

Abstract
Cadmium is a toxic heavy metal which is environmentally and occupationally relevant. The mechanisms underlying cadmium-induced autophagy are not yet completely understood. The present study shows that cadmium induces autophagy, as demonstrated by the increase of LC3-II formation and the GFP-LC3 puncta cells. The induction of autophagosomes was directly visualized by electron microscopy in cadmium-exposed skin epidermal cells. Blockage of LKB1 or AMPK by siRNA transfection suppressed cadmium-induced autophagy. Cadmium-induced autophagy was inhibited in dominant-negative AMPK-transfected cells, whereas it was accelerated in cells transfected with the constitutively active form of AMPK. mTOR signaling, a negative regulator of autophagy, was downregulated in cadmium-exposed cells. In addition, cadmium generated reactive oxygen species (ROS) at relatively low levels, and caused poly(ADP-ribose) polymerase-1 (PARP) activation and ATP depletion. Inhibition of PARP by pharmacological inhibitors or its siRNA transfection suppressed ATP reduction and autophagy in cadmium-exposed cells. Furthermore, cadmium-induced autophagy signaling was attenuated by either exogenous addition of catalase and superoxide dismutase, or by overexpression of these enzymes. Consequently, these results suggest that cadmium-mediated ROS generation causes PARP activation and energy depletion, and eventually induces autophagy through the activation of LKB1-AMPK signaling and the down-regulation of mTOR in skin epidermal cells.
Keywords: Cadmium, Autophagy, LKB1, AMPK, mTOR, ROS
Introduction
Cell death is generally classified into three categories: apoptosis, autophagy, and necrosis (Kitanaka and Kuchino, 1999). Apoptosis and autophagy are controlled tightly by a regulatory mechanism and these types of cell death play a central role in tissue homeostasis, development, and disease. The best known mode of cell death is apoptosis (Shimada et al., 1998; Waalkes et al., 2000), characterized by cellular shrinkage, nuclear condensation, and DNA fragmentation (Kroemer and Reed, 2000; Li et al., 1997; Susin et al., 1999). Biochemical changes such as the activation of caspases and/or endonucleases are also important characteristics of apoptosis (Arends et al., 1990; Patel et al., 1996). Autophagy is a cellular defense process in which cytosolic components, organelles, and invading bacteria are transported by autophagosomes to lysosomes for degradation (Dice, 2007; Levine and Klionsky, 2004; Mizushima, 2007; Muller et al., 2000). Thus, autophagy is evidenced by the early appearance of large inclusions in the cytoplasm derived from autophagic vacuoles or autolysosomes. Such autophagy could be caused by starvation, cytoplasmic renewal, elimination of intracellular components and pathogens, innate and acquired immune responses, and programmed cell death (Hara et al., 2006; Komatsu et al., 2006; Nakagawa et al., 2004; Paludan et al., 2005; Pattingre et al., 2005).
Beclin 1 and microtuble-associated protein 1 light chain 3 (LC3) are the critical components in autophagy. Beclin 1 is the mammalian orthologue of the yeast Atg6/Vps 30, and is involved in the regulation of autophagy (Liang et al., 1999; Qu et al., 2003; Tassa et al., 2003; Yue et al., 2003; Zeng et al., 2006). LC3 is the mammalian homologue of yeast Atg8 and localizes to autophagosomal membranes after post-translational modifications. LC3 exists in two molecular forms; LC3-I (18 kD) is cytosolic form, whereas LC3-II (16 kD) binds to autophagosomes (Kabeya et al., 2000; Mizushima et al., 2001). The amount of LC3-II directly correlates with the number of autophagosomes (Kabeya et al., 2000).
The energy-sensing LKB1-AMP-activated protein kinase (AMPK) pathway regulates cell survival under energy deprivation which increases AMP: ATP ratio (Lizcano et al., 2004; Shaw et al., 2004). AMPK is a heterotrimeric protein complex consisting of AMPK-α, AMPK-β, and AMPK-γ subunits. AMPK is downstream of LKB1 in a signaling pathway that regulates energy homeostasis (Hardie, 2004; Shaw et al., 2004; Woods et al., 2003). LKB1 was identified as the gene mutated in human Peutz-Jeghers syndrome (PJS) (Hemminki et al., 1998; Jenne et al., 1998) and necessary for the activation of AMPK (Hawley et al., 2003; Shaw et al., 2004). Changes in cellular AMP/ATP ratios promote allosteric interaction between AMP and AMPK-γ subunit, which promotes phosphorylation of AMPK-α subunit at T172 and activation of AMPK signaling (Andersson et al., 2004; Hardie, 2004; Sanders et al., 2007). Moreover, AMPK has been implicated in many aspects of cell proliferation, apoptosis, and autophagy (Liang et al., 2007; Luo et al., 2005; Motoshima et al., 2006).
Cadmium is a toxic heavy metal and human carcinogen. Food chain, cigarette smoke, and cadmium mining industry are the main sources of cadmium exposure to humans (Jarup, 2003). Cadmium induces either apoptosis or carcinogenesis depending on the conditions such as concentrations and times exposed. It was recently reported that cadmium induced autophagy in mesangial cells (Wang et al., 2008; Wang et al., 2009). Further, cadmium-induced increase of intracellular ROS is involved in cell death caused by this metal (Son et al., 2010b). These findings suggest that in addition to the induction of apoptosis, cadmium leads to autophagic cell death. However, the cellular mechanisms by which cadmium causes autophagy have not been extensively explored. Little information is available about the relationship between intracellular ROS generation and autophagy. We used mouse JB6 epidermal cell lines to study the autophagy response to cadmium. JB6 cells are widely used and well studied in carcinogenesis induced by tumor promoters and metals as well as oxidative stress (Dhar et al., 2002). Therefore, the present study using these cells will contribute to further understanding of cadmium mediated toxicity and carcinogenicity. The concentrations of cadmium which were used in our experiments were 1 to 10 μM. These concentrations are 10 to 100 times higher than the blood levels (15 μg/l) of industrial workers who are occupationally exposed to cadmium (Glahn et al., 2008). However, cadmium has a long biological half-life (15 – 20 years) and accumulates in various organs such as the liver, kidneys, lung, bone, and eyes (Henson and Chedrese, 2004; Jin et al., 1998). Thus, the concentrations used in the present study are highly relevant to occupational exposure.
The present study examined whether cadmium actually causes autophagic cell death using the mouse epidermal cell line, JB6. We also determined the possible roles of intracellular ROS generated by cadmium on the process of autophagy. In addition, we investigated the molecular mechanisms involved in cadmium-induced autophagy. Here we demonstrate the critical roles of ROS-mediated signaling and the involvement of LKB1-AMPK signaling pathways in cadmium-induced autophagy.
Materials and methods
Chemicals and laboratory wares
Unless specified otherwise, all chemicals and laboratory wares were purchased from Sigma Chemical Co. (St. Louis, MO) and Falcon Labware (Bectone-Dickinson, Franklin Lakes, NJ), respectively. Eagle’s minimal essential medium (EMEM), fetal bovine serum (FBS), gentamicin, geneticin, and L-glutamine were purchased from Gibco Co. (Gibco BRL, NY). 3,4-Dihydro-5[4-(1-peperindinyl)butoxy]-1(2H)-isoquinoline (DPQ) and 3-Methyladenine (3-MA) were purchased from Calbiochem (San Diego, CA).
Cell culture and treatment
JB6 mouse epidermal cells (ATCC, CRL-2010) were cultured in EMEM supplemented with 5% FBS, 2 mM L-glutamine, and 50 μg/ml of gentamicin. GFP-LC3 JB6 cells generated by stable trasfection of an expression vector encoding for the LC3-GFP fusion protein were maintained in EMEM with 5% FBS and 800 μg/ml of G418. The cells were grown at 37 °C in a 5% CO2 atmosphere. One million cells per milliliter were resuspended in the growing medium and divided into 60 mm culture dishes or 96-well plates. When the cells reached to 80% confluence, the medium was replaced with a fresh EMEM containing various concentrations of CdCl2 (0–10 μM) and chemicals.
Plasmids and transfection
GFP-LC3 plasmid was provided from Dr. Jia Luo (University of Kentucky). Constitutively active forms of AMPKα (CA-AMPK) and dominant negative forms of AMPKα (DN-AMPK) constructs were gifted from Dr. J. Suttles (University of Louisville). CAT-Myc-DDK-, SOD1-Myc-DDK-tagged plasmids were purchased from Origene (Rockville, MD) and SOD2-EGFP-tagged plasmid came from Addgene (Cambridge, MA). Transfections were performed using Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA) according to the manufacture’s protocol. Briefly, JB6 cells were seeded in 6-well culture plates and transfected with 4 μg of plasmids at approximately 50% confluency. Transfected cells with GFP-LC3 were examined by fluorescence microscope. Overexpressed or knockdown cellular levels of the proteins specific for AMPKα, catalase (CAT), superoxide dismutase-1 (SOD1), and SOD2 were checked by immunoblotting, and the cells were maintained using G418 for stable cell lines.
Electron microscopy
To morphologically observe the induction of autophagy in cadmium-treated JB6 cells, we performed electron microscopy analysis as described elsewhere (Paglin et al., 2001). After being treated with cadmium (10 μM) for 24 h, cells were washed twice with PBS and fixed with ice-cold glutaraldehyde (3.5% in 0.1 M Sorenson’s buffer, pH 7.4) for 30 min at 4 °C. Cells were postfixed in 1% osmium tetroxide (OsO4) for 30 min at 4 °C and embedded in LX 122 before being cut and stained with uranyl acetate/lead citrate. Finally the cells were observed using a Hitachi H600 electron microscope (Hitachi Instrument, Tokyo, Japan).
GFP-LC3 localization
The GFP-LC3 puncta were quantified by counting the number of cells as described elsewhere (Alexander et al., 2010). Briefly, JB6 cells stably transfected with GFP-LC3 construct were divided on coverslips plated on 6-well plates (0.2 × 106/coverslip). Cells were exposed to cadmium (0–10 μM) with or without various activators or inhibitors for 24 h and fixed in ice-cold methanol. Total 50 GFP positive cells were counted under a fluorescence microscopy (Carl Zeiss, Germany), and cells with more than five puncta were considered autophagic GFP-LC3 puncta cells.
Western blot analysis
Cell lysates were made in a lysis buffer (50 mM Tris-Cl, pH 7.4, 1 mM EDTA, 150 mM NaCl, 1% NP-40, 0.25% Na-deoxycholate, and 1 μg/ml of aprotinin, leupeptin, and pepstatin). Equal amounts (30 μg/sample) of protein were separated by the NuPAGE Bis-Tris electrophoresis system (Invitrogen, Carlsbad, CA) and blotted onto a nitrocellulose membrane (Whatman, Dassel, Germany). Blots were probed with primary antibodies for either 2 h at room temperature or overnight at 4 °C before incubation with horseradish peroxidase-conjugated anti-IgG in a blocking buffer for 1 h. Monoclonal antibodies specific for β-actin (SC-47778), 8-OHdG (SC-66036), and polyclonal antibody specific to STRAD (SC-55052) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The polyclonal antibody specific to LC3 (PD014) and monoclonal antibody specific to PARP (SA-249) were purchased from BML (Woburn, MA) and Biomol (Plymouth meeting, PA), respectively. LKB1 (3047), p-LKB1 (Ser428; 3482), AMPKα (2532), p-AMPKα (Thr172; 2535), mTOR (2972), p-mTOR (Ser2448; 2971), p70 S6 kinase (9202), p-p70 S6 kinase (Thr421/Ser424; 9204), ACC (3662), p-ACC (3661), and MO25α (2716) polyclonal antibodies were purchased from Cell Signaling (Beverly, MA). PAR (AM80) was purchased from Calbiochem (San Diego, CA). Secondary antibodies and enhanced chemiluminescence substrate were from Pierce (Rockford, IL). The blots were exposed to Hyperfilm (Amersham Pharmacia Biotech).
Small interfering RNA transfection
Silencer pre-designed small interference RNA (siRNA) for mouse AMPKα (siRNA ID: S98536), LKB1 (siRNA ID: S74499), PARP-1 (siRNA ID: S62055), and negative control siRNA (AM4611) were obtained from Ambion (Austin, TX). JB6 cells were seeded in 96- or 6-well culture plates and transfected at approximately 50% confluency with the siRNA duplexes using Lipofectamine™ RNAi MAX (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Cellular levels of the proteins specific for the siRNA transfection were checked by immunoblotting, and all experiments were performed 24 h after transfection.
ATP assay
Intracellular ATP levels were determined using Molecular Probes’ ATP determination kit (A22066) as described elsewhere (Son et al., 2009b). Briefly, cadmium-exposed cells (2×106 cells/ml) were resuspended in a buffer (250 μl) containing 10 mM KH2PO4 and 4 mM MgSO4 (pH 7.4) before heating at 98 °C for 4 min. ATP levels were determined by using luciferase and its substrate D-luciferin. Finally, light emission was quantified in a Glomax™ 96 microplate luminometer (Promega, Madison, WI).
Electron spin resonance (ESR) assay
All ESR measurements were conducted using a Bruker EMX spectrometer (Bruker Instruments, Billerica, MA) and a flat cell assembly, as described previously (Son et al., 2010a). A spin trap, 5,5-dimethyl-1-pyrroline-1-oxide (DMPO) was charcoal purified and distilled to remove all ESR detectable impurities before use. PBS was also purified with Chelex 100 to protect from transition metal ion contamination. The Acquisit program was used for data acquisitions and analyses (Bruker Instruments). The cadmium-exposed cells were harvested and mixed with DMPO (100 mM). The samples were then transferred to a flat cell for ESR measurement. Experiments were performed at room temperature and under ambient air.
Statistical analysis
All the data are expressed as mean ± standard error (SE). One-way analysis of variance (ANOVA) using SPSS ver. 10.0 software was used for the multiple comparisons. A value of P < 0.05 was considered statistically significant.
Results
Cadmium induces autophagy in JB6 cells
Cadmium treatment increased the protein levels of LC3 in JB6 cells in a dose- and time-dependent manner (Figs. 1A and B). A dramatic accumulation of LC3-II, a hallmark of autophagy, was observed in the cells after 12 h of cadmium treatment (10 μM) and was further augmented at 24 h after the treatment. JB6 cells stably transfected with GFP-LC3 exhibited an increase fluorescence intensity of puncta (autophagic vesicles) when treated with cadmium (Fig. 1C). The total number of GFP-LC3 puncta positive cells also increased dependent on the dose and the time of exposure to cadmium (Figs. 1D and E). The occurrence of autophagy by cadmium was supported by direct observation of the formation of autophagosomes using electron microscopy (Fig. 1F). As shown in this figure, the control cells exhibited normal nuclei with uniform and finely dispersed chromatin, whereas abundant autophagosomes in the cytoplasm were produced in cadmium-exposed cells. Cadmium-induced autophagy was further examined by staining with acridine orange and monodansylcadaverine (MDC), where many cells showed acidic compartment and mature autophagosomes depending on the concentrations of cadmium (Suppl. 1A and B). However, the number of cells stained with acridine orange or MDC was reduced in the presence of 3-methyladenine (3-MA), an autophagy inhibitor (Suppl. 1C). Furthermore, we found that cadmium-induced increase in the both fluorescence intensity of GFP-LC3 and puncta positive cells was attenuated significantly by treatment with 3-MA (Figs. 1G and H).
Fig. 1.


Cadmium induces autophagy in JB6 cells. (A) The cells were exposed to the increasing concentrations (0–10 μM) of cadmium for 24 h. (B) Cells were exposed to 10 μM cadmium for various times (0–24 h). Protein lysates were analyzed by a NuPAGE Bis-Tris electrophoresis system, and the levels of LC3-I and LC3-II were detected by Western blot analysis. (C) JB6 cells stably transfected with GFP-LC3 were treated with cadmium for various concentrations (0–10 μM) and visualized by microscopy for fluorescent puncta. The number of GFP-LC3 puncta positive cells were counted and presented at indicated concentrations (D) and times (E). (F) JB6 cells treated with or without cadmium for 24 h were analyzed on electron microscopy. Arrows indicate autophagosomes. In addition, before treated with cadmium (10 μM), the cell were pre-incubated with 3-MA (5 mM) for 1 h. After incubation 24 h, a fluorescence of GFP-LC3 puncta cells were visualized by microscopy (G) and the number of GFP-LC3 puncta positive cells were counted and presented on graph (H). β-actin was used as an internal control. The results are shown as the mean ± SE of three separate experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 versus the control value or the cadmium treatment alone (ANOVA, Scheffe’s test).
The energy sensing LKB1-AMPK pathway is involved in cadmium-induced autophagy in JB6 cells
Cadmium treatment induced the phosphorylation of LKB1 in a dose-dependent manner (Fig. 2A). This induction started 1h after cadmium treatment and sustained to 24 h (Fig. 2B). However, there were no increases in the protein levels of MO25α and STRAD in cadmium-exposed cells. To determine the role of LKB1 in cadmium-induced autophagy, the expression of LKB1 was knocked down with siRNA transfection to the cells. The cadmium-increased formation of LC3-II was down-regulated in si-LKB1 transfected cells, indicating the involvement of LKB1 in cadmium-induced autophagy mechanisms (Fig. 2C). This result was further supported by the findings that the si-LKB1 transfection attenuated significantly (p < 0.01) the number of puncta positive cells when compared with the control siRNA transfected cadmium-exposed cells (Fig. 2D). Figure 3 shows that AMPKα, which is activated by LKB1 and functions as an intracellular energy sensor regulating metabolism and cell proliferation through phosphorylation of acetyl CoA carboxylase (ACC), is also involved in cadmium-induced autophagy. The phosphorylated levels of AMPKα and ACC were dramatically increased by cadmium in a dose- and time-dependent manner (Figs. 3A and B). However, the phosphorylation of AMPKβ or AMPKγ was not apparent in cadmium-exposed cells (data not shown). Subsequently, siRNA specific to AMPKα a dominant negative form of AMPKα (DN-AMPK), or a constitutively active form of AMPKα (CA-AMPK) were transfected to the cells. Cadmium-induced LC3-II formation and GFP-LC3 puncta cells were attenuated by si-AMPKα or DN-AMPK transfection (Figs. 3C–F). In contrast, cadmium-induced autophagy was accelerated in the cells transfected with CA-AMPK, as evidenced by the significant increases LC3-II formation and GFP-LC3 puncta cells compared with vector control (Figs. 3E and F). The involvement of AMPKα in cadmium-mediated autophagy was further supported by the finding that a pharmacological inhibitor or activator. For example, the treatment with AMPKα inhibitor, compound C, the formation of LC3-II and GFP-LC3 puncta cells by cadmium were attenuated. In contrast, the treatment with AMPKα activator, AICAR, exerted opposite effects (Suppl. 2A–C).
Fig. 2.
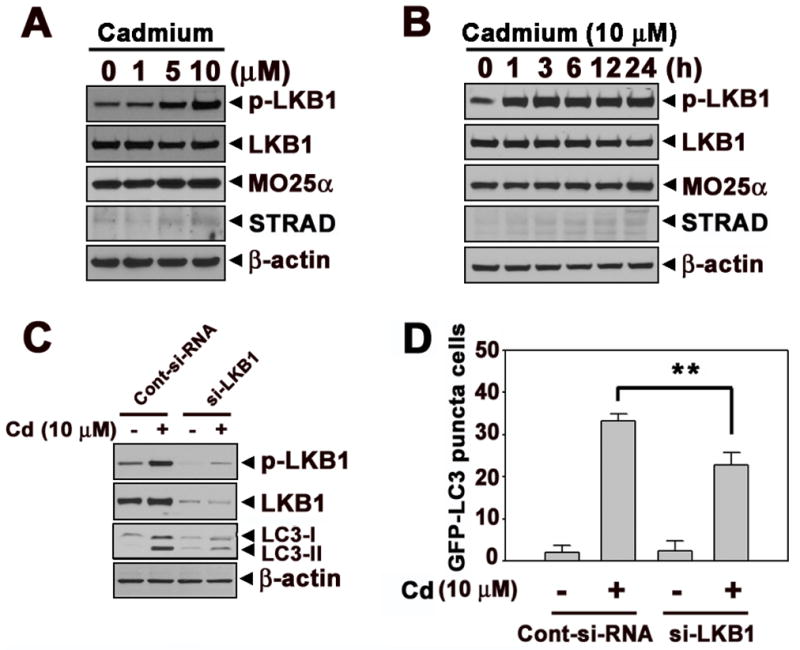
LKB1 signaling is involved in cadmium-induced autophagy. Cellular proteins were prepared from cells exposed to the indicated concentrations of cadmium for 24 h (A) or indicated times at 10 μM cadmium (B) and then processed for Western blot analysis. JB6 cells were transfected with siRNA specific to LKB1. After 24 h incubation, the cells were exposed to 10 μM cadmium for additional 24 h. (C) The expression levels of LKB1 and LC3 were analyzed by Western blot. (D) Quantification of GFP-LC3 puncta cells were shown from GFP-positive cells. The results represent the mean ± SE of three independent experiments. **P < 0.01 represents significant difference between the experiments. β-actin was used as loading control.
Fig. 3.
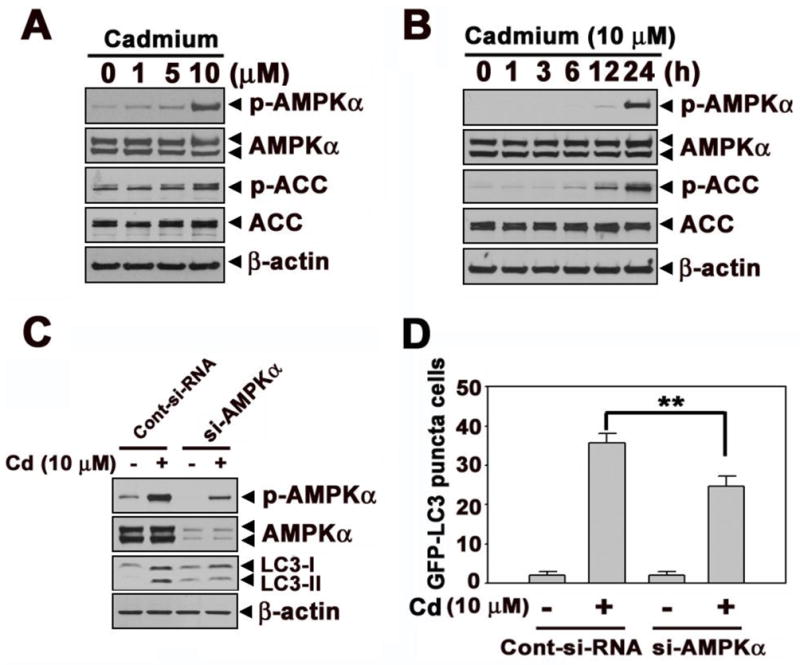

AMPKα play an important role in cadmium-induced autophagy. Cell lysates were prepared from cells subjected to the various doses (0–10 μM) of cadmium for 24 h (A) various times (0–24 h) at 10 μM cadmium (B) and then analyzed by NuPAGE Bis-Tris electrophoresis system followed by immunoblot analysis. In addition, JB6 cells were transfected with siRNA specific for AMPKα, and the cells were exposed to 10 μM cadmium.After 24 h incubation, the expression levels of AMPKα and LC3 were analyzed by Western blot assay (C). Quantification puncta positive cells were shown in Figure (D). Furthermore, dominant negative AMPKα (DN-AMPK) or constantly active form of AMPKα (CA-AMPK) was transfected to the JB6 cells. The transfected cells were incubated with in the absence or presence of cadmium for 24 h. Afterward, the expression levels of p-ACC, ACC, and LC3 were analyzed by Western blot (E) and The number of GFP-LC3 puncta cells were counted and presented in graph (F). *P < 0.05 represents significant difference between the experiments.
Cadmium induces autophagy through mTOR signaling in JB6 cells
mTOR, which negatively mediates autophagy, is important in autophagy processing. We examined whether mTOR signaling is involved in cadmium-induced LKB1-AMPKα mediated autophagy. Western blot analysis showed that phosphorylation of mTOR and total mTOR levels were down-regulated by exposing cells with cadmium in a dose- and time-dependent manner (Figs. 4A and B). In addition, phosphorylation of p70S6K which is downstream target of mTOR was down-regulated by cadmium (Figs. 4A and B). These results demonstrated that mTOR activity was down-regulated in cadmium-induced autophagy process. The blockage of either LKB1 or AMPKα by siRNA transfection prevented the cadmium-induced decrease in p-mTOR levels (Figs. 4C and D). Similarly, pretreatment with compound C or transfection with DN-AMPK suppressed the cadmium-induced reduction of p-mTOR levels, whereas CA-AMPK transfecion or AICAR pretreatment facilitated the down-regulation of mTOR in cadmium-treated cells (Suppl. 3A and B). These results indicated that mTOR signaling is an important mediator in cadmium-mediated autophagy of JB6 cells, where LKB1-AMPK signaling is upstream of mTOR signaling.
Fig. 4.

mTOR signaling is downstream of LKB1-AMPK pathway in cadmium-exposed cells. (A) Cells were incubated with various cadmium concentrations (0–10 μM) for 24 h. (B) Cells were also exposed various times (0–24 h) at 10 μM cadmium. The proteins levels were analyzed by Western blot. The phosphorylated mTOR and total mTOR levels were analyzed after transfection with siRNA specific for LKB1(C) or AMPKα (D) to the cells. β-actin was used as loading control.
ATP depletion through PARP-1 activation leads to autophagy in cadmium-exposed cells
The LKB1-AMPK cascade is sensitive to cellular energy status. AMPK is activated by stresses such as metabolic poisoning, oxidative stress, hypoxia, and nutrition deprivation that lead to the depletion in cellular ATP (Hardie, 2004). PARP-1 activation is also a rapid inducer of energy depletion through poly(ADP-ribosyl)ation (Hong et al., 2004; Yu et al., 2002). Cadmium treatment induced the formation of PAR in a dose- and time-dependent manner (Figs. 5A and B). The formation of PAR polymers by cadmium also increased in a dose- and time-dependent manner (Suppl. 4A and B). These results suggested that cadmium activate PARP-1 in JB6 cells. In addition, intracellular ATP levels decreased gradually according to the doses and the times exposed to cadmium (Figs. 5C and D). The cadmium-mediated decrease in ATP levels was significantly prevented either by si-PARP transfection or by treatment with a pharmacological PAPR inhibitor, DPQ (Fig. 5E). Moreover, the suppression of PARP-1 by treating DPQ or by its specific siRNA transfection transparently diminished the cellular levels of PAR, p-LKB1, p-AMPKα, and LC3-II proteins that had been increased in cadmium-treated cells (Figs. 5F–G). Furthermore, the number of GFP-LC3 puncta cells was significantly reduced by inhibiting the activation of PARP-1 (Fig. 5H).
Fig. 5.

PARP-1 activation and intracellular ATP depletion by cadmium are involved in LKB1-AMPK-mediated autophagy. JB6 cells were exposed to the indicated concentrations of cadmium for 24 h or 10 μM of cadmium treated to the cells for various times (0–24 h), after which they were assayed by Western blot analysis (A) (B) and ATP level analysis (C) (D). The pretreated cells with DPQ (30 μM) for 1 h or transfeced cells with siRNA specific for PARP were exposed to 10 μM cadmium. After 24 h exposed to cadmium, intracellular ATP levels (E), GFP-LC3 puncta cells (H), and protein levels (F) (G) were analyzed. Results are shown as the mean ± SE of at least three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 versus the control or the cadmium treatment alone.
Consistent generation of ROS at low levels leads to autophagy signaling in cadmium-exposed JB6 cells
In order to verify the relation between ROS and autophagy signaling in cadmium-exposed cells, we measured free radical generation using ESR spin trapping (Fig. 6A). As shown in the figure, the vehicle control did not show a detectable ESR signal, whereas cadmium treatment generated a 1:2:2:1 quartet ESR signal. The ROS levels generated by cadmium were quite low, while the generation was maintained up to 24 h after treatment (Figs. 6B and C). Moreover, over-expression of antioxidant enzymes such as CAT, SOD1, and SOD2 in JB6 cells reduced the cadmium-induced increases in the p-LKB1, p-AMPKα, and LC3-II levels (Fig. 6D). These results suggest that intracellular ROS generation leads to autophagy via LKB1-AMPKα pathway in cadmium-exposed cells.
Fig. 6.

The generation of ROS by cadmium mediates autophagy signaling in JB6 cells. Electron spin resonance (ESR) spectra were recorded after the treatment of cadmium (0 – 10 μM) for 24 h (A). The dose-(B) and time-course (C) signal intensity of DMPO-OH was expressed. (D) Catalase (CAT), SOD1, and SOD2 overexpressed stable cell lines were established. After 24 h exposed with cadmium (0 – 10 μM) to the overexpressed cells, the protein expression levels were analyzed by Western blot. The results are shown as the mean ± SE of three separate experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 versus the unexposed control (ANOVA, Scheffe’s test). The ESR spectrometer settings were as follows: frequency, 9.8 GHz; power, 39.91 mW; modulation frequency, 100 kHz; receiver gain, 5.02 × 105; time constant, 40.96 ms; modulation amplitude, 1.00 G, scan time, 60 s; and magnetic field, 3,451 ± 100 G. All spectra shown are an accumulation of 16 scans.
Discussion
Autophagy may help cell survival by purging the cells damaged from toxic metabolites and intracellular pathogens. However, autophagy may also promote cell death through excessive self-digestion and degradation of essential cellular constituents. Despite recent advances in understanding its molecular mechanisms and biological functions (Levine and Klionsky, 2004), it is unclear whether autophagy acts fundamentally as a cell survival or cell death pathway. In the absence of nutrients, autophagy allows cell survival, whereas prolonged autophagy can lead to non-apoptotic type-II programmed cell death (Kondo et al., 2005; Lum et al., 2005; Reggiori and Klionsky, 2005). Dysregulation of autophagy contributes to age-related pathologies, neurodegenerative diseases, and carcinogenesis (Kondo et al., 2005; Lee et al., 2010; Yue et al., 2003). In the present study, we investigated the mechanisms by which cadmium induces autophagy in mouse skin epidermal JB6 cells and demonstrated that cadmium induces autophagy of the cells through ROS-dependent activation of LKB-AMPK signaling pathways.
Cadmium affects a diverse range of cellular events such as proliferation, differentiations, and apoptosis. Here we initially showed the induction of autophagy in cadmium-exposed JB6 cells, as proven by the appearance of autophagosomes, the formation of LC3-II, and the increase of GFP-LC3 functa cells. Cadmium-induced formation of autophagocytic GFP-LC3 puncta supports the typical induction of autophagy by a heavy metal. Cadmium-induced autophagy was further supported by the observation that the protein levels of Beclin 1, which is also an important marker of autophagy, were increased after cadmium treatment in a dose- and time-dependent manner (Suppl. 5A and B). Consequently, these results support that cadmium induces autophagy in skin epidermal cells.
As the role of autophagy in influencing cell survival or death is controversial and dependent upon the exposure conditions, the function of autophagy in cadmium-exposed cells can differ. It was reported that autophagy did not protect cells from cadmium-induced toxicity (Wang et al., 2008), but a recent investigation supported a protective effect of autophagy on cadmium-induced cell death (Lim et al., 2010). Our current findings suggest that autophagy involves cell death mechanisms in cadmium-exposed cells. This is supported by the results showing the 3-MA-mediated decreases in the number of acridine orange- or MDC-positive cells as well as of GFP-LC3 puncta cells (Suppl. 1C). In addition, cadmium treatment decreased viability of JB6 cells in a dose- and time-dependent manner (Suppl. 6A), which was significantly inhibited by treating the cells with 3-MA (Suppl. 6B) or DPQ (Suppl. 6C). Collectively, these results reveal that cadmium-induced autophagy predominantly leads to cytotoxicity in skin epidermal cells.
Recent studies have demonstrated that calcium signaling is a major regulator of cadmium-induced autophagy in mesangial cells (Wang et al., 2008; Wang et al., 2009). This is partially consistent with our recent findings where calcium-activated signaling was closely associated with cadmium-induced cell death in JB6 cells (Son et al., 2010b). By contrast, the present study suggests that constantly generated ROS by cadmium plays a critical role in cadmium-induced autophagy in skin epidermal cells. Accumulated evidence suggest the involvement of the LKB1-AMPK energy-sensing pathway in the process of autophagy (Alexander et al., 2010; Huang and Shen, 2009; Liang et al., 2007). AMPK is also activated by stresses such as oxidative stress, hypoxia, and nutrient deprivation (Hardie, 2004). Here we found that cadmium induces the phosphorylation of LKB1, AMPKα, and ACC in a dose- and time-dependent manner. Knockdown of LKB1 or AMPKα and transfection with DN-AMPK attenuated the cadmium-induced autophagy. Moreover, our current findings revealed that cadmium-induced autophagy was affected sensitively by changing AMPKα activity in that AMPKα inhibitor, compound C, and AMPKα activator, AICAR, have opposite roles on the autophagy. Considering these results and the previous reports, we suggest that LKB1-AMPK signaling is tightly involved in cadmium-induced autophagy in JB6 cells.
The mammalian target of rapamycin (mTOR) kinase is a major negative regulator of autophagy. It has been known that mTOR signaling is frequently dysregulated in cancer, where LKB/AMPK signaling can act as the upstream of mTOR (Ruggero and Pandolfi, 2003; Sabatini, 2006). Our present data reveal that mTOR signaling is down-regulated in cadmium-exposed JB6 cells in a dose- and time-dependent manner and this is attenuated by transfecting the cells with either si-LKB1 or si-AMPKα. In parallel with these results, the cadmium-mediated decrease in p-mTOR levels was suppressed by compound C treatment or DN-AMPK transfection (Suppl. 3A and B). These results support the involvement of mTOR signaling as the down-stream effecter of LKB1-AMPK pathway in cadmium-induced autophagy.
We previously reported that continuously-generated low levels of ROS caused DNA damage and PARP activation, eventually leading to ATP depletion (Son et al., 2009a; Son et al., 2009b). It is known that PARP-1 regulates autophagy under oxidative stress (Huang and Shen, 2009). It has also been reported that cadmium is capable of producing hydrogen peroxide and that this is a critical step for cell death in cadmium-exposed JB6 cells (Son et al., 2010b). In this regard, we determined whether or not ROS participate in the upstream signaling of LKB1-AMPK pathway in cadmium-exposed cells. As demonstrated by the cadmium-mediated PAR formation and its inhibition either by si-PARP transfection or DPQ treatment with the subsequent increase of cellular ATP levels, our findings suggested a critical role for ROS in a cadmium-induced autophagic pathway. The involvement of ROS in cadmium-induced autophagy was directly supported by the findings from the ESR spectrum and the reduction of p-LKB1, p-AMPKα, and LC3-II levels in the cells over-expressing CAT, SOD1, or SOD2. Similarly, exogenous addition of CAT or SOD to cadmium-treated cells attenuated the phosphorylated levels of these signaling molecules (Suppl. 7A and B). In addition, cadmium-induced ROS-dependent and AMPKα-mediated autophagy signaling were shown by other cell types such as MEF (mouse embryo fibroblasts cells), BEAS-2B (human bronchial epithelial cells), and Jurkat (human T lymphoma cells) (Suppl. 8A–F). The phosphorylation of AMPKα was upregulated by cadmium, corresponding with LC3-II formation in these cells. However, the increasing of AMPKα and LC3-II by cadmium was dramatically attenuated in the CAT, SOD, and SOD2 over-expressing MEF, BEAS-2B and Jurkat cells. These results further supported the critical role of ROS and AMPKα signaling in cadmium-induced autophagy process. Overall, mitochondrial alteration is considered to be involved in cadmium-induced autophagy in that cadmium treatment induced significantly the disruption of mitochondrial membrane potential (MMP) (Suppl. 9A and B).
In summary, the present study demonstrates that cadmium induces autophagy in a dose- and time-dependent manner, where the LKB1-AMPKα signaling pathway plays a critical role. In particular, cadmium generates intracellular ROS at low levels and this leads to PARP activation, resulting in cellular energy depletion, which are the upstream events required for LKB1-AMPK-mediated autophagy in cadmium-exposed epidermal cells. By contrast, mTOR signaling influences cadmium-induced autophagy by acting as the down-stream effector of LKB-AMPK signaling. This study provides a novel finding of autophagy signaling by cadmium and may contribute to further understanding of cadmium-mediated diseases.
Supplementary Material
Highlights.
Cadmium, a toxic heavy metal, induces autophagic cell death through ROS-dependent activat ion of the LKB1-AMPK signaling. Cadmium generates intracellular ROS at low levels and this leads to severe DNA damage and PARP activation, resulting in ATP depletion, which are the upstream events of LKB1-AMPK-mediated autophagy. This novel finding may contribute to further understanding of cadmium-mediated diseases.
Acknowledgments
We thank Dr. Jia Luo (University of Kentucky) for GFP-LC3 plasmid, Dr. J. Suttles (University of Louisville) for CA-AMPKα and DN-AMPKα constructs, and Hong Lin for technical help. This research was supported by NIH grants (R01ES015518, 1R01CA119028, R01ES015375, and 1R01CA116697).
Abbreviations
- ROS
reactive oxygen species
- PARP
poly (ADP-ribose) polymerase
- LC3
microtuble-associated protein 1 light chain 3
- AMPK
AMP-activated protein kinase
- PJS
Peutz-Jeghers syndrome
- EMEM
Eagle’s minimal essential medium
- FBS
fetal bovine serum
- DPQ
3,4-Dihydro-5[4-(1-peperindinyl)butoxy]-1(2H)-isoquinoline
- 3-MA
3-Methyladenine
- CA-AMPK
constitutively active forms of AMPKα
- DN-AMPK
dominant negative forms of AMPKα
- SOD
superoxide dismutase
- CAT
catalase
- si-RNA
small interference RNA
- SSB
single-strand break
- 8-OHdG
8-hydrodeoxyguanosine
- PI
propidium iodide
- ESR
electron spin resonance
- DMPO
5,5-dimethyl-1-pyrroline-1-oxide
- MDC
monodansylcadaverine
- ACC
acetyl CoA carboxylase
- mTOR
mammalian target of rapamycin
- MMP
mitochondrial membrane potential
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, Kusewitt D, Mills GB, Kastan MB, Walker CL. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010;107:4153–4158. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, Carling D, Small CJ. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem. 2004;279:12005–12008. doi: 10.1074/jbc.C300557200. [DOI] [PubMed] [Google Scholar]
- Arends MJ, Morris RG, Wyllie AH. Apoptosis. The role of the endonuclease. Am J Pathol. 1990;136:593–608. [PMC free article] [PubMed] [Google Scholar]
- Dhar A, Young MR, Colburn NH. The role of AP-1, NF-kappaB and ROS/NOS in skin carcinogenesis: the JB6 model is predictive. Mol Cell Biochem. 2002:234–235. 185–193. [PubMed] [Google Scholar]
- Dice JF. Chaperone-mediated autophagy. Autophagy. 2007;3:295–299. doi: 10.4161/auto.4144. [DOI] [PubMed] [Google Scholar]
- Glahn F, Schmidt-Heck W, Zellmer S, Guthke R, Wiese J, Golka K, Hergenroder R, Degen GH, Lehmann T, Hermes M, Schormann W, Brulport M, Bauer A, Bedawy E, Gebhardt R, Hengstler JG, Foth H. Cadmium, cobalt and lead cause stress response, cell cycle deregulation and increased steroid as well as xenobiotic metabolism in primary normal human bronchial epithelial cells which is coordinated by at least nine transcription factors. Arch Toxicol. 2008;82:513–524. doi: 10.1007/s00204-008-0331-9. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hardie DG. The AMP-activated protein kinase pathway--new players upstream and downstream. J Cell Sci. 2004;117:5479–5487. doi: 10.1242/jcs.01540. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, Jarvinen H, Kristo P, Pelin K, Ridanpaa M, Salovaara R, Toro T, Bodmer W, Olschwang S, Olsen AS, Stratton MR, de la Chapelle A, Aaltonen LA. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- Henson MC, Chedrese PJ. Endocrine disruption by cadmium, a common environmental toxicant with paradoxical effects on reproduction. Exp Biol Med (Maywood) 2004;229:383–392. doi: 10.1177/153537020422900506. [DOI] [PubMed] [Google Scholar]
- Hong SJ, Dawson TM, Dawson VL. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol Sci. 2004;25:259–264. doi: 10.1016/j.tips.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Huang Q, Shen HM. To die or to live: the dual role of poly(ADP-ribose) polymerase-1 in autophagy and necrosis under oxidative stress and DNA damage. Autophagy. 2009;5:273–276. doi: 10.4161/auto.5.2.7640. [DOI] [PubMed] [Google Scholar]
- Jarup L. Hazards of heavy metal contamination. Br Med Bull. 2003;68:167–182. doi: 10.1093/bmb/ldg032. [DOI] [PubMed] [Google Scholar]
- Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, Muller O, Back W, Zimmer M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18:38–43. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- Jin T, Lu J, Nordberg M. Toxicokinetics and biochemistry of cadmium with special emphasis on the role of metallothionein. Neurotoxicology. 1998;19:529–535. [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitanaka C, Kuchino Y. Caspase-independent programmed cell death with necrotic morphology. Cell Death Differ. 1999;6:508–515. doi: 10.1038/sj.cdd.4400526. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- Lee JH, Budanov AV, Park EJ, Birse R, Kim TE, Perkins GA, Ocorr K, Ellisman MH, Bodmer R, Bier E, Karin M. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science. 2010;327:1223–1228. doi: 10.1126/science.1182228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM, Mills GB. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- Lim SC, Hahm KS, Lee SH, Oh SH. Autophagy involvement in cadmium resistance through induction of multidrug resistance-associated protein and counterbalance of endoplasmic reticulum stress WI38 lung epithelial fibroblast cells. Toxicology. 2010;276:18–26. doi: 10.1016/j.tox.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, Alessi DR. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–448. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- Luo Z, Saha AK, Xiang X, Ruderman NB. AMPK, the metabolic syndrome and cancer. Trends Pharmacol Sci. 2005;26:69–76. doi: 10.1016/j.tips.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Martin D, Salinas M, Fujita N, Tsuruo T, Cuadrado A. Ceramide and reactive oxygen species generated by H2O2 induce caspase-3-independent degradation of Akt/protein kinase B. J Biol Chem. 2002;277:42943–42952. doi: 10.1074/jbc.M201070200. [DOI] [PubMed] [Google Scholar]
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoshima H, Goldstein BJ, Igata M, Araki E. AMPK and cell proliferation--AMPK as a therapeutic target for atherosclerosis and cancer. J Physiol. 2006;574:63–71. doi: 10.1113/jphysiol.2006.108324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller O, Sattler T, Flotenmeyer M, Schwarz H, Plattner H, Mayer A. Autophagic tubes: vacuolar invaginations involved in lateral membrane sorting and inverse vesicle budding. J Cell Biol. 2000;151:519–528. doi: 10.1083/jcb.151.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, Hamada S, Yoshimori T. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D, Yahalom J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61:439–444. [PubMed] [Google Scholar]
- Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Munz C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–596. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- Patel T, Gores GJ, Kaufmann SH. The role of proteases during apoptosis. FASEB J. 1996;10:587–597. doi: 10.1096/fasebj.10.5.8621058. [DOI] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiori F, Klionsky DJ. Autophagosomes: biogenesis from scratch? Curr Opin Cell Biol. 2005;17:415–422. doi: 10.1016/j.ceb.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3:179–192. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403:139–148. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada H, Shiao YH, Shibata M, Waalkes MP. Cadmium suppresses apoptosis induced by chromium. J Toxicol Environ Health A. 1998;54:159–168. doi: 10.1080/009841098158980. [DOI] [PubMed] [Google Scholar]
- Son YO, Hitron JA, Wang X, Chang Q, Pan J, Zhang Z, Liu J, Wang S, Lee JC, Shi X. Cr(VI) induces mitochondrial-mediated and caspase-dependent apoptosis through reactive oxygen species-mediated p53 activation in JB6 Cl41 cells. Toxicol Appl Pharmacol. 2010a;245:226–235. doi: 10.1016/j.taap.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son YO, Jang YS, Heo JS, Chung WT, Choi KC, Lee JC. Apoptosis-inducing factor plays a critical role in caspase-independent, pyknotic cell death in hydrogen peroxide-exposed cells. Apoptosis. 2009a;14:796–808. doi: 10.1007/s10495-009-0353-7. [DOI] [PubMed] [Google Scholar]
- Son YO, Kook SH, Jang YS, Shi X, Lee JC. Critical role of poly(ADP-ribose) polymerase-1 in modulating the mode of cell death caused by continuous oxidative stress. J Cell Biochem. 2009b;108:989–997. doi: 10.1002/jcb.22332. [DOI] [PubMed] [Google Scholar]
- Son YO, Lee JC, Hitron JA, Pan J, Zhang Z, Shi X. Cadmium induces intracellular Ca2+- and H2O2-dependent apoptosis through JNK- and p53-mediated pathways in skin epidermal cell line. Toxicol Sci. 2010b;113:127–137. doi: 10.1093/toxsci/kfp259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Brenner C, Larochette N, Prevost MC, Alzari PM, Kroemer G. Mitochondrial release of caspase-2 and -9 during the apoptotic process. J Exp Med. 1999;189:381–394. doi: 10.1084/jem.189.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassa A, Roux MP, Attaix D, Bechet DM. Class III phosphoinositide 3-kinase--Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem J. 2003;376:577–586. doi: 10.1042/BJ20030826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waalkes MP, Fox DA, States JC, Patierno SR, McCabe MJ., Jr Metals and disorders of cell accumulation: modulation of apoptosis and cell proliferation. Toxicol Sci. 2000;56:255–261. doi: 10.1093/toxsci/56.2.255. [DOI] [PubMed] [Google Scholar]
- Wang SH, Shih YL, Ko WC, Wei YH, Shih CM. Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol Life Sci. 2008;65:3640–3652. doi: 10.1007/s00018-008-8383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SH, Shih YL, Kuo TC, Ko WC, Shih CM. Cadmium toxicity toward autophagy through ROS-activated GSK-3beta in mesangial cells. Toxicol Sci. 2009;108:124–131. doi: 10.1093/toxsci/kfn266. [DOI] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Overmeyer JH, Maltese WA. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J Cell Sci. 2006;119:259–270. doi: 10.1242/jcs.02735. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.