

Abstract
The direct, by-product–free conversion of basic feedstocks to products of medicinal and agricultural relevance is a broad goal of chemical research. Butadiene is a product of petroleum cracking and is produced on an enormous scale (about 12 × 106 metric tons annually). Here, with the use of a ruthenium catalyst modified by a chiral phosphate counterion, we report the direct redox-triggered carbon-carbon coupling of alcohols and butadiene to form products of carbonyl crotylation with high levels of anti-diastereoselectivity and enantioselectivity in the absence of stoichiometric by-products.
Although many important methods for catalytic asymmetric carbon-carbon bond formation exist, much of this technology is not well suited for implementation on a large scale. Consequently, there is a need to discover and develop carbon-carbon–forming reactions that embody the principal characteristics of process relevance; in particular, the ability to transform abundant, ideally renewable, feedstocks to value-added products in the absence of stoichiometric by-products (1–4). This quality is embodied by alkene hydroformylation, the prototypical carbon-carbon bond–forming hydrogenation and largest-volume application of homogenous catalysis (5, 6). Accordingly, systematic efforts toward the discovery and development of carbon-carbon bond–forming hydrogenations were initiated in our laboratory (7, 8). We have found that diverse π-unsaturated reactants reductively couple to carbonyl compounds and imines under hydrogenation conditions or transfer hydrogenation conditions employing a sacrificial reductant (such as isopropanol), offering an alternative to the use of stoichiometric organometallic reagents. Most significantly, under transfer hydrogenation conditions, primary alcohols serve dually as hydrogen donors and aldehyde precursors, enabling carbonyl addition directly from the alcohol oxidation level in the absence of stoichiometric by-products.
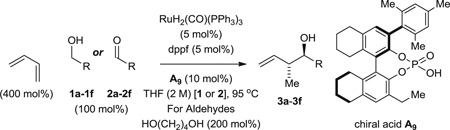
In the course of advancing hydrogenative methods for polyketide construction, anti-diastereoselective and enantioselective carbonyl crotylations from the alcohol or aldehyde oxidation level were developed, using α-methyl allyl acetate as the crotyl donor (9–11). Butadiene-alcohol carbon-carbon coupling potentially enables by-product–free access to identical crotylation products. However, although catalytic systems displaying the essential reactivity were defined (12–14), stereocontrolled hydrohydroxyalkylation of butadiene has proven elusive when both iridium (12) and ruthenium (13, 14) catalysts were used. Here we report that ruthenium catalysts bearing C1-symmetric 1,1'-bi-2-naphthol (BINOL)–derived phosphate counterions (15–22) promote anti-diastereoselective and enantioselective carbonyl crotylations from the alcohol or aldehyde oxidation level. This protocol bypasses the use of premetallated reagents for carbonyl crotylation, which are prepared from 2-butene or butadiene itself (23–28) (Fig. 1).
Fig. 1.

Direct and indirect carbonyl crotylation. IPC, (−)-isopinocampheyl; DBU, 1,8 diazabicyclo[5.4.0]undec-7-ene; dppf, 1,1,-bis(diphenylphosphino)ferrocene, TBAF, tetrabutylammonium fluoride; MW, molecular weight; dr, diastereomeric ratio.
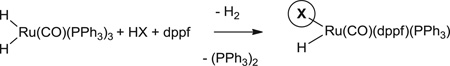 |
Eq. 1 |
In ruthenium-catalyzed carbonyl syn-crotylation from the alcohol or aldehyde oxidation level (14), 2-silyl-butadienes were required to enforce the intervention of a single geometrical isomer at the stage of the transient σ-crotylruthenium species, which appears to engage in stereospecific carbonyl addition through a closed transition structure. Although the (E)- and (Z)-σ-crotylruthenium intermediates obtained upon butadiene hydrometallation are considerably closer in energy and thus harder to discriminate, increasing steric congestion at the ruthenium center should bias formation of the thermodynamically more stable (E)-isomer. Based on this reasoning, we further postulated that ruthenium complexes bearing counterions of variable size could be prepared in situ through the acid-base reaction of H2Ru(CO)(PPh3)3 and HX (Eq. 1) (29, 30), enabling a systematic evaluation of diastereoselectivity in response to the steric demand of the counterion. As revealed in the hydrohydroxyalkylation of butadiene employing ruthenium catalysts modified by benzenesulfonate, mesitylsulfonate (mesityl = 2,4,6-trimethylphenyl), and trisylsulfonate (trisyl = triisopropylphenyl) counterions, diastereoselectivity increases with the increasing size of the counterion. In the case of the trisylsulfonate, concentration-dependent diastereoselectivity is attributed to a steric inhibition of the acid-base reaction (Fig. 2).
Fig. 2.

Enhanced anti-diastereoselectivity in response to increasing steric demand of the ruthenium counterion in the hydrohydroxyalkylation of benzylic alcohol 1b. Yields are of material isolated by silica gel chromatography. Diastereomeric ratios were determined by 1H NMR analysis of crude reaction mixtures. See the supplementary materials for further details.
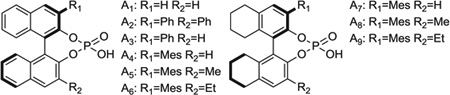
These data suggested the feasibility of directing both relative and absolute stereochemistry, using BINOL-derived phosphate counterions. Whereas the parent (R)-BINOL phosphoric acid A1 conferred poor stereoselectivity, the corresponding 3,3′-diphenyl derivative A2 promoted promising levels of diastereoselectivity and a 57:43 enantiomeric ratio (ER) (Table 1, entries 1 and 2). The related C1-symmetric phosphoric acid, which incorporates only a single phenyl moiety at the 3-position A3, displayed higher enantioselectivity than A1 or A2 (Table 1, entries 1 to 3). Based on this observation, the 3-mesityl phosphoric acid A4 was prepared and assayed, revealing an ER of 87:13 (Table 1, entry 4). The loading of butadiene can be decreased to 100 mol % with little impact on yield and stereoselectivity, although 400 mol % loadings provided optimal results and facilitated transfer of the volatile reagent on a small scale (Table 1, entries 4 to 7). At higher loadings of butadiene (800 mol %), catalytic efficiency is decreased, presumably due to nonproductive coordination of butadiene to coordination sites on the catalyst required for binding of the transient aldehyde. Increasing the size of the 3-aryl substituent did not improve stereoselectivity (see the supplementary materials for additional experiments); however, enhanced diastereoselectivity was observed in connection with the 3-mesityl-3′-methyl phosphoric acid A5 and 3-mesityl-3′-ethyl phosphoric acid A6 (Table 1, entries 8 and 9). Further, we found that the 3-mesityl-H8-BINOL A7 promoted higher enantioselectivity (89:11 ER) than the corresponding H4-derivative A4 (87:13 ER) (Table 1, entry 10). An improvement in diastereoselectivity was observed in connection with the 3′-methyl and 3′-ethyl H8-derivatives A8 and A9, yet a slight decrease in enantioselectivity was observed (Table 1, entries 11 and 12). Attributing decreased enantioselectivity to an incomplete acid-base reaction for such sterically demanding acids, we explored higher loadings of acid (10 mol %), using the H4-derivative A6 and the H8-derivatives A7 and A9 (Table 1, entries 13 to 15), which ultimately led to identification of A9 as the chiral acid of choice (Table 1, entry 16).
Table 1.
Optimization of anti-diastereoselectivity and enantioselectivity in the hydrohydroxyalkylation of benzylic alcohol 1b using BINOL-derived phosphate counterions. Yields are of material isolated by silica gel chromatography. Diastereomeric ratios were determined by 1H NMR analysis of crude reaction mixtures. ERs (er) were determined by chiral stationary-phase high-performance liquid chromatography analysis. These data represent only a small sampling of conditions and phosphoric acids that were screened. For additional details, see supplementary materials text.
 | |||||
|---|---|---|---|---|---|
| Entry | Acid (mol%) | Butadiene | Time (hrs) | Yield% | er (dr) |
| 1 | A1 (5) | 400 mol% | 19 | 45 | 52:48 (1:1) |
| 2 | A2 (5) | 400 mol% | 19 | 91 | 57:43 (5:1) |
| 3 | A3 (5) | 400 mol% | 19 | 83 | 69:31 (2:1) |
| 4 | A4 (5) | 400 mol% | 19 | 84 | 87:13 (3:1) |
| 5 | A4 (5) | 100 mol% | 19 | 72 | 87:13 (3:1) |
| 6 | A4 (5) | 200 mol% | 19 | 80 | 87:13 (3:1) |
| 7 | A4 (5) | 800 mol% | 19 | 50 | 87:13 (3:1) |
| 8 | A5 (5) | 400 mol% | 19 | 15 | 85:15 (6:1) |
| 9 | A6 (5) | 400 mol% | 19 | 21 | 87:15 (5:1) |
| 10 | A7 (5) | 400 mol% | 19 | 45 | 89:11 (3:1) |
| 11 | A8 (5) | 400 mol% | 19 | 15 | 86:14 (5:1) |
| 12 | A9 (5) | 400 mol% | 19 | 29 | 86:14 (4:1) |
| 13 | A6 (10) | 400 mol% | 19 | 33 | 92:8 (7:1) |
| 14 | A7 (5) | 400 mol% | 19 | 48 | 90:10 (3:1) |
| 15 | A9 (10) | 400 mol% | 19 | 50 | 94:6 (7:1) |
| ⇨16 | A9 (10) | 400 mol% | 48 | 80 | 94:6 (7:1) |
 | |||||
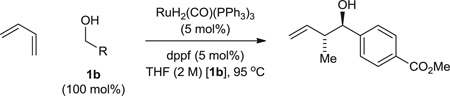
Using the chiral acid A9, benzylic alcohols 1a to 1f were assayed in the hydrohydroxyalkylation of butadiene to form the products of carbonyl crotylation 3a to 3f. The purity of alcohols 1a to 1f proved to be important, because trace quantities of carboxylic acid contribute to a racemic background reaction. Also, 31P nuclear magnetic resonance (NMR) analysis was essential in terms of evaluating the purity of the chiral acid A9, because 1H NMR was ineffective in this regard. With attention to these precautions, crotylation proceeds in good yield with anti-diastereoselectivities ranging from 5:1 to 9:1 and ERs ranging from 93:7 to 96:4 (Table 2). An identical set of adducts 3a to 3f are accessible from aromatic aldehydes 2a to 2f upon the use of 1,4-butanediol (31) (200 mol %) as the terminal reductant under otherwise identical conditions. Comparable anti-diastereoselectivities (4:1 to 8:1) and ERs (94:6 to 93:7) are observed (Table 2).
Table 2.
Direct anti-diastereoselective and enantioselective carbonyl crotylation via hydrohydroxyalkylation of butadiene and related aldehyde-reductive couplings using the BINOL-derived phosphate counterion A9. Characterization methods were as described in Table 1.
 | ||||
|---|---|---|---|---|
| Entry | Product | [o] Level | Y [%] 4a (dr) | er |
| 1 | 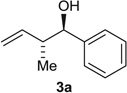 |
Alcohol Aldehyde |
86 (8:1) 74 (8:1) |
95:5 94:6 |
| 2 | 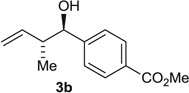 |
Alcohol Aldehyde |
80 (7:1) 80 (6:1) |
94:6 93:7 |
| 3 | 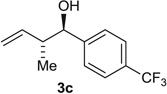 |
Alcohol Aldehyde |
83 (7:1) 63 (6:1) |
96:4 94:6 |
| 4 | 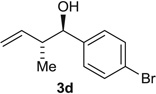 |
Alcohol Aldehyde |
97 (5:1) 78 (5:1) |
93:7 93:7 |
| 5 | 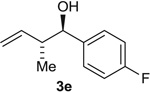 |
Alcohol Aldehyde |
85 (6:1) 80 (5:1) |
93:7 94:6 |
| 6 | 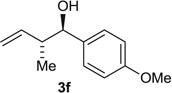 |
Alcohol Aldehyde |
72 (6:1) 66 (6:1) |
93:7 94:6 |
| 7 | 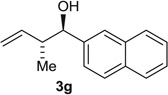 |
Alcohol Aldehyde |
95 (9:1) 79 (4:1) |
94:6 93:7 |
A plausible catalytic mechanism is depicted in Fig. 3. Hydrometallation of butadiene, as observed in stoichiometric reactions of RuHCl(CO)(PPh3)3 with 1,2- and 1,3-dienes (32, 33), delivers the π-allylruthenium complex. Counterion-dependent partitioning of the (E)- and (Z)-σ-crotylruthenium isomers precedes stereospecific carbonyl addition by way of the σ-crotylruthenium haptomer through a closed transition structure. The resulting homoallylic ruthenium alkoxide, which resists dehydrogenation because all coordination sites at the metal center are occupied, participates in alkoxide exchange with a reactant alcohol to release the product of crotylation and provide a pentacoordinate ruthenium alkoxide. The vacant coordination site at this stage enables dehydrogenation to form an aldehyde and regenerate the ruthenium hydride to close the catalytic cycle (Fig. 3).
Fig. 3.

Proposed mechanism for ruthenium-catalyzed hydrohydroxyalkylation of butadiene, illustrating the counterion-dependent partitioning of (E)- and (Z)-σ-crotylruthenium isomers.
Because organic molecules are compounds composed of carbon and hydrogen, the formation of carbon-carbon bonds under hydrogenation and transfer hydrogenation conditions is a natural end point in the evolution of strategies for organic synthesis. As illustrated here in the case of carbonyl crotylation, alcohol-butadiene hydrohydroxyalkylations enhance synthetic efficiency by removing the degrees of separation between reagent and feedstock, while bypassing discrete alcohol oxidation and the generation of stoichiometric by-products. These initial findings set the stage for the development of catalysts that exhibit enhanced stereoselectivities and substrate scope.
Supplementary Material
Acknowledgments
The Robert A. Welch Foundation (grant F-0038) and the NIH’s National Institute of General Medical Sciences (grant RO1-GM069445) are acknowledged for financial support.
Footnotes
Supplementary Materials
www.sciencemag.org/cgi/content/full/336/6079/324/DC1
Materials and Methods
Tables S1 to S4
References (34–42)
References and Notes
- 1.Butters M, et al. Chem. Rev. 2006;106:3002. doi: 10.1021/cr050982w. [DOI] [PubMed] [Google Scholar]
- 2.Sheldon RA, The E. Green Chem. 2007;9:1273. [Google Scholar]
- 3.Busacca CA, Fandrick DR, Song JJ, Senanayake CH. Adv. Synth. Catal. 2011;353:1825. [Google Scholar]
- 4.Magano J, Dunetz JR. Chem. Rev. 2011;111:2177. doi: 10.1021/cr100346g. [DOI] [PubMed] [Google Scholar]
- 5.Frohning CD, Kohlpaintner CW. In: Applied Homogeneous Catalysis with Organometallic Compounds. Cornils B, Herrmann WA, editors. Weinheim, Germany: Wiley-VCH; 1996. pp. 29–104. [Google Scholar]
- 6.van Leeuwen PWNM. Homogeneous Catalysis: Understanding the Art. Kluwer, Dordrecht, Netherlands: 2004. pp. 139–174. [Google Scholar]
- 7.Bower JF, Krische MJ. Top. Organomet. Chem. 2011;34:107. doi: 10.1007/978-3-642-15334-1_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hassan A, Krische MJ. Org. Process Res. Dev. 2011;15:1236. doi: 10.1021/op200195m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim IS, Han SB, Krische MJ. J. Am. Chem. Soc. 2009;131:2514. doi: 10.1021/ja808857w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao X, Townsend IA, Krische MJ. J. Org. Chem. 2011;76:2350. doi: 10.1021/jo200068q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao X, Han H, Krische MJ. J. Am. Chem. Soc. 2011;133:12795. doi: 10.1021/ja204570w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zbieg JR, Fukuzumi T, Krische MJ. Adv. Synth. Catal. 2010;352:2416. doi: 10.1002/adsc.201000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:6338. doi: 10.1021/ja801213x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zbieg JR, Moran J, Krische MJ. J. Am. Chem. Soc. 2011;133:10582. doi: 10.1021/ja2046028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alper H, Hamel N. J. Am. Chem. Soc. 1990;112:2803. [Google Scholar]
- 16.Komanduri V, Krische MJ. J. Am. Chem. Soc. 2006;128:16448. doi: 10.1021/ja0673027. [DOI] [PubMed] [Google Scholar]
- 17.Hamilton GL, Kang EJ, Mba M, Toste FD. Science. 2007;317:496. doi: 10.1126/science.1145229. [DOI] [PubMed] [Google Scholar]
- 18.Rueping M, Antonchick AP, Brinkmann C. Angew. Chem. Int. Ed. 2007;46:6903. doi: 10.1002/anie.200702439. [DOI] [PubMed] [Google Scholar]
- 19.Mukherjee S, List B. J. Am. Chem. Soc. 2007;129:11336. doi: 10.1021/ja074678r. [DOI] [PubMed] [Google Scholar]
- 20.Liao S, List B. Angew. Chem. Int. Ed. 2010;49:628. doi: 10.1002/anie.200905332. [DOI] [PubMed] [Google Scholar]
- 21.Jiang G, List B. Chem. Commun. (Camb.) 2011;47:10022. doi: 10.1039/c1cc12499d. [DOI] [PubMed] [Google Scholar]
- 22.Xu B, Zhu S-F, Xie X-L, Shen J-J, Zhou Q-L. Angew. Chem. Int. Ed. 2011;50:11483. doi: 10.1002/anie.201105485. [DOI] [PubMed] [Google Scholar]
- 23.Denmark SE, Fu J. Chem. Rev. 2003;103:2763. doi: 10.1021/cr020050h. [DOI] [PubMed] [Google Scholar]
- 24.Yus M, González-Gómez JC, Foubelo F. Chem. Rev. 2011;111:7774. doi: 10.1021/cr1004474. [DOI] [PubMed] [Google Scholar]
- 25.Hoffmann RW, Ladner W. Tetrahedron Lett. 1979;20:4653. [Google Scholar]
- 26.Brown HC, Bhat KS. J. Am. Chem. Soc. 1986;108:293. doi: 10.1021/ja00279a042. [DOI] [PubMed] [Google Scholar]
- 27.Brown HC, Bhat KS. J. Am. Chem. Soc. 1986;108:5919. doi: 10.1021/ja00279a042. [DOI] [PubMed] [Google Scholar]
- 28.Kim H, Ho S, Leighton JL. J. Am. Chem. Soc. 2011;133:6517. doi: 10.1021/ja200712f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobson A, Robinson SR, Uttley MF. J. Chem. Soc., Dalton Trans. 1975;1975:370. [Google Scholar]
- 30.Zbieg JR, McInturff EL, Leung JC, Krische MJ. J. Am. Chem. Soc. 2011;133:1141. doi: 10.1021/ja1104156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maytum HC, Tavassoli B, Williams JMJ. Org. Lett. 2007;9:4387. doi: 10.1021/ol702029n. [DOI] [PubMed] [Google Scholar]
- 32.Hiraki K, Ochi N, Sasada Y, Hayashida H, Fuchita Y, Yamanaka SJ. Chem. Soc. Dalton Trans. 1985;1985:873. [Google Scholar]
- 33.Xue P, et al. Organometallics. 2004;23:4735. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.