

Abstract
Organic chemists are now able to synthesize small quantities of almost any known natural product, given sufficient time, resources and effort. However, translation of the academic successes in total synthesis to the large-scale construction of complex natural products and the development of large collections of biologically relevant molecules present significant challenges to synthetic chemists. Here we show that the application of two nature-inspired techniques, namely organocascade catalysis and collective natural product synthesis, can facilitate the preparation of useful quantities of a range of structurally diverse natural products from a common molecular scaffold. The power of this concept has been demonstrated through the expedient, asymmetric total syntheses of six well-known alkaloid natural products: strychnine, aspidospermidine, vincadifformine, akuammicine, kopsanone and kopsinine.
The field of natural product total synthesis has evolved dramatically over the past 60 years owing substantially to the efforts of strategy-based organic chemists along with remarkable improvements in our bond-forming capabilities. However, two critical challenges that remain for this field are those of translating laboratory-level academic success in total synthesis to the large-scale assembly of biologically important molecules1 and building large collections of natural product families (or their analogues) for use as biological probes or in medicinal chemistry2.
To address these two fundamental challenges in small-molecule synthesis, we have been inspired by the strategies used in nature to solve these problems. For example, as chemists, we typically use ‘stop-and-go’ synthetic protocols, whereby individual transformations are conducted as stepwise processes punctuated by the isolation and purification of intermediates at each stage of the sequence3. By contrast, in nature the rapid conversion of simple starting materials to complex molecular scaffolds is accomplished through the use of transformation-specific enzymes, which mediate a continuous series of highly regulated catalytic cascades in what amounts to be a highly efficient ‘biochemical assembly line’4,5.Moreover, biosynthesis in nature provides an appealing alternative to the traditional ‘single-target’ approach to chemical synthesis in that it typically involves the construction of natural product collections through the assembly of a common intermediate (Fig. 1).
Figure 1. Cascade catalysis in biosynthesis.

In nature, transform-specific enzymes in continuous catalytic cascades rapidly produce common biosynthetic intermediates and natural products. Preakuammicine serves as a biosynthetic precursor to a range of structurally diverse members of the Strychnos, Aspidosperma and Kopsia alkaloid families, including strychnine and vincadifformine. Et, ethyl; Gluc, glucose; Me, methyl.
Design plan
We recently sought to develop a novel asymmetric approach to total synthesis based on the application of these two nature-inspired concepts, namely collective total synthesis and organocascade catalysis3,4. Whereas syntheses targeting an advanced core structure applicable to the synthesis of closely related natural products within a family have been frequently reported6, much less common is the preparation of an intermediate endowed with functionality amenable to the preparation of structurally diverse natural products in different families—a strategy we term collective total synthesis7,8. We predicted that the hybridization of the strategies of collective natural product synthesis and enantioselective organocascade catalysis9 should rapidly give access to useful quantities of single-enantiomer natural product collections or families with unprecedented levels of ease and efficiency.
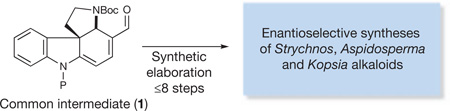
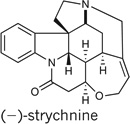
To demonstrate this approach, we targeted for total synthesis six well-known, structurally complex members of the Strychnos, Aspidosperma and Kopsia families of alkaloids, in particular strychnine, the best-known member of the Strychnos family, which has been the focus of intense synthetic interest for over 50 years. Moreover, this target has served as a key metric for the success of our nature-inspired dual techniques10,11.
Importantly, strychnine is believed to share a biosynthetic precursor with a range of prominent Strychnos, Aspidosperma and Kopsia alkaloids12. This common intermediate, preakuammicine, arises biosynthetically through a controlled enzymatic cascade involving a coupling of the precursors tryptamine and secologanin, followed by skeletal rearrangement (Fig. 1). Thus, in a prime example of natural economy, a single intermediate is exploited in the construction of a diverse collection of complex molecular products.
We expected the preparation of intermediate 1, which incorporates the requisite functionality for expedient conversion to each of the target natural products (Fig. 2), to be a central element of our design strategy. The key tetracyclic precursor 1 would itself be accessed through a one-flask, asymmetric Diels–Alder/elimination/conjugate addition organocascade sequence commencing with a simple tryptamine-derived substrate13.
Figure 2. Collective natural product synthesis: nature-inspired application of cascade catalysis.

Synthesis of six structurally diverse Strychnos, Aspidosperma and Kopsia alkaloids is expected to proceed from a common intermediate tetracycle prepared by means of organocascade catalysis. Boc, tert-butoxycarbonyl; Im, iminium catalysis.
In detail, we hoped that exposure of the 2-(vinyl-1-selenomethyl) tryptamine system2 to propynal in the presence of an imidazolidinone catalyst (3) would set in motion a Diels–Alder [4 + 2] addition13 (Fig. 3). As a key element of enantiocontrol, the acetylenic functionality of the catalyst-bound propynal would be expected to partition away from the bulky tert-butyl (t-Bu) group, leaving the naphthyl group effectively to shield the bottom face of the reacting alkyne. The activated dienophile would then undergo endo-selective Diels–Alder cycloaddition with the substituted 2-vinyl indole 2. By contrast with our previous studies using methyl-sulphide-substituted 2-vinyl indoles, as demonstrated in our synthesis of minfiensine13, we felt a conceptually unique series of cascade cycles might arise through the incorporation of organoselenide substitution on the diene, leading to a novel, architecturally complex system. More specifically, following the Diels–Alder cycloaddition, the cycloadduct 4 would be poised to undergo facile β-elimination of methyl selenide to furnish the unsaturated iminium ion 5. We proposed this change in mechanism owing to the higher propensity of selenides to undergo β-elimination in comparison with sulphides14.
Figure 3. Proposed mechanism of organocascade cycles for the generation of a common tetracyclic intermediate (1).

An organocascade reaction between 2-vinyl indole 2 and propynal is expected to proceed through an organocatalytic Diels–Alder/β-elimination/amine conjugate addition sequence along path A, involving iminium ion catalysis, or path B, involving Brønsted acid catalysis. 1-Nap, 1-naphthyl; SeMe, selenomethyl.
In the second cycle, iminium-catalysed 5-exo-heterocyclization of the pendant carbamate was expected to occur at the δ-position to the indolinium ion (5 → 6; Fig. 3, path A, X = NR2) to deliver, following hydrolysis, the enantioenriched spiroindoline core (1). We considered the possibility of an alternative second cycle wherein iminium 5 might undergo facile cyclization at the indoline carbon to generate pyrroloindoline 7 transiently (Fig. 3, path B). In this situation, we recognized that amine or Brønsted acid catalysis might thereafter induce the necessary 5-exo-heterocyclization of the pendant carbamate to also furnish 6. Importantly, either cascade sequence could allow for the rapid and enantioselective production of the complex tetracyclic spiroindoline 1 from simple tryptamine-derived and ynal substrates in a single operation.
Experimental results
The feasibility of the proposed organocascade sequence was first evaluated in the context of a total synthesis of strychnine. The requisite 2-vinyl indole 10 was prepared in three steps from 9 according to the standard procedures outlined in Fig. 4 (ref. 15). The crucial organocascade addition–cyclization was accomplished with the use of 1-naphthyl-substituted imidazolidinone catalyst 3 in the presence of 20 mol% tribromoacetic acid (TBA) co-catalyst, forming the complex spiroindoline 11 in 82% yield and with excellent levels of enantioinduction (97%e.e.). Notably, we have now gathered evidence that path B of the cascade sequence is operational. Specifically, when the same protocol was performed in the presence of stoichiometric catalyst at −78 °C and quenched after 10 min with Et3N, an 84% yield of pyrroloindoline 7 (protecting group, PMB) was obtained. Moreover, exposure of pyrroloindoline 7 (protecting group, PMB) to catalytic 3 ·TBA and, separately, N-methyl 3 ·TBA (incapable of undergoing iminium formation) facilitates the conversion to the spiroindoline 11 at comparable rates.
Figure 4. Twelve-step enantioselective total synthesis of (−)-strychnine.

Reagents and conditions are as follows. a, NaH, PMBCl, dimethylformamide (DMF), 0 °C. PMB, para-methoxybenzyl. b, SeO2, dioxane, H2O, 100 °C. c, (EtO)2P(O)CH2SeMe, 18-crown-6, potassium bis(trimethylsilyl)amide (KHMDS), tetrahydrofuran (THF), −78 °C to room temperature (RT, 23 °C). e.e., enantiomeric excess. d, (Ph3P)3RhCl, toluene, PhCN, 120 °C. e, COCl2, Et3N, toluene, −45 °C to RT, then MeOH,−30 °C to RT. f, DIBAL-H,CH2Cl2, −78 °C to RT, then trifluoroacetic acid (TFA), −78 °C to RT. g, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), K2CO3, DMF, (Z)-4-bromo-3-iodobut-2-enyl acetate (13), RT. h, DIBAL-H, CH2Cl2, −78 °C. i, 25 mol% Pd(OAc)2, Bu4NCl, NaHCO3, EtOAc, RT. j, PhSH, TFA, 45 °C. k, NaOAc, Ac2O, AcOH, malonic acid, 120 °C. Ac, acetyl.
The product of the key organocascade sequence, tetracyclic spiroindoline 11, was advanced to strychnine in only eight additional steps (Fig. 4). In detail, decarbonylation was achieved through the use of Wilkinson’s catalyst. Subsequent treatment with phosgene and methanol16 served to introduce a carbomethoxy group at the dienamine α-position. Next, on exposure to DIBAL-H, the enamine unsaturation was reduced and the requisite tertiary indoline stereocentre was installed to provide the unsaturated ester 12 (existing as an inconsequential mixture of alkene isomers) in 62% overall yield for the three steps. We converted intermediate 12 to vinyl iodide 14 through a two-step 76%-yield protocol involving allylation with the substituted allyl bromide 13 and concomitant alkene isomerization, followed by DIBAL-H-mediated reduction of both ester functionalities.
As a second key step, we predicted direct conversion of the vinyl iodide 14 to the protected Wieland–Gumlich aldehyde 15 through a cascade Jeffery–Heck cyclization/lactol formation sequence17. Insertion of palladium into the vinyl iodide and subsequent carbopalladation would forge the six-membered ring and produce an alkyl palladium intermediate, which would then undergo β-hydride elimination to provide an enol that would rapidly engage in lactol formation with the proximal alcohol-bearing side chain.
Following extensive investigations, we discovered an optimized set of conditions to allow successful Jeffery–Heck cyclization/lactol formation, using vinyl iodide 14 to form the PMB-protected Wieland–Gumlich aldehyde 15 in 58% yield18. Early studies showed that the PMB protecting group was critical in facilitating regioselective β-hydride elimination away from the indoline ring methine (and productively towards the alcohol). Such a high degree of regiocontrol presumably arises from the allylic strain that accompanies formation of the N-PMB-substituted enamine, a destabilizing element that is absent from the corresponding enol formation step.
Finally, the synthetic sequence was completed by TFA-mediated removal of the PMB group13 to furnish the Wieland–Gumlich aldehyde (16), which, on heating in a mixture of malonic acid, acetic anhydride and sodium acetate18, delivered enantioenriched (−)-strychnine in 12 steps and 6.4% overall yield from commercial materials. To our knowledge, this asymmetric organocascade-based sequence is the shortest route to enantioenriched strychnine that has been accomplished so far19–22.
We next turned to the construction of related alkaloids of the Strychnos, Aspidosperma, and Kopsia families based on the strategy of collective total synthesis outlined above. The total synthesis of (−)-akuammicine11 (Fig. 5) was achieved starting with unsaturated ester 12 (prepared in the course of the synthesis of strychnine; see Fig. 4). Treatment of the PMB-protected spiroindoline 12 with TFA and thiophenol at 60 °C resulted in the cleavage of the PMB protecting group as well as isomerization of the alkene into conjugation with the ester to give diamine 17 in 91% yield. Allylation of the pyrrolidine nitrogen with functionalized allyl bromide 18 (ref. 23) gave vinyl iodide 19, a precursor to akuammicine, by means of a Heck cyclization11. We expected insertion of palladium into the vinyl iodide followed by carbopalladation of the α, β-unsaturated ester to give an alkyl palladium intermediate. β-hydride elimination would then furnish the natural product. In the event, treatment of vinyl iodide 19 with palladium acetate under Jeffery conditions24 gave (−)-akuammicine, prepared in a total of ten steps and 10% overall yield.
Figure 5. Ten-step enantioselective synthesis of (−)-akuammicine.

Reagents and conditions are as follows. a, TFA, PhSH, 60 °C. b, (Z)-1-bromo-2-iodobut-2-ene (18), K2CO3, DMF, RT. c, 20 mol% Pd(OAc)2, NaHCO3, Bu4NCl, MeCN, 65 °C.
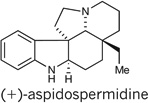
The alkaloids aspidospermidine25 and vincadifformine26 are among the most highly sought Aspidosperma alkaloid targets. Aspidospermidine, in particular, has been the subject of extensive investigation, having been synthesized by over 30 research groups27,28. Application of our cascade catalysis approach allowed access to both of these alkaloids according to the following sequence. Conversion of the cascade product 21 into the vinyl iodide 23 was achieved in a three-step sequence as outlined in Fig. 6. After optimization, we found that a Heck cyclization of the vinyl iodide onto the tri-substituted double bond could be used to form triene 24 in 65% yield, thus completing the pentacyclic core of aspidospermidine. We achieved a simultaneous global hydrogenation/debenzylation of triene 24 using palladium hydroxide on carbon under hydrogen pressure, to afford (+)-aspidospermidine in nine linear steps and 23% overall yield (Fig. 6), which constitutes the shortest enantioselective synthesis of aspidospermidine reported so far29,30. Finally, we recognized the potential to synthesize (+)-vincadifformine from (+)-aspidospermidine by means of an oxidation and carbomethoxylation sequence30. Indeed, Swern oxidation of aspidospermidine leads to the formation of imine 25, which can be treated with n-butyllithium followed by methyl cyanoformate to obtain (+)-vincadifformine (Fig. 6) in 11 steps and 8.9% overall yield31.
Figure 6. Enantioselective total syntheses of (+)-aspidospermidine and (+)-vincadifformine.

Reagents and conditions are as follows. a, NaH, DMF, BnBr, RT. b, SeO2, dioxane, H2O, 100 °C. c, (EtO)2P(O)CH2SeMe, 18-crown-6, KHMDS, THF, −78 °C to RT. ent-, enantio-. d, Ph3PCH3I, n-butyllithium, THF, 0 °C, then AcOH, NaCNBH3, 0 °C. Bn, benzyl. e, TFA, CH2Cl2, RT. f, (Z)-3-bromo-1-iodoprop-1-ene (22), K2CO3, DMF, RT. g, (Ph3P)4Pd, Et3N, toluene, 80 °C. h, Pd(OH)2, H2 (200 p.s.i.), MeOH, EtOAc, RT. i, CH2Cl2, DMSO, (COCl)2. j, n-butyllithium, NCCO2Me, THF, −78 °C to RT.
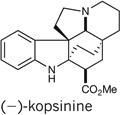
We further extended our strategy of collective total synthesis to the synthesis of kopsinine32–35 and the related compound kopsanone33,36. A unified approach to producing both alkaloids was implemented, allowing for a two-step conversion of kopsinine to kopsanone by means of a biomimetic thermocyclization33,37 (Fig. 7). Kopsinine was synthesized by first treating enantio-21 with trimethylsilyl iodide and then by vinyl triphenylphosphonium bromide to induce a deprotection/conjugate addition. Further treatment with KOt-Bu promoted a Wittig olefination to form the cyclic alkene found in triene 26. An enamine α-carbomethoxylation was then accomplished with phosgene–methanol16 to afford an intermediate ester, which was selectively reduced to diene 27 by treatment with palladium on carbon (Pd/C) and H2 in 69% over two steps.
Figure 7. Enantioselective total syntheses of (−)-kopsinine and (−)-kopsanone.

Reagents and conditions are as follows. a, Et3N, CH2Cl2, Me3SiI, 0 °C, then MeOH, H2C=CHPPh3Br, 40 °C, then CH2Cl2, THF, KOt-Bu, 0 °C. b, COCl2, Et3N, toluene, −45 °C to RT, then MeOH, −30 °C to RT. c, Pd/C,H2, EtOAc, EtOH, 0 °C. d, H2C=CHSO2Ph, benzene, 100 °C. e, Raney Ni, EtOH, 78 °C. f, 1N HCl, 130°C.
Dienes such as 27 can undergo [4 + 2] cycloadditions with a range of dienophiles, including vinyl sulfones33,38. We were able to obtain cycloadduct 28 in 83% yield through treatment of diene 27 with phenylvinyl sulfone in refluxing benzene. Furthermore, we were able to obtain (−)-kopsinine by performing a simultaneous desulfonylation, benzyl hydrogenolysis and diastereoselective alkene reduction of sulfone 28 with Raney nickel33 to give (−)-kopsinine in only nine steps, which is a significant improvement over the previous 19-step chiral-auxiliary-mediated approach32. Our attempts to directly convert (−)-kopsinine to (−)-kopsanone thermally according to the method of ref. 33 were unsuccessful. However, simple acid-mediated hydrolysis to give kopsinic acid (29), and subsequent heating of this material without solvent37, furnished (−)-kopsanone in only 11 chemical steps.
As anticipated, application of collective total synthesis to each of the target compounds—strychnine, akuammicine, aspidospermidine, vincadifformine, kopsinine and kopsanone—was readily accomplished with unprecedented levels of efficiency (Table 1). Perhaps most notably, these collective asymmetric syntheses took a total of 34 steps for the six natural products described (in comparison with 76 total steps in previous studies).
Table 1.
Enantioselective synthesis of six well-known indole alkaloid
 | ||||
|---|---|---|---|---|
| Compound | No. steps here* |
Overall yield (%) |
PSAC steps |
PSCA steps |
 |
12 | 6.4 | 25 (refs 19,20) | 16 (ref. 21) |
 |
9 | 24 | 13 (ref. 30) | 11 (ref. 29) |
 |
9 | 14 | NA | 19 (ref. 32) |
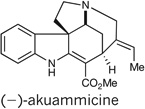 |
10 | 10 | NA | NA |
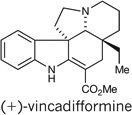 |
11 | 8.9 | NA | 10 (ref. 31) |
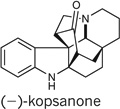 |
11 | 10 | NA | NA |
Step counts represent the longest linear sequence from commercially available 9. NA, not applicable. PSAC, previous shortest asymmetric catalytic synthesis; PSCA, previous shortest chiral auxiliary or chiral pool synthesis.
See Supplementary Information for details of these syntheses.
Conclusion
We have demonstrated the capabilities of collective total synthesis in combination with organocascade catalysis, a synthetic strategy that provides researchers with the tools to gain ready access to large collections of complex molecular architectures. In particular, we describe the shortest asymmetric synthesis of (−)-strychnine, the best-known member of the Strychnos alkaloid family. We hope to describe the value of this approach in terms of other natural product and medicinal agent families in the near future.
METHODS SUMMARY
All reactions were performed under an inert atmosphere using dry solvents in anhydrous conditions, unless otherwise noted. Full experimental details and characterization data for all new compounds are included in Supplementary Information.
Supplementary Material
Acknowledgments
Financial support was provided by NIHGMS (R01 GM078201-05) and gifts from Merck, Bristol-Myers Squibb and Abbott. S.B.J. and B.S. thank Bristol-Myers Squibb and Merck, respectively, for graduate fellowships.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions S.B.J., B.S. and A.M. participated in the performance and analysis of the experiments. S.B.J., B.S., A.M. and D.W.C.M. designed the experiments. S.B.J. and D.W.C.M. wrote the paper.
The authors declare no competing financial interests.
References
- 1.Walji A, MacMillan DWC. Strategies to bypass the Taxol problem. Enantioselective cascade catalysis, a new approach for the efficient construction of molecular complexity. Synlett. 2007:1477–1489. [Google Scholar]
- 2.Va P, Campbell EL, Robertson WM, Boger DL. Total synthesis and evaluation of a key series of C5-substituted vinblastine derivatives. J. Am. Chem. Soc. 2010;132:8489–8495. doi: 10.1021/ja1027748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang Y, Walji AM, Larsen CH, MacMillan DWC. Enantioselective organocascade catalysis. J. Am. Chem. Soc. 2005;127:15051–15053. doi: 10.1021/ja055545d. [DOI] [PubMed] [Google Scholar]
- 4.Simmons B, Walji A, MacMillan DWC. Cycle-specific organocascade catalysis: application to olefin hydroamination, hydro-oxidation, and amino-oxidation, and to natural product synthesis. Angew. Chem. Int. Ed. 2009;48:4349–4353. doi: 10.1002/anie.200900220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dewick PM. Medicinal Natural Products: A Biosynthetic Approach. 3rd edn. Wiley; 2008. [Google Scholar]
- 6.Corey EJ, Imai N, Pikul S. Catalytic enantioselective synthesis of a key intermediate for the synthesis of prostanoids. Tetrahedr. Lett. 1991;32:7517–7520. [Google Scholar]
- 7.Kuehne ME, Wang T, Seraphin D. The total synthesis of (±)-mossambine. Synlett. 1995:557–558. [Google Scholar]
- 8.Bandarage UK, Kuehne ME, Glick SD. Total syntheses of racemic albifloranine and its anti-addictive congeners, including 18-methoxycoronaridine. Tetrahedron. 1999;55:9405–9424. [Google Scholar]
- 9.Grondal C, Jeanty M, Enders D. Organocatalytic cascade reactions as a new tool in total synthesis. Nature Chem. 2010;2:167–178. doi: 10.1038/nchem.539. [DOI] [PubMed] [Google Scholar]
- 10.Bonjoch J, Sole D. Synthesis of strychnine. Chem. Rev. 2000;100:3455–3482. doi: 10.1021/cr9902547. [DOI] [PubMed] [Google Scholar]
- 11.Sirasani G, Paul T, Dougherty W, Kassel S, Andrade RB. Concise total syntheses of (±)-strychnine and (±)-akuammicine. J. Org. Chem. 2010;75:3529–3532. doi: 10.1021/jo100516g. [DOI] [PubMed] [Google Scholar]
- 12.Hudlicky T, Reed JW. The Way of Synthesis: Evolution of Design and Methods for Natural Products. Wiley-VCH; 2007. [Google Scholar]
- 13.Jones SB, Simmons B, MacMillan DWC. Nine-step enantioselective total synthesis of (+)-minfiensine. J. Am. Chem. Soc. 2009;131:13606–13607. doi: 10.1021/ja906472m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas PJ, Stirling CJM. Elimination and addition reactions. Part 34. The effect of activating group and medium on leaving group rank in elimination from carbanions. J. Chem. Soc. Perkin Trans. II. 1978;11:1130–1134. [Google Scholar]
- 15.Gatta F, Misiti D. Selenium dioxide oxidation of tetrahydro-β-carboline derivatives. J. Heterocycl. Chem. 1987;24:1183–1187. [Google Scholar]
- 16.Prashad M, Lavecchia L, Prasad K, Repic O. A convenient synthesis of 3-substituted 1H-indoles. Synth. Commun. 1995;25:95–100. [Google Scholar]
- 17.Oestreich M, editor. The Mizoroki–Heck Reaction. Wiley; 2009. [Google Scholar]
- 18.Anet FAL, Robinson R. Conversion of the Wieland–Gumlich aldehyde into strychnine. Chem. Ind. 1953:245. [Google Scholar]
- 19.Knight SD, Overman LE. Enantioselective total synthesis of (−)-strychnine. J. Am. Chem. Soc. 1993;115:9293–9294. [Google Scholar]
- 20.Mori M, Nakanishi M, Kajishima D, Sato Y. A novel and general synthetic pathway to Strychnos indole alkaloids: total syntheses of (−)-tubifoline, (−)-dehydrotubifoline, and (−)-strychnine using palladium-catalyzed asymmetric allylic substitution. J. Am. Chem. Soc. 2003;125:9801–9807. doi: 10.1021/ja029382u. [DOI] [PubMed] [Google Scholar]
- 21.Sole D, et al. Total synthesis of (−)-strychnine via the Wieland–Gumlich aldehyde. Angew. Chem. Int. Ed. 1999;38:395–397. doi: 10.1002/(SICI)1521-3773(19990201)38:3<395::AID-ANIE395>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 22.Martin DB, Vanderwal CD. A synthesis of strychnine by a longest linear sequence of six steps. Chem. Sci. 2011;2:649–651. [Google Scholar]
- 23.Sole D, Diaba F, Bonjoch J. Nitrogen heterocycles by palladium-catalyzed cyclization of amino-tethered vinyl halides and ketone enolates. J. Org. Chem. 2003;68:5746–5749. doi: 10.1021/jo034299q. [DOI] [PubMed] [Google Scholar]
- 24.Jeffery T. On the efficiency of tetraalkylammonium salts in Heck type reactions. Tetrahedron. 1996;52:10113–10130. [Google Scholar]
- 25.Marino JP, Rubio MB, Cao GF, de Dios A. Total synthesis of (+)-aspidospermidine: a new strategy for the enantiospecific synthesis of Aspidosperma alkaloids. J. Am. Chem. Soc. 2002;124:13398–13399. doi: 10.1021/ja026357f. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi S, Peng G, Fukuyama T. Efficient total syntheses of (±)-vincadifformine and (−)-tabersonine. Tetrahedr. Lett. 1999;40:1519–1522. [Google Scholar]
- 27.Hajicek J. A review on recent developments in syntheses of the post-secodine indole alkaloids. Part I: The primary alkaloid types. Collect. Czech. Chem. Commun. 2004;69:1681–1767. [Google Scholar]
- 28.Cho H-K, Tam NT, Cho CG. Total synthesis of (±) aspidospermidine starting from 3-ethyl-5-bromo-2-pyrone. Bull. Korean Chem. Soc. 2010;31:3382–3384. [Google Scholar]
- 29.Gnecco D, et al. Synthesis of an aspidosperma alkaloid precursor: synthesis of (+)-aspidospermidine. Arkivoc. 2003;2003(xi):185–192. [Google Scholar]
- 30.Kozmin SA, Iwama T, Huang Y, Rawal VH. An efficient approach to Aspidosperma alkaloids via [4 + 2] cycloadditions of aminosiloxydienes: Stereocontrolled total synthesis of (±)-tabersonine. Gram-scale catalytic asymmetric syntheses of (+)-tabersonine and (+)-16-methoxytabersonine. Asymmetric syntheses of (+)-aspidospermidine and (−)-quebrachamine. J. Am. Chem. Soc. 2002;124:4628–4641. doi: 10.1021/ja017863s. [DOI] [PubMed] [Google Scholar]
- 31.Kuehne M, et al. Application of ferrocenylalkyl chiral auxiliaries to syntheses of indolenine alkaloids: enantioselective syntheses of vincadifformine, ψ- and 20-epi-ψ-vincadifformines, tabersonine, ibophyllidine, and mossambine. J. Org.Chem. 1998;63:2172–2183. [Google Scholar]
- 32.Magnus P, Brown P. Total synthesis of (−)-kopsinilam, (−)-kopsinine, and the bis-indole alkaloids (−)-norpleiomutine and (−)-pleiomutine. J. Chem. Soc. Chem. Commun. 1985:184–186. [Google Scholar]
- 33.Kuehne ME, Seaton PJ. Studies in biomimetic alkaloid syntheses. 13. Total syntheses of racemic aspidofractine, pleiocarpine, pleiocarpinine, kopsinine, N-methylkopsanone, and kopsanone. J. Org. Chem. 1985;50:4790–4796. [Google Scholar]
- 34.Wenkert E, Pestchanker MJ. A formal total synthesis of kopsinine. J. Org. Chem. 1988;53:4875–4877. [Google Scholar]
- 35.Ogawa M, Kitagawa Y, Natsume M. A high-yield cyclization reaction for the framework of aspidosperma alkaloids synthesis of (±)-kopsinine and its related alkaloids. Tetrahedr. Lett. 1987;28:3985–3986. [Google Scholar]
- 36.Gallagher T, Magnus P. Synthesis of (±)-kopsanone and (±)-10,22-dioxokopsane, heptacyclic indole alkaloids. J. Am. Chem. Soc. 1983;105:2086–2087. [Google Scholar]
- 37.Kump C, Dugan JJ, Schmid H. Ringschlussreaktionen an Pleiocarpa-Alkaloiden. Helv. Chim. Acta. 1966;49:1237–1243. [Google Scholar]
- 38.Magnus P, Payne AH, Hobson L. Synthesis of the kopsia alkaloids (±)-11,12-demethoxylahadinine B, (±)-kopsidasine and (±)-kopsidasine-N-oxide. Tetrahedr. Lett. 2000;41:2077–2081. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.