

Abstract
Circulating fetal 3,3′,5-tri-iodo-l-thyronine (T3 ) is maintained at very low levels until a dramatic prepartum surge. 3,3′,5-Tri-iodo-l-thyronine inhibits serum-stimulated proliferation in near-term ovine cardiomyocytes, but it is not known whether midgestation myocytes are also inhibited. Because early cessation of cardiomyocyte mitosis would result in an underendowed heart, we hypothesized that 0.67 gestation (100 of 145 days gestation) ovine cardiomyocytes would be insensitive to suppressive growth effects of T3 . These younger cardiomyocytes were grown with T3 in 10% serum-enriched media for 24 hours. Physiological (0.37, 0.75, and 1.5 nmol/L) concentrations of T3 dramatically suppressed mitotic activity in cardiomyocytes (P < .001). 3,3′,5-Tri-iodo-l-thyronine stimulated phosphorylation of extracellular signal-regulated kinase and AKT (also known as Protein Kinase B [PKB]) signaling pathways. Nevertheless, the protein content of the cell cycle suppressor, p21, increased 2-fold (P < .05), and promoter, cyclin D1, decreased by 50%. Contrary to our hypothesis, elevated levels of T3 powerfully inhibit proliferation of midgestation fetal cardiomyocytes. Thus, midgestation maternal hyperthyroidism might lead to an underendowed fetal myocardium.
Keywords: fetal heart, cardiomyocyte proliferation, thyroid hormone, p21, cyclin D1
Introduction
At birth, nearly 60% to 70% of ovine cardiomyocytes have gained a second nucleus and no longer divide.1 Their loss of mitotic potential occurs through a process known as terminal differentiation,2,3 which begins at 110 days of gestation and continues until parturition at about 145 days. Before 110 days, mononucleated fetal cardiomyocytes divide rapidly to build the cellular endowment of the growing myocardium.1 The process of terminal cardiomyocyte maturation in sheep is predominantly prenatal and is thought to more closely represent human heart development.4 This precocious growth pattern differs from the pattern in the altricial rat and mouse heart, in which the terminal differentiation process does not begin until after birth; only about 7% of cardiomyocytes are binucleated at birth but more than 95% of cardiomyocytes are binucleated by 2 weeks of postnatal age.5
An adequate endowment of mature cardiomyocytes is important for the postnatal transition when left ventricular cardiomyocytes contract against a sudden doubling of arterial pressure.6 Beyond birth, the heart enlarges through the hypertrophic enlargement of individual cardiomyocytes and the expansion of the extracellular matrix with only a small contribution by proliferation.7,8 The number of cardiomyocytes contained within the heart at birth can vary according to prenatal hormonal conditions.4,9 Fetal growth factors like insulin-like growth factor 1,10 angiotensin II (Ang II),11 and cortisol12 independently stimulate proliferation rates of near-term ovine cardiomyocytes, whereas the rates are suppressed by atrial natriuretic peptide13 and thyroid hormone.14 In the living sheep fetus, proliferation rates are also suppressed under conditions of placental insufficiency.15
The ovine thyroid gland begins to secrete thyroxine (T4) around 50 days gestational age (dGA)16,17 and the concentration of T4 in plasma slowly rises over the first 2/3 of gestation. However, T4 itself has a minor influence on cardiomyocyte growth and function because of its low potency. The affinity of T4 for alpha and β-thyroid hormone receptors (TRα and TRβ) is nearly 10 to 15 times less than for 3,3′,5-tri-iodo-l-thyronine (T3).18 3,3′,5-Tri-iodo-l-thyronine is highly effective in binding thyroid receptors (TRα1 and TRβ1). Over the last few weeks of gestation, fetal T3 levels increase by nearly10-fold as T4 is converted to T3 under the influence of ever increasing deiodinase activity in peripheral tissues.19–21 Deiodinase gene expression increases under the influence of cortisol, which is elevated dramatically in the last few weeks of gestation consequent to the maturation of steroid synthetic activity in the adrenal gland.21,22
We previously reported that physiological concentrations of T3 are powerful suppressants of serum-stimulated proliferation in cardiomyocytes isolated from late-gestation fetal sheep hearts (∼0.9 gestation)14 and that the elevations of T3 in the near-term fetus stimulate maturation of cardiomyocytes.23 At this stage of development, plasma levels of T3 are rising; consequently by the time of parturition, nearly 70% of cardiomyocytes will have exited the cell cycle.1 However, it is not clear whether younger more rapidly dividing cardiomyocytes are as sensitive to elevations in T3 as older ones.
It was once believed that maternal thyroid hormones did not cross the placenta. However, recent evidence proves the contrary.24–26 It is now well-accepted that maternal thyroid levels influence fetal levels even before the fetal thyroid is active.24,27 There are a number of putative transporters for thyroid hormone in the placenta but monocarboxylate transporter 8 (MCT8) is thought to be the most important.28 Thus, if T3 levels became abnormally elevated in fetuses and if cardiomyocytes were sensitive to T3, the resulting suppression of cardiomyocyte proliferation would lead to a heart with an inadequate endowment of cardiomyocytes at birth; such a heart might suffer compromised function later in life.
We hypothesized that cardiomyocytes from 100 dGA (0.6 gestation) fetal hearts would be less sensitive to mitotic suppression with T3 exposure than 135 dGA fetal cardiomyocytes previously studied. Resistance to a T3 suppressive effect would be a protective mechanism to prevent harm that could be caused by inadequate proliferation at a stage when building the myocardium is crucial. In keeping with the hypothesis, we also reasoned that unlike older myocytes, the expression levels of the cell cycle inhibitory protein, p21, would not be elevated by T3 nor would the protein levels of the cell cycle promoter, cyclin D1, be suppressed. We supposed that signaling cascades associated with proliferation, including the extracellular signal-regulated kinase (ERK) branch of the mitogen-activated protein kinase (MAPK) cascade and the phosphoinositol-3 kinase [PI3K]/AKT pathways, would, likewise, not be altered by T3. To test the hypothesis, we isolated cardiac myocytes from sheep fetuses that were ∼100 days gestation and determined the degree to which T3 affected mitosis and associated cell signaling.
Materials and Methods
Animals
Animals were studied and cared for in accordance with the Institutional Animal Care and Use Committee at the Oregon Health and Science University (Portland, Oregon). Primary cultures of cardiomyocytes were obtained from noninstrumented, control fetal sheep (Ovis aries; mixed western breed) at 102 ± 3 dGA, where term is ∼145 dGA. The fetal cardiac myocytes that comprise the myocardium at ∼100 dGA are phenotypically homogeneous and are mononucleated.1
Materials
3,3′,5-Tri-iodo-l-thyronine (thyroid hormone), 5-bromo-2′-deoxyuridine (BrdU), insulin–transferrin–sodium selenite (ITSS), penicillin–streptomycin–amphotericin B (PSA), Type XIV protease, and laminin were obtained from Sigma-Aldrich (St Louis, Missouri). Type II collagenase was obtained from Worthington Biochemicals (Lakewood, New Jersey).
Antibodies for Western Blot Analyses
The following antibodies were obtained from Cell Signaling (Danvers, Massachusetts). Phospho-p44/42 MAPK (ERK1/2) rabbit polyclonal antibody detects endogenous levels of p44 and p42 MAPK (ERK1 and ERK2) when phosphorylated either individually or dually at Thr202 and Tyr204. Phospho-AKT (Ser473; D9E) XP rabbit monoclonal antibody detects endogenous levels of AKT only when phosphorylated at Ser473. AKT rabbit polyclonal antibody detects endogenous levels of total AKT1, AKT2, and AKT3 proteins. Rabbit monoclonal α-tubulin detects endogenous levels of total α-tubulin protein and does not cross-react with recombinant β-tubulin. Mouse monoclonal p21 Waf1/Cip1 (DCS60) antibody detects endogenous levels of total p21 protein. The antibody does not cross-react with other cyclin-dependent kinase inhibitors and recognizes the amino-terminal portion of p21. The remaining antibodies were obtained from Santa Cruz Biotech, Inc. (Santa Cruz, California). Mouse monoclonal cyclin D1 recognizes full-length peptide and does not cross-react with cyclin D3. Mouse monoclonal ERK2 antibody recognizes C-terminus of MAPK p42. Mouse monoclonal TRα1/β1 recognizes the ligand binding domain of each receptor (TRα1: 47 kDa; TRβ1: 58 kDa). Horseradish peroxidase linked secondary antibodies (goat anti-rabbit IgG and horse anti-mouse IgG) were obtained from Cell Signaling.
Cardiac Myocyte Isolation
Ewes were euthanized by intravenous injection of a commercial solution of sodium pentobarbital (SomnaSol, ∼80 mg/kg, Butler Schein Animal Health, Dublin, Ohio). Fetuses received a bolus dose of heparin (5000 U, Baxter, Deerfield, Illinois), followed by 5 mL of saturated potassium chloride (KCl) into the umbilical vein to arrest the heart in diastole. Fetuses were weighed and the heart excised, trimmed in a standard way, blotted dry, and weighed. Hearts were enzymatically dissociated as previously described by our laboratory.29 Briefly, hearts were perfused in a retrograde manner with gassed solutions (95% O2 and 5% CO2, 39°C); tyrodes buffer for 5 minutes ([no calcium added]; 140 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2.6H2O, 10 mmol/L glucose, and 10 mmol/L 4-(2-hydroxyethyl)-1-piperazineethane sulfonic acid (HEPES); pH adjusted to 7.35 with NaOH) until the vessels were clear of blood; 2 minutes with 160 U/mL type II and 0.78 U/mL type XIV protease in tyrodes buffer to digest the tissue; finally, 5 minutes with a high potassium, calcium-free Kraftbrühe (KB) solution (74 mmol/L glutamic acid, 30 mmol/L KCl, 30 mmol/L KH2PO4, 20 mmol/L taurine, 3 mmol/L MgSO4, 0.5 mmol/L ethylene glycol tetraacetic acid (EGTA), 10 mmol/L HEPES, and 10 mmol/L glucose; pH adjusted to 7.37 using KOH). The left ventriclular (LV) and right ventricular (RV) free walls were dissected from the heart and placed into separate tubes containing 20 mL KB solution. The tissue was gently agitated to release cardiomyocytes.
Cardiomyocyte Cultures
Cardiomyocytes were cultured as previously described by our laboratory.13,14 The freshly isolated slurry of myocytes rested for 30 to 60 minutes at room temperature (RT) before centrifugation and resuspension in sterile serum media (10% fetal bovine serum; Invitrogen, Carlsbad, California); Dulbecco's Minimum Eagle Medium (DMEM) low glucose: 5.56 mmol/L d-glucose, 4 mmol/L l-glutamine, 1 mmol/L sodium pyruvate, 5.33 mmol/L KCl, 0.4 mmol/L glycine, pH 7.4 (Invitrogen, #11885-084) supplemented with 10 mg/L of ITSS, and 10 mL/L PSA. All cultures were performed at 39°C and 5% CO2. Cells were preplated twice to remove non-myocyte cells (2 hours each time). Myocytes were then seeded on 22 × 22 mm2 glass coverslips (for measurement of proliferation) or in 6-well plates (for protein analysis). Coverslips and 6-well plates were coated with laminin (4 mg/mL) for at least 4 hours and aspirated just before plating at a density of 1.5 × 105 cells per coverslip and 5 × 105 cells per well. Cardiomyocytes were incubated in serum media for 24 hours. Cells were then incubated in serum free (SF) media for 48 hours, and the media was changed again to fresh SF media for 24 hours before treatment; treatment commenced on culture day 5 for all experiments.
Proliferation Studies (BrdU Uptake)
In order to determine the effects of T3 on cardiomyocyte proliferation, we measured BrdU incorporation (10 μmol/L) as an index of cell proliferation in 2 groups of cultured fetal cardiomyocytes: (1) 48 hour in SF medium with increasing T3 doses and (2) 48 hour of serum medium with increasing T3 concentrations. We chose to treat with media containing T3 with and without serum because we wanted to compare both conditions with our previous studies in near-term fetal sheep. Concentrations of T3 ranged from those occurring normally in fetal sheep during gestation (0.37, 0.75, and 1.5 nmol/L)19 to those that might occur under pathological conditions (3, 10, and 100 nmol/L).14,30,31 The total T3 levels in the last one third of gestation was represented as 0.37 nmol/L, approximating the normal circulating levels near 100 dGA, and 1.5 nmol/L, the concentration found just prior to birth.32,33 Many in vitro studies describing T3 actions use only the higher concentrations of T3.30,31,34 In our study, we used the higher conventional concentrations exclusively to determine T3's effect on cell cycle proteins to provide comparisons with previously published studies. At the end of the study, cardiomyocytes were fixed in ice-cold ethanol for staining procedures.
5-Bromo-2′-deoxyuridine Uptake Analysis
Cells were permeabilized by incubating with 5 μg/mL DNase as described previously11 for 30 minutes at 37°C. Fixed cultured cardiomyocytes were then incubated with mouse anti-myosin heavy chain α/β (1:5000, ab15, Abcam, Cambridge, Massachusetts) and rat anti-BrdU (1:500, ab6326, Abcam) antibodies overnight at 4°C. Following phosphate-buffered saline (PBS) washes, cells were incubated in anti-mouse rhodamine red (1:200, Jackson ImmunoResearch, West Grove, Pennsylvania) and anti-rat fluorescein isothiocyanate (FITC; 1:200, Jackson ImmunoResearch, West Grove, Pennsylvania) secondary antibodies for 2 hours at RT. Coverslips were mounted onto slides using Vectashield Hardset mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, California) and stored in the dark overnight at 4°C to allow the mounting medium to set.14 The portion of BrdU-positive myocytes was determined in a random sample of 300 (minimum) myocytes using fluorescence microscopy (×400 magnification; Zeiss Axiophot, Bartels & Stout, Bellevue, Washington) with FITC (excitation: 450-490 nm and emission: 515-565 nm) and TRITC filters (tetramethylrhodamine isothiocyanate, excitation: 485 nm and emission: 515-530 nm). 5-Bromo-2′-deoxyuridine-positive cardiomyocytes were identified as those that stained positive for both myosin (rhodamine) and punctuate nuclei (FITC), indicating BrdU incorporation.
Thyroid Hormone Signaling
Fetal cardiomyocytes were prepared in 6-well plates as described above for signaling studies on day 5 of culture. In order to isolate the signaling cascades triggered by T3 alone, we performed these studies in SF media to remove the influence of serum.30,31,35 The first group of cells was incubated with increasing concentrations of T3 in SF media (1.5, 10, and 100 nmol/L) for 24 hours at 39°C. These were used to determine whether T3 affected p21 (cell cycle inhibitor) and cyclin D1 (cell cycle promoter) expression. These same lysates were used to determine whether the thyroid hormone receptor isoforms (TRα1 and TRβ1) are altered by media conditions or concentration of T3 used. We also determined the rapid activation profile of ERK and AKT by phosphorylation (pERK and pAKT) at the same concentrations following a 10-minute stimulation. Cell lysates were prepared for protein analysis as described below.
Western Blot Analysis
After the appropriate incubation period, medium was aspirated, cardiomyocytes were rinsed in ice-cold 1× PBS, lysed (5 mmol/L Tris-HCl, 5 mmol/L EGTA, 5 mmol/L EDTA, 0.06% sodium dodecyl sulfate (SDS), protease inhibitor mini-complete tablet [Roche, Indiana], and phosphatase inhibitor cocktail I and II [Sigma-Aldrich]), and collected into prechilled tubes. Protein concentration was quantified by BCA assay (Pierce, Rockford, Illinois). Equal amounts of total protein/sample (10 μg) were separated by SDS polyacrylamide gel electrophoresis (PAGE) on a 10% Tris-glycine gel and transferred to a nitrocellulose membrane (Optitran BA-S 83, Whatman, New Jersey). Membranes were blocked with 5% milk in 1× Tris-buffered saline + 0.01% Tween 20 (TBS-T) buffer for 1 hour at RT. Membranes were incubated with primary antibodies (1:1000) overnight at 4°C in 4% bovine serum albumin (Sigma-Aldrich) in 1× TBS-T buffer. Membranes were washed in large volumes of TBS-T before exposure to the secondary antibody (1:5000) in 5% milk–TBS-T for 1 hour at RT. Antibody binding was detected using chemiluminescence (SuperSignal, Pierce, Illinois); protein expression was quantified from a digitized image of the blot using NIH Image J (Version 1.4; NIH). Protein density was normalized to total protein and expressed as phospho-protein/total protein (ERK and AKT) or protein/α-tubulin (TRα1/β1, p21, and cyclin D1).
Statistical Analysis
Only data from left ventricular cells are reported because responses in the RV were similar to those from the LV. On average, BrdU incorporation was slightly lower in RV, but the trends between ventricles were otherwise similar. A minimum of 300 cells per treatment group were assessed for BrdU incorporation and reported as a percentage of total number of cardiomyocytes counted. One-way analysis of variance (ANOVA) was used to analyze BrdU uptake as well as Western blot density. If justified by ANOVA, differences were further analyzed by Tukey multiple comparison post hoc test for the differences between treatment groups. Statistical significance was set at P < .05. Data are presented as mean ± SEM.
Results
5-Bromo-2′-deoxyuridine Uptake
Figure 1 shows a single 100 day cardiomyocyte positively labeled with BrdU and several other unlabeled cardiomyocytes with myofibrils stained for myosin heavy chain α and β (Figure 1). 3,3′,5-Tri-iodo-l-thyronine did not affect BrdU incorporation rates under SF culture conditions, which were on the order of 12% (Figure 2A), and much higher than seen later in gestation.14,36 Proliferation rates rose dramatically to ∼25% when serum was added to the medium (Figure 2B). However, contrary to our original hypothesis, T3 (at all concentrations tested) reduced BrdU incorporation rates in the stimulatory environment of serum-rich media (P < .001, Figure 2B). Also of note, there was a dose-dependent decrease in BrdU uptake at lower T3 concentrations; proliferation was decreased between serum (0.0 nmol/L T3) and 0.37 nmol/L T3 (P < .01), which was further decreased by 0.75 nmol/L T3 (P < .05). There was no further statistical reduction in BrdU uptake when compared with 1.5 nmol/L T3; this latter concentration appeared to represent a plateau in the suppression of proliferation with no further inhibition at higher doses.
Figure 1.
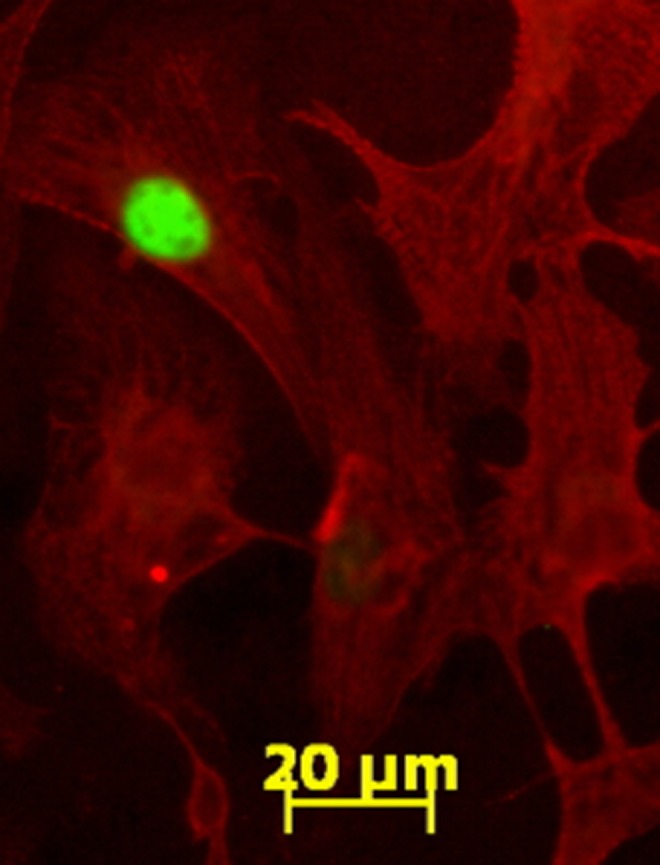
5-Bromo-2′-deoxyuridine (BrdU) uptake in proliferating fetal sheep cardiomyocytes in vitro. After 48-hour incubation with BrdU, positive myocytes are marked by fluorescein isothiocyanate (green) staining in the nuclei of cells that also stained for myosin (red). Image shown is at ×400 magnification.
Figure 2.

5-Bromo-2′-deoxyuridine (BrdU; 10 μmol/L) incorporation in left ventricular cardiomyocytes from 100 dGA fetal sheep. A, T3 has no effect on BrdU incorporation of cells treated in serum free media but (B) inhibits BrdU uptake stimulated by 10% fetal bovine serum at all concentrations of T3 used. Data are mean ± standard error of mean, each bar n = 7, ##P < .01, ###P < .001 versus serum control, *P < .05 versus T3 0.37 nmol/L.
Roles of p21 and Cyclin D1
Protein levels of TRα1 or TRβ1 were determined by Western blot analysis of lysates of cells incubated for 24 hours in SF media or T3 (1.5, 10, and 100 nmol/L) in SF media (Figure 3). Protein levels were also measured in cells grown in serum-rich media. Interestingly, there was no difference in TRα1 or TRβ1 levels between SF and serum-rich conditions (Figure 3). There was no effect of T3 concentration on receptor expression of either major isoform of the thyroid receptor. The cell cycle inhibitor p21 was upregulated in SF conditions after 24 hours of T3 exposure at increasing doses (Figure 4, P < .05). 3,3′,5-Tri-iodo-l-thyronine (1.5 nmol/L) stimulated p21 protein expression in a dose response fashion. Cyclin D1 levels were significantly decreased by T3 in cardiomyocytes compared with cells in SF control (Figure 5). The expression of p21 and cyclin D1 was not different between SF and serum-rich conditions alone.
Figure 3.

Effect of culture conditions on thyroid receptor expression. Both TRα1 and TRβ1 protein levels are not different between serum and serum free conditions. Addition of T3 did not alter the receptor expression. Data are mean ± standard error of mean, each bar n = 7.
Figure 4.

Dose-dependent protein expression of p21 to T3. Exposure to T3 for 24 hours resulted in dose-dependently increased protein levels of the cell cycle inhibitor p21. Data are mean ± standard error of mean, each bar n = 7, *P < .05, and **P < .01 versus serum free control.
Figure 5.

Dose-dependent protein expression of cyclin D1 to T3. Exposure to T3 for 24 hours resulted in dose dependently decreased protein levels of the cell cycle promoter cyclin D1. Data are mean ± standard error of mean, each bar n = 7, *P < .05, and **P < .01 versus serum free control.
3,3′,5-Tri-iodo-l-thyronine Signaling: Role of ERK and AKT
Phosphorylated ERK (pERK) and Akt (pAKT) were determined by Western blot analysis of lysates of cardiomyocytes that were exposed to a 10-minute bout of T3 at 3 different concentrations: 1.5, 10, and 100 nmol/L (Figure 6). pERK expression increased with increasing T3 concentration (Figure 6A; P < .05), while pAKT was elevated equally without an upward trend with increasing T3 doses (Figure 6B, P < .05). Serum-free levels of both pERK and pAKT were significantly lower than levels in the presence of serum (P < .05).
Figure 6.

Activation of ERK and AKT (also known as Protein Kinase B [PKB]) by T3. Following an acute exposure to T3, ERK phosphorylation is greater with increasing T3 concentrations (A). AKT phosphorylation, however, increases equally with all three doses of T3. Data are mean ± standard error of mean, each bar n = 7, *P < .05, and **P < .01 versus serum free control. ERK indicates extracellular signal-related kinase; T3, 3,3′,5-tri-iodo-l-thyronine.
Discussion
Our findings demonstrate that 100 dGA ovine cardiomyocytes are not protected from the growth suppressive actions of thyroid hormone. This was shown in 2 ways: (1) BrdU uptake was decreased with increasing T3 levels in a dose-response manner, and (2) BrdU uptake was maximally reduced by 70% in the presence of 1.5 nmol/L T3 in the more immature 100 days myocyte and about the same degree of suppression of 74% at 1.5 nmol/L T3 in the older fetal cardiomyocytes. This finding did not support our hypothesis. We had reasoned that the myocardium in the younger fetus would have been more protected from mitosis suppression by T3. Such resistance would prevent cardiomyocyte replication at a stage when cardiomyocytes are not ordinarily exposed to significant levels of T3 and when myocyte numbers are rapidly increasing.
Baseline BrdU uptake rates, without serum enrichment, were 10% to 12% in 100 dGA cardiomyocytes compared with 1% to 2% in 135 dGA cardiomyocyte experiments previously published.14 The younger cardiomyocytes showed serum-stimulated BrdU uptake rates of ∼25% when compared with ∼10% in the 135 dGA cells.14 The age difference in proliferative activity was expected because myocytes from the 100 dGA time point are nearly all mononucleated and are normally proliferating at a higher rate in vivo. As term approaches, myocyte cell cycle activity is known to decrease, an increasing number of myocytes are terminally differentiated (binucleated) and have exited the cell cycle.1
3,3′,5-Tri-iodo-l-thyronine did not suppress the rates of BrdU uptake among cardiomyocytes under SF conditions, even at the highest doses tested. A similar effect was previously found in near-term ovine cardiomyocytes.14 Thus, a sustained level of mitosis, as measured by BrdU uptake, continued in the presence of T3 even though the cell cycle suppressant protein, p21, was upregulated and the cell cycle promoter, cyclin D1, was depressed in the presence of T3 at all concentrations. These are not apparently due to an altered expression pattern of the 2 most common receptors, TRα1 and TRβ1. The retention of a basal level of proliferation, while interesting, is not likely to be physiologically important because SF conditions are not encountered in the living fetal heart. We speculate that under SF conditions T3 stimulates a known insulin-sensitive cardiomyocyte survival pathway in which the PI3K/AKT phosphorylation pathway is activated.37,38
We found that T3 activates both ERK and AKT in fetal cardiomyocytes. Activation of the ERK and often, AKT, pathway constituents is associated with pro-proliferative actions. However, in our study, we found that ERKs were activated simultaneously with a suppression of cell cycle activity. We cannot explain this finding. However, others have reported that ERK activation can be associated with elevations in p21 when cell cycle activity is suppressed.39,40 Thus, the immature cardiomyocyte appears to display a similar behavior.
In humans, but not in sheep, elevated levels of thyroid hormone in maternal plasma lead to elevated fetal thyroid hormone levels throughout gestation.24–26,41 Thus, fetal hyperthyroidism is associated with maternal hyperthyroidism. The monocarboxylate transporter, which transports T3 and T4, has been identified in human placentas28 and appears to be the primary placental transporter for thyroid hormones. Maternal levels of circulating total T4 (∼110 ng/mL) and circulating total T3 (∼2.0 ng/mL) are elevated during normal pregnancy.42,43 Normal human umbilical artery levels of T4 (90.5 ± 0.02 ng/mL) and T3 (1.9 ± 0.96 ng/mL) at birth are similar to fetal sheep levels.19,44
We have recently reported that in 130 days ovine fetuses, exogenous elevation of T3 to levels normally found at term (145 dGA) stimulates the maturation of cardiomyocytes by suppressing proliferation, increasing binucleation and stimulating the sarcoplasmic reticulum transport protein SERCA2a.23 In these experiments, p21 levels were elevated and cyclin D1 levels suppressed as predicted by the effects of T3 on isolated 135 dGA cardiomyocytes.14 Thus, it is likely that the in vitro studies are reliable indicators of expected responses to thyroid conditions in the womb. The role of p21 and cyclin D1 in regulating the cell cycle under the influence of thyroid hormone remains uncertain even though these 2 proteins change in a direction predictive of their regulatory function.
The results of our studies suggest that elevated T3 would suppress cardiomyocyte proliferation in vivo even in cardiomyocytes that are normally dividing rapidly. We predict that the rates of cell division in the immature cardiomyocytes of mammals would be deleteriously suppressed by hyperthyroid conditions. If the suppressive effect of T3 on ovine cardiomyocyte cell cycle activity also occurs in human cardiomyocytes, elevated levels of maternal T3 at midgestation might lead to fetal hearts with a reduced endowment of cardiomyocytes. Such an effect could compromise the myocardium for life.
Conclusions
The last third of ovine gestation is a critical window of development for the myocardium because it is the period of time when cardiac myocytes mature and their numbers are set at birth. During this period, both mononucleated and binucleated myocyte populations are sensitive to nutritional, hormonal, and hemodynamic modification.4,10,12 This report shows that cardiomyocytes that have not yet begun to enter the terminal differential phase and nearly 30 to 40 days away from seeing the naturally occurring prepartum T3 increase are nevertheless very sensitive to the mitotic suppression effects of T3. The findings suggest that the myocardium would be negatively impacted if thyroid levels were to exceed normal concentrations at midgestation. If midgestation human cardiomyocytes are as sensitive to elevated thyroid levels as the sheep cardiomyocytes in this study, maternal hyperthyroid levels could lead to an underendowed myocardium at birth.
Acknowledgments
The authors thank Drs Stephen Back and Roger Hohimer for the generous donations of control fetal sheep tissue for this study as well as Mr Robert Webber and Ms Loni Socha for excellent technical assistance. Drs Perrie O’Tierney and Sonnet Jonker provided sound advice.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: supported by NIH grants NICHD P01 HD 34430, NINDS R37, NS045737, NHLBI R21 HL093617, and R01 HL102763; NNC was supported by NHLBI training grant T32HL094294 and KLT was supported by the M. Lowell Edwards Endowment.
References
- 1. Jonker SS, Zhang L, Louey S, Giraud GD, Thornburg KL, Faber JJ. Myocyte enlargement, differentiation, and proliferation kinetics in the fetal sheep heart. J Appl Physiol. 2007;102(3):1130–1142 [DOI] [PubMed] [Google Scholar]
- 2. Rakusan K. Cardiac growth, maturation, and aging. In: Zak R, ed. Growth of the Heart in Health and Disease. New York, NY: Raven Press; 1984:131–164 [Google Scholar]
- 3. Clubb FJ, Jr, Bishop SP. Formation of binucleated myocardial cells in the neonatal rat. An index for growth hypertrophy. Lab Invest. 1984;50(5):571–577 [PubMed] [Google Scholar]
- 4. Barbera A, Giraud GD, Reller MD, Maylie J, Morton MJ, Thornburg KL. Right ventricular systolic pressure load alters myocyte maturation in fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2000;279(4):R1157–R1164 [DOI] [PubMed] [Google Scholar]
- 5. Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol. 1996;271(5 pt 2):H2183–H2189 [DOI] [PubMed] [Google Scholar]
- 6. Smolich JJ, Walker AM, Campbell GR, Adamson TM. Left and right ventricular myocardial morphometry in fetal, neonatal, and adult sheep. Am J Physiol. 1989;257(1 pt 2):H1–H9 [DOI] [PubMed] [Google Scholar]
- 7. Bugaisky L, Zak R. Cellular growth of cardiac muscle after birth. Tex Rep Biol Med. 1979;39:123–138 [PubMed] [Google Scholar]
- 8. Zak R, Kizu A, Bugaisky L. Cardiac hypertrophy: its characteristics as a growth process. Am J Cardiol. 1979;44(5):941–946 [DOI] [PubMed] [Google Scholar]
- 9. Zhang L. Prenatal hypoxia and cardiac programming. J Soc Gynecol Investig. 2005;12(1):2–13 [DOI] [PubMed] [Google Scholar]
- 10. Sundgren NC, Giraud GD, Schultz JM, Lasarev MR, Stork PJ, Thornburg KL. Extracellular signal-regulated kinase and phosphoinositol-3 kinase mediate IGF-1 induced proliferation of fetal sheep cardiomyocytes. Am J Physiol Regul Integr Comp Physiol. 2003;285(6):R1481–R1489 [DOI] [PubMed] [Google Scholar]
- 11. Sundgren NC, Giraud GD, Stork PJ, Maylie JG, Thornburg KL. Angiotensin II stimulates hyperplasia but not hypertrophy in immature ovine cardiomyocytes. J Physiol. 2003;548(pt 3):881–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Giraud GD, Louey S, Jonker S, Schultz J, Thornburg KL. Cortisol stimulates cell cycle activity in the cardiomyocyte of the sheep fetus. Endocrinology. 2006;147(8):3643–3649 [DOI] [PubMed] [Google Scholar]
- 13. O'Tierney PF, Chattergoon NN, Louey S, Giraud GD, Thornburg KL. Atrial Natriuretic Peptide Inhibits Angiotensin II-Stimulated Proliferation in Fetal Cardiomyocytes. J Physiol. 2010;558(pt 15):2879–2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chattergoon NN, Giraud GD, Thornburg KL. Thyroid hormone inhibits proliferation of fetal cardiac myocytes in vitro. J Endocrinol. 2007;192(2):R1–R8 [DOI] [PubMed] [Google Scholar]
- 15. Louey S, Jonker SS, Giraud GD, Thornburg KL. Placental insufficiency decreases cell cycle activity and terminal maturation in fetal sheep cardiomyocytes. J Physiol. 2007;580(pt 2):639–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hopkins PS, Thorburn GD. The effects of foetal thyroidectomy on the development of the ovine foetus. J Endocrinol. 1972;54(1):55–66 [DOI] [PubMed] [Google Scholar]
- 17. Thorburn GD, Hopkins PS. Thyroid function in the foetal lamb. In: Comline KS, Cross KW, Dawes GS, Nathanielsz PW, eds. Foetal and Neonatal Physiology: Proceedings of the Sir Joseph Barcroft Centenary Symposium Held at the Physiological Laboratory Cambridge . Texas: Cambridge University Press; 1973:488–507 [Google Scholar]
- 18. Chopra IJ, Carlson HE, Solomon DH. Comparison of inhibitory effects of 3,5,3'-triiodothyronine (T3), thyroxine (T4), 3,3,',5'-triiodothyronine (rT3), and 3,3'-diiodothyronine (T2) on thyrotropin-releasing hormone-induced release of thyrotropin in the rat in vitro. Endocrinology. 1978;103(2):393–402 [DOI] [PubMed] [Google Scholar]
- 19. Polk DH. Thyroid hormone metabolism during development. Reprod Fertil Dev. 1995;7(3):469–477 [DOI] [PubMed] [Google Scholar]
- 20. Fowden AL, Mundy L, Silver M. Developmental regulation of glucogenesis in the sheep fetus during late gestation. J Physiol. 1998;508(pt 3):937–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Forhead AJ, Curtis K, Kaptein E, Visser TJ, Fowden AL. Developmental control of iodothyronine deiodinases by cortisol in the ovine fetus and placenta near term. Endocrinology. 2006;147(12):5988–5994 [DOI] [PubMed] [Google Scholar]
- 22. Thomas AL, Krane EJ, Nathanielsz PW. Changes in the fetal thyroid axis after induction of premature parturition by low dose continuous intravascular cortisol infusion to the fetal sheep at 130 days of gestation. Endocrinology. 1978;103(1):17–23 [DOI] [PubMed] [Google Scholar]
- 23. Chattergoon NN, Giraud GD, Louey S, Stork P, Fowden AL, Thornburg KL. Thyroid hormone drives fetal cardiomyocyte maturation. FASEB J. 2012;26(1):397–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chan SY, Vasilopoulou E, Kilby MD. The role of the placenta in thyroid hormone delivery to the fetus. Nat Clin Pract Endocrinol Metab. 2009;5(1):45–54 [DOI] [PubMed] [Google Scholar]
- 25. Higuchi R, Kumagai T, Kobayashi M, Minami T, Koyama H, Ishii Y. Short-term hyperthyroidism followed by transient pituitary hypothyroidism in a very low birth weight infant born to a mother with uncontrolled Graves' disease. Pediatrics. 2001;107(4):E57. [DOI] [PubMed] [Google Scholar]
- 26. Calvo R, Obregon MJ, Escobar DR, Morreale DE. The rat placenta and the transfer of thyroid hormones from the mother to the fetus. Effects of maternal thyroid status. Endocrinology. 1992;131(1):357–365 [DOI] [PubMed] [Google Scholar]
- 27. Kilby MD, Barber K, Hobbs E, Franklyn JA. Thyroid hormone action in the placenta. Placenta. 2005;26(2-3):105–113 [DOI] [PubMed] [Google Scholar]
- 28. Chan SY, Franklyn JA, Pemberton HN, et al. Monocarboxylate transporter 8 expression in the human placenta: the effects of severe intrauterine growth restriction. J Endocrinol. 2006;189(3):465–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jonker SS, Faber JJ, Anderson DF, Thornburg KL, Louey S, Giraud GD. Sequential growth of fetal sheep cardiac myocytes in response to simultaneous arterial and venous hypertension. Am J Physiol Regul Integr Comp Physiol. 2007;292(2):R913–R919 [DOI] [PubMed] [Google Scholar]
- 30. Burton PB, Raff MC, Kerr P, Yacoub MH, Barton PJ. An intrinsic timer that controls cell-cycle withdrawal in cultured cardiac myocytes. Dev Biol. 1999;216(2):659–670 [DOI] [PubMed] [Google Scholar]
- 31. Kuzman JA, Gerdes AM, Kobayashi S, Liang Q. Thyroid hormone activates Akt and prevents serum starvation-induced cell death in neonatal rat cardiomyocytes. J Mol Cell Cardiol. 2005;39(5):841–844 [DOI] [PubMed] [Google Scholar]
- 32. Wu S, Polk D, Wong S, Reviczky A, Vu R, Fisher DA. Thyroxine sulfate is a major thyroid hormone metabolite and a potential intermediate in the monodeiodination pathways in fetal sheep. Endocrinology. 1992;131(4):1751–1756 [DOI] [PubMed] [Google Scholar]
- 33. Polk DH, Wu SY, Wright C, Reviczky AL, Fisher DA. Ontogeny of thyroid hormone effect on tissue 5'-monodeiodinase activity in fetal sheep. Am J Physiol. 1988;254(3 pt 1):E337–E341 [DOI] [PubMed] [Google Scholar]
- 34. Lin HY, Sun M, Tang HY, et al. L-Thyroxine vs. 3,5,3'-triiodo-l-thyronine and cell proliferation: activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Am J Physiol Cell Physiol. 2009;296(5):C980–C991 [DOI] [PubMed] [Google Scholar]
- 35. Hu LW, Benvenuti LA, Liberti EA, Carneiro-Ramos MS, Barreto-Chaves ML. Thyroxine-induced cardiac hypertrophy: influence of adrenergic nervous system versus renin-angiotensin system on myocyte remodeling. Am J Physiol Regul Integr Comp Physiol. 2003;285(6):R1473–R1480 [DOI] [PubMed] [Google Scholar]
- 36. Polk DH, Callegari CC, Newnham J, et al. Effect of fetal thyroidectomy on newborn thermogenesis in lambs. Pediatr Res. 1987;21(5):453–457 [DOI] [PubMed] [Google Scholar]
- 37. Matsui T, Nagoshi T, Rosenzweig A. Akt and PI 3-kinase signaling in cardiomyocyte hypertrophy and survival. Cell Cycle. 2003;2(3):220–223 [PubMed] [Google Scholar]
- 38. Kato T, Muraski J, Chen Y, et al. Atrial natriuretic peptide promotes cardiomyocyte survival by cGMP-dependent nuclear accumulation of zyxin and Akt. J Clin Invest. 2005;115(10):2716–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen D, Heath V, O'Garra A, Johnston J, McMahon M. Sustained activation of the raf-MEK-ERK pathway elicits cytokine unresponsiveness in T cells. J Immunol. 1999;163(11):5796–5805 [PubMed] [Google Scholar]
- 40. Dangi S, Chen FM, Shapiro P. Activation of extracellular signal-regulated kinase (ERK) in G2 phase delays mitotic entry through p21CIP1. Cell Prolif. 2006;39(4):261–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Haddow JE, Palomaki GE, Allan WC, et al. Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. N Engl J Med. 1999;341(8):549–555 [DOI] [PubMed] [Google Scholar]
- 42. Niswander KR, Gordon M, The Collaborative Perinatal Study of the National Institute of Neurological Disease and Stroke: The Women and Their Babies. Report. Philadelphia, PA: W.B. Saunders Company; 1972: 246–249 [Google Scholar]
- 43. Seely BL, Burrow GN. Thyroid disease and pregnancy. In: Creasy RK, Resnik R, eds. Maternal-Fetal Medicine: Principles and Practice. Philadelphia, PA: W.B. Saunders Company; 1994 [Google Scholar]
- 44. Hume R, Simpson J, Delahunty C, et al. Human fetal and cord serum thyroid hormones: developmental trends and interrelationships. J Clin Endocrinol Metab. 2004;89(8):4097–4103 [DOI] [PubMed] [Google Scholar]