.
Abbreviations
- NK
natural killer
- APC
antigen-presenting cell
- ICAM-1
intercellular adhesion molecule-1
- TCR
T cell antigen receptor
- LFA-1
lymphocyte function-associated antigen-1
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Monks C R F, Freiberg B A, Kupfer H, Sciaky N, Kupfer A. Nature (London) 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 2.Grakoui A, Bromley S K, Sumen C, Davis M M, Shaw A S, Allen P M, Dustin M L. Science. 1999;285:221–227. [PubMed] [Google Scholar]
- 3.Davis D M, Chiu I, Fassett M, Cohen G B, Mandelboim O, Strominger J L. Proc Natl Acad Sci USA. 1999;96:15062–15067. doi: 10.1073/pnas.96.26.15062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dustin M L, Cooper J A. Nat Immunol. 2000;1:23–29. doi: 10.1038/76877. [DOI] [PubMed] [Google Scholar]
- 5.Wulfing C, Davis M M. Science. 1998;282:2266–2269. doi: 10.1126/science.282.5397.2266. [DOI] [PubMed] [Google Scholar]
- 6.Wulfing C, Sjaastad M D, Davis M M. Proc Natl Acad Sci USA. 1998;95:6302–6307. doi: 10.1073/pnas.95.11.6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dustin M L, Olszowy M W, Holdorf A D, Li J, Bromley S, Desai N, Widder P, Rosenberger F, van der Merwe P A, Allen P M, Shaw A S. Cell. 1998;94:667–677. doi: 10.1016/s0092-8674(00)81608-6. [DOI] [PubMed] [Google Scholar]
- 8.Paul W E, Seder R A. Cell. 1994;76:241–251. doi: 10.1016/0092-8674(94)90332-8. [DOI] [PubMed] [Google Scholar]
- 9.Wulfing C, Chien Y H, Davis M M. Immunol Cell Biol. 1999;77:186–187. doi: 10.1046/j.1440-1711.1999.00813.x. [DOI] [PubMed] [Google Scholar]
- 10.Dustin M L, Ferguson L M, Chan P Y, Springer T A, Golan D E. J Cell Biol. 1996;132:465–474. doi: 10.1083/jcb.132.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis M M, Boniface J J, Reich Z, Lyons D, Hampl J, Arden B, Chien Y. Annu Rev Immunol. 1998;16:523–544. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- 12.Dustin M L, Shaw A S. Science. 1999;283:649–650. doi: 10.1126/science.283.5402.649. [DOI] [PubMed] [Google Scholar]
- 13.Garboczi D N, Ghosh P, Utz U, Fan Q R, Biddison W E, Wiley D C. Nature (London) 1996;384:134–141. doi: 10.1038/384134a0. [DOI] [PubMed] [Google Scholar]
- 14.Safran S. Statistical Thermodynamics of Surfaces, Interfaces, and Membranes. Reading, MA: Addison–Wesley; 1994. [Google Scholar]
- 15.Valitutti S, Muller S, Cella M, Padovan E, Lanzavecchia A. Nature (London) 1995;375:148–151. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- 16.Valitutti S, Lanzavecchia A. Immunol Today. 1997;18:299–304. [PubMed] [Google Scholar]
- 17.Van Kampen N G. Stochastic Processes in Physics and Chemistry. Amsterdam: North–Holland; 1987. [Google Scholar]
- 18.Hohenberg P C, Halperin B I. Rev Mod Phys. 1977;49:435–479. [Google Scholar]
- 19.Shaw A S, Dustin M L. Immunity. 1997;6:361–369. doi: 10.1016/s1074-7613(00)80279-4. [DOI] [PubMed] [Google Scholar]
- 20.Matsui K, Boniface J J, Steffner P, Reay P A, Davis M M. Proc Natl Acad Sci USA. 1994;91:862–866. doi: 10.1073/pnas.91.26.12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tominaga Y, Kita Y, Satoh A, Asai S, Kato K, Ishikawa K, Horiuchi T, Takashi T. J Immunol. 1998;161:4016–4022. [PubMed] [Google Scholar]
- 22.Simson R, Wallraff E, Faix J, Niewohner J, Gerisch G, Sackmann E. Biophys J. 1998;74:514–522. doi: 10.1016/S0006-3495(98)77808-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bruinsma R, Behrisch A, Sackmann E. Phys Rev E. 2000;61:4253–4267. doi: 10.1103/physreve.61.4253. [DOI] [PubMed] [Google Scholar]
- 24.Fein M, Unkeless J, Chuang F Y, Sassaroli M, da Costa R, Vaananen H, Eisinger J. J Membr Biol. 1993;135:83–92. doi: 10.1007/BF00234654. [DOI] [PubMed] [Google Scholar]
- 25.Groves J T, Wulfing C, Boxer S G. Biophys J. 1996;71:2716–2723. doi: 10.1016/S0006-3495(96)79462-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sloan-Lancaster J, Presley J, Ellenberg J, Yamazaki T, Lippincott-Schwartz J, Samelson L E. J Cell Biol. 1998;143:613–624. doi: 10.1083/jcb.143.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montixi C, Langlet C, Bernard A M, Thimonier J, Dubois C, Wurbel M A, Chauvin J P, Pierres M, He H T. EMBO J. 1998;17:5334–5348. doi: 10.1093/emboj/17.18.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xavier R, Brennan T, Li Q Q, McCormack C, Seed B. Immunity. 1998;8:723–732. doi: 10.1016/s1074-7613(00)80577-4. [DOI] [PubMed] [Google Scholar]
- 29.Jacobson K A, Moore S E, Yang B, Doherty P, Gordon G W, Walsh F S. Biochim Biophys Acta. 1997;1330:138–144. doi: 10.1016/s0005-2736(97)00128-4. [DOI] [PubMed] [Google Scholar]
- 30.Zhu C. J Biomech. 2000;33:23–30. doi: 10.1016/s0021-9290(99)00163-3. [DOI] [PubMed] [Google Scholar]
- 31.Viola A, Lanzavecchia A. Acta Pathol Microbiol Immunol Scand. 1999;107:615–623. doi: 10.1111/j.1699-0463.1999.tb01450.x. [DOI] [PubMed] [Google Scholar]
- 32.Sykulev Y, Brunmark A, Tsomides T J, Kageyama S, Jackson M, Peterson P A, Eisen H N. Proc Natl Acad Sci USA. 1994;91:11487–11491. doi: 10.1073/pnas.91.24.11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valez-Gomez M, Reyborn H, Strominger J. Hum Immunol. 2000;61:28–38. doi: 10.1016/s0198-8859(99)00159-7. [DOI] [PubMed] [Google Scholar]