

Abstract
Rationale
Tramadol is a prescription analgesic that activates mu opioid and monoamine receptor systems. Tramadol is thought to have limited abuse potential compared to mu opioid agonists, but laboratory data indicate that it shares some of their pharmacodynamic effects.
Objectives
This study evaluated the effect of mu opioid receptor blockade with naltrexone on the pharmacodynamic action of tramadol in humans.
Methods
This inpatient, double-blind, randomized, within-subject study examined the effects of oral placebo, tramadol (87.5, 175 and 350 mg) and hydromorphone (4 and 16 mg; positive control) after 1 hr pretreatment with oral naltrexone (0 and 50 mg). Ten recreational opioid users completed the study. Pharmacodynamic effects were measured before and for 7 hr after initial drug administration.
Results
Lower doses of tramadol and hydromorphone were generally placebo-like. Hydromorphone (16 mg) produced prototypic mu opioid agonist-like effects that were blocked by naltrexone. Tramadol (350 mg) produced miosis and increased ratings of “Good Effects” and “Liking ,” but also increased ratings of “Bad Effects.” Naltrexone reversed tramadol-induced physiological effects and mydriasis emerged, but unlike results with hydromorphone, naltrexone only partially attenuated tramadol’s positive subjective effects and actually enhanced several unpleasant subjective ratings.
Conclusions
Naltrexone can be used to disentangle the mixed neuropharmacological actions of tramadol. High dose tramadol produces a mixed profile of effects. These data suggest that both mu and non-mu opioid actions play a role in tramadol’s subjective profile of action.
Keywords: Tramadol, Naltrexone, Hydromorphone, Subjective Effects
Introduction
Illicit use of prescription pain relievers is a persistent public health problem in the United States (U.S.). More individuals over the age of 12 reported past month nonmedical use of prescription pain relievers (5.1 million people) than cocaine (1.5 million people), methamphetamine (353,000 people) or heroin (200,000 people) in 2010 (Substance Abuse and Mental Health Services Administration [SAMHSA], 2011). Most prescription pain relievers used for nonmedical reasons are mu opioid agonists (e.g., (Manchikanti et al., 2010; Mello et al., 1981; SAMHSA, 2007; 2009; Sullivan et al., 2006; Walsh et al., 1996).
Tramadol produces its analgesic effects, in part, through its monaminergic activity in contrast to prototypic opioid analgesics (Epstein et al., 2006; Grond and Sablotzki, 2004). Prior to the U.S. Food and Drug Administration (FDA) approval of tramadol for use in mild-to-moderate pain, it had widespread use in Germany with few cases of reported abuse (Radbruch et al., 1996). An early human laboratory abuse liability study with parenteral tramadol also indicated that it had limited abuse potential compared to morphine (Preston et al., 1991). These data and the German experience likely played a significant role in the U.S. decision to approve tramadol as an unscheduled substance. However, since tramadol’s approval in the U.S. there have been reports of its abuse and misuse (Cicero et al., 2005; Schneider et al., 2009). These data have led to some individual states, including Kentucky and Arkansas, placing tramadol on the list of controlled substances. Tramadol is a racemic mixture. The + and − enantiomers of tramadol inhibit serotonin and norepinephrine reuptake, respectively (Matthiesen et al., 1998). These isomers bind only weakly to the mu opioid receptor, but are metabolized by cytochrome P450 2D6 (CYP2D6) enzymes into o-desmethyltramadol (M1; Driessen et al., 1993; Frink et al., 1996; Gillen et al., 2000; Raffa et al., 1992; Stamer et al., 2007; Volpe et al., 2011). M1 has a comparatively higher affinity for the mu receptor than tramadol (Raffa et al., 1992; Volpe et al., 2011).
To date, pre-clinical studies suggest that the abuse-related behavioral effects of tramadol are similar to those of other full mu opioid receptor agonists, but generally of lesser magnitude. For example, rats and rhesus monkeys self-administer tramadol to a greater degree than vehicle, albeit less than has been observed with prototypic opioids (O’Connor and Mead, 2010; Yanagita, 1978). Other preclinical research has shown that tramadol engenders significant conditioned place preference like morphine (Sprague et al., 2002; Tzschentke et al., 2002) and drug discrimination studies show cross-generalization between morphine and tramadol (Filip et al., 2004; Ren and Zheng, 2000; Swedberg et al., 1988; 1992).
Clinical studies show similar results in that orally administered tramadol also produces some effects that overlap with prototypic opioid effects (Carroll et al., 2006; Duke et al., 2011; Jasinski et al., 1993; Lanier et al., 2010; Lofwall et al., 2007; Zacny, 2005). For instance, tramadol and oxycodone produced comparable increases in ratings of “Liking” (Jasinski et al., 1993), single oral tramadol doses (200 and 400 mg) decreased opioid withdrawal among subjects physically dependent on morphine (Lofwall et al. 2007), naloxone precipitated opioid withdrawal symptoms among subjects maintained on oral tramadol (Lanier et al. 2010) and higher (but not lower) doses of tramadol substituted for hydromorphone in a drug discrimination procedure, but did not significantly increase positive subjective ratings (e.g., “Liking,” “Good Effects”) as did hydromorphone (Duke et al., 2011).
The similarities between tramadol and prototypic mu opioids implicate the mu opioid receptor in the behavioral effects of tramadol. To determine more fully how activation of the mu opioid receptor influences its behavioral pharmacology, preclinical studies have tested the impact of mu opioid receptor blockade with naloxone or naltrexone on the effects of tramadol (Filip et al., 2004; Ren and Zheng, 2000; Tzschentke et al., 2002). Those studies showed that mu opioid receptor blockade attenuated discriminative-stimulus and conditioned place preference effects of tramadol, suggesting that mu opioid receptor activation is primarily responsible for the behavioral effects of tramadol. To date, there are no published studies that have tested mu opioid receptor blockade on the acute pharmacodynamic effects of tramadol in non-dependent humans. This study sought to determine how naltrexone pretreatment influences the pharmacodynamic actions of tramadol. Thus, ten non-opioid dependent adults with recent histories of recreational opioid use completed an inpatient, double-blind, randomized, within-subject protocol whereby the effects of oral tramadol (0, 87.5, 175 and 350 mg) were assessed after pretreatment with oral placebo and naltrexone (50 mg). Oral hydromorphone (4 and 16 mg) served as a positive control. It was hypothesized that tramadol would produce effects comparable to hydromorphone but of lower magnitude and that naltrexone administration would attenuate the effects of both tramadol and hydromorphone.
Methods
Participants
Non-physically dependent adult recreational opioid users were recruited and compensated for participation. Individuals seeking treatment for substance abuse or successfully sustaining abstinence were excluded. All participants were in good health verified by medical history and physical examination, an electrocardiogram and laboratory tests. Persons with seizure disorders, asthma or other respiratory disorders, head injury, hypertension, cardiovascular disease or an abnormal ECG were excluded. All participants reported illicit use of opioids, which was confirmed by urinalysis. This University of Kentucky Institutional Review Board approved the study. Participants gave their written informed consent; the consent document stated that oral placebo, tramadol, hydromorphone and naltrexone would be administered. A Certificate of Confidentiality was obtained from the National Institute on Drug Abuse. The study was conducted in accordance with the Helsinki guidelines.
This inpatient study lasted approximately 6 weeks. Sessions were conducted at least 72 hr apart (e.g., Monday and Thursday). Prior to each session, urine specimens and breath samples were obtained and tested for illicit drugs to ensure the absence of unauthorized substance use. Females were tested for pregnancy prior to each session. Participants fasted for 2 hr and abstained from smoking cigarettes for 1 hr prior to the start of sessions; they were not allowed to eat or smoke during sessions.
Drugs
All drugs were administered in random order. Tramadol (87.5, 175 and 350 mg; UDL Labs, Rockford, IL), hydromorphone (4 and 16 mg; Halo Pharmaceuticals, Whippany, NJ) and naltrexone (50 mg; Barr Labs, Pomona, NY) were prepared by over-encapsulating drug with cornstarch filler in opaque gelatin capsules. Placebo capsules contained only cornstarch. The intermediate and high doses of tramadol were selected to match those used in a previous study (Jasinski et al., 1993); the low dose was selected so that a four-fold range of doses could be tested, including a dose in the therapeutic range. The hydromorphone doses were selected to be roughly equipotent to the low and high tramadol doses based on previous research (Jasinski et al., 1993; Walsh et al., 2008a). The naltrexone dose was selected because it provides near complete mu opioid blockade (Lee et al., 1988; Walsh et al., 1996). The first dose containing placebo or naltrexone was administered at approximately 9:00 a.m. (i.e., time point 0; Table 1). The second dose containing placebo, tramadol or hydromorphone was administered 1 h later (i.e., time point 60; Table 1).
Table 1.
Data collection schedule during experimental sessions.
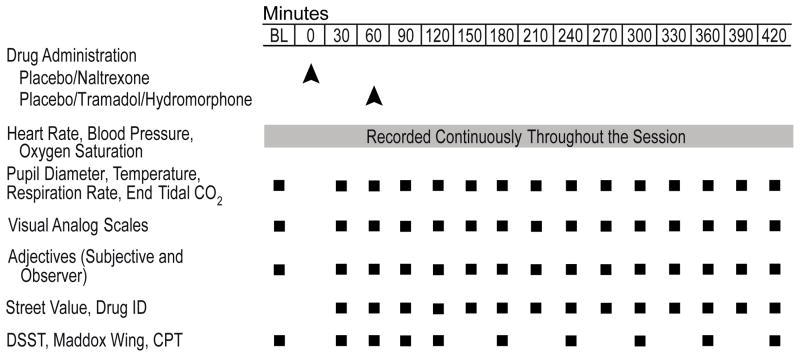
|
Study design
A double-blind, within-subject, placebo-controlled design was used. Twelve sessions were conducted at the University of Kentucky Clinical Research Development and Operations Center (CRDOC) during which participants received one of the twelve possible dose combinations (i.e., placebo or 50 mg naltrexone combined with placebo, 87.5, 175 or 350 mg tramadol or 4 or 16 mg hydromorphone). Baseline data were collected for 30 min before (i.e., 8:30 a.m.) and continued for 7 hr after drug administration (see Table 1).
Experimental sessions
Data collection techniques were similar to those described previously (Stoops et al., 2010). Table 1 illustrates the data collection timeline for all measures. The dosing intervals and data collection time points were selected 1) to allow naltrexone to reach Tmax prior to the onset of expected opioid agonist effects and 2) to capture the onset, peak and offset of effects of tramadol, M1 and hydromorphone.
Physiological measures
Oxygen saturation, blood pressure and heart rate were collected every min, except during the Continuous Performance Task, using an automatic monitoring device (Scholar III model 507ELC2, Criticare Systems INC, Waukesha, WI). Pupil diameter was determined with a pupillometer (PLR-200, NeurOptics, Irvine, CA) in constant room lighting. Expired CO2 (mm Hg) and respiratory rate (breaths/min) were measured using a capnograph (N85, Nellcor, Boulder, CO).
Subject- and observer-rated measures
Subject-rated measures included a visual analog scale (VAS; Walsh et al., 2008a), a Street Value Questionnaire, a Drug Identification Questionnaire and a 51-item adjective checklist that encompassed an Opioid Agonist Scale and Withdrawal Scale (see Walsh et al., 1995 for those scale descriptions). Items sensitive to the effects of norepinephrine reuptake inhibitors (i.e., atomoxetine and desipramine; Heil et al., 2002; Jasinski et al., 2008) and serotonin reuptake inhibitors (Vanderkooy et al., 2002) also were included in this checklist. The items were “Abdominal Pain,” “Agitated,” “Bad,” “Blurred Vision,” “Decreasing Your Appetite,” “Dizzy,” “Drowsy,” “Dry Mouth,” “Fatigued or Lazy,” “Flushing,” “Headache,” “Heavy or Sluggish Feeling,” “Nervous,” “Relaxed,” “Sick,” “Sleepy,” “Stimulated,” “Sweating,” “Turning of Stomach,” “Twitch,” and “Weak,” scored from 0 to 4 on a Likert-type scale. An observer-rated Opioid Agonist adjective rating scale also was used (Walsh et al., 2008a).
Ocular and performance tasks
The Maddox Wing ocular task (see Walsh et al., 2008a), computerized Digit Symbol Substitution Task (DSST; McLeod et al., 1982) and Continuous Performance Task (CPT; Martin et al., 2007) also were completed.
Statistical Analysis
All measures were analyzed initially as raw time course data using a two-factor analysis of variance (ANOVA with Proc Mixed to account for any missing data; dose × time). SAS 9.2 software for Windows was used and results were considered significant when p < 0.05. The online physiological measures, collected at 1-min intervals, were averaged across time to yield 30-min intervals. Dunnett's post hoc tests were used to compare scores between active doses and placebo across time points. When a significant interaction of dose and time was observed, significant main effects are not reported. Outcomes with only a significant main effect of time also are not reported.
In addition, individual peak scores analyses (either nadir or maximum depending upon the direction of effects, with the exception of pupil diameter, which was analyzed as both nadir and maximum) were completed using a one-factor ANOVA (dose). Raw values were used to calculate peak, with the exception of oxygen saturation, heart rate, blood pressure, respiration rate and expired CO2, which were calculated as change from baseline. Dunnett's post hoc tests were used to compare peak scores between active doses and placebo.
Drug identification data were analyzed by determining the most commonly identified drug (i.e., the mode) for each dose condition. If the drug was identified as an opioid, the mode for the type of opioid also was determined.
Results
Participants
Fifteen participants screened for the study, thirteen were qualified and admitted and ten completed the study. One of those screened was lost to follow up and one failed to meet inclusion/exclusion criteria due to opioid physical dependence. Three participants left early after admission for reasons unrelated to the protocol: one was medically discharged due to development of a toothache, one left due to craving opioids and one left due to childcare problems. Only data from the ten completers (six male, four female) were analyzed. Nine were Caucasian and one was of mixed race (Asian and Caucasian). Participants were 37 ± 8 (mean ± SD) years old. All reported current recreational use of prescription opioids with a mean of 9 days using per month (± 2). The most common opioids used were oxycodone and hydrocodone. One participant also had used heroin in the month preceding screening. Eight participants were daily cigarette smokers and five reported current alcohol use (10 ± 7 days out of the past 30). Other drug use in the 30 days prior to screening reported on the Addiction Severity Index and other screening instruments was common. Seven participants reported use of benzodiazepines (3 ± 3 days out of the past 30), five reported marijuana use (19 ± 14 days out of the past 30), four reported cocaine use (7 ± 4 days out of the past 30) and one reported amphetamine use (4 days out of the past 30). No subject was physically dependent on any drug requiring detoxification.
Time Course
Physiological Measures
As shown in the top panels of Figure 1, tramadol and hydromorphone produced dose related decreases in pupil diameter, with the highest dose of tramadol and both doses of hydromorphone decreasing pupil diameter significantly compared to placebo. Miosis was generally evident within 1–1.5 hr of hydromorphone administration (i.e., at times 120–150), but appeared later after the higher doses of tramadol (i.e., 2.5 hr after dosing at time 210); in both cases, pupil constriction persisted throughout the 6-hr post dosing period of the sessions. The reductions in pupil diameter following tramadol were of much smaller magnitude than those produced by hydromorphone. As shown in the bottom panels of Figure 1, naltrexone blocked the miosis produced by 350 mg tramadol and 16 mg hydromorphone and, while not statistically significant, tramadol actually increased pupil diameter (i.e., mydriasis) after naltrexone pretreatment compared to placebo. Naltrexone also blocked the miosis produced by the lower tramadol dose and hydromorphone dose (data not shown).
Figure 1.

Data are shown for mean values (n=10) for pupil diameter for placebo (PLB) and tramadol (TRAM) doses following PLB pretreatment (top left panel), PLB and 350 mg TRAM following PLB and naltrexone (NTX) pretreatment (bottom left panel), PLB and hydromorphone (HM) doses following PLB pretreatment (top right panel) and PLB and 16 mg HM following PLB and NTX pretreatment (bottom right panel) as a function of time (X-axis) since drug administration in the 7-hr session. NTX/PLB pretreatment was administered at Time 0 and the arrows denote when PLB/TRAM/HM was administered. Time course analysis revealed a significant interaction of dose and time condition for pupil diameter (F154,1384 = 1.3, p = 0.01). Filled symbols indicate a significant difference from the corresponding PLB time point.
Significant main effects of dose (F11,99 = 2.0, p = 0.03) and time (F14,126 = 3.5, p < 0.0001) were observed for respiration rate but there were no consistent differences detected in post hoc testing between placebo and active doses (data not shown). There were no significant findings for the other physiological measures. However, one notable finding, which was not analyzed statistically, was that subjects vomited more often during sessions testing higher doses of tramadol compared to sessions testing hydromorphone. Specifically, across all 120 sessions in the study, subjects vomited in a total of twelve sessions. Vomiting occurred in seven sessions following the two higher tramadol doses combined with naltrexone, four sessions following the two higher tramadol doses combined with placebo and one session following the high hydromorphone dose combined with placebo.
Subject- and observer-rated measures
Time-dependent increases were observed on VAS ratings of “Liking” after administration of the high tramadol and hydromorphone doses relative to placebo, although the effects of tramadol were of lower magnitude than those of hydromorphone (Figure 2-top left panel). These effects were evident 1 hr after dosing (i.e., at time 120) and ratings were elevated throughout the remainder of the session. Ratings produced by 350 mg tramadol were generally no longer statistically different from placebo with naltrexone pretreatment; however, the absolute time course and magnitude of ratings were quite similar to the effects of 350 mg tramadol in combination with placebo. The lack of a statistically significant effect for the combination of 350 mg tramadol and naltrexone could be due to increased inter-individual variability. Naltrexone completely blocked ratings of “Liking” produced by 16 mg hydromorphone. The lower tramadol and hydromorphone doses were placebo-like after placebo and naltrexone pretreatment (data not shown). A similar pattern of effects was observed for 350 mg tramadol combined with naltrexone for ratings of “Drug Effect” (data not shown), “High” and “Good Effects” as well as Street Value (Figure 2).
Figure 2.

Data are shown for mean values (n=10) for ratings of “Liking” (top left panel), “Good Effects” (bottom left panel), “High” (top right panel) (maximum = 100 for those items) and Street Value estimates (bottom right panel) for PLB (circles), 350 mg TRAM and 16 mg HM following PLB (squares for TRAM; flat-bottomed triangles for HM) and NTX (diamonds for NTX alone; hexagons for TRAM; flat-topped triangles for HM) pretreatment as a function of time (X-axis) since drug administration in the 7 hr session. Time course analysis revealed significant main effects of dose (F11,99 values > 2.3, p values < 0.05) and time (F14,126 values > 5.5, p values < 0.0001) for “Liking,” “Good Effects” and “High”. A significant interaction of dose and time (F143,1287 = 1.62, P < 0.0001) was observed for Street Value. All other details are the same as for Figure 1.
An interaction of dose and time (F154,1386 = 1.3, p < 0.01) was observed for “Bad Effect”ratings with high dose tramadol transiently increasing “Bad Effect” ratings relative to placebo after placebo pretreatment, but generally not after naltrexone pretreatment (data not shown). “Bad Effect” ratings also were elevated relative to placebo when the intermediate dose of tramadol was combined with naltrexone (data not shown).
Statisically significant outcomes were observed on the Withdrawal Scale (main effects of dose and time; F11,99 = 2.2, p = 0.02 and F14,126 =5.9, p < 0.0001, respectively) and ratings of “Abdominal Pain” (main effect of dose F11,99 = 2.7, p = 0.004), “Bad,” “Sick,” “Heavy or Sluggish Feeling,” “Dry Mouth” (main effects of dose and time; F11,99 values > 2.1, p values < 0.05 and F14,126 values > 2.3, p values < 0.05, respectively), “Flushing,” “Twitch,” and “Turning of Stomach” (interaction of dose and time; F154,1385 values > 1.4, p values ≤ 0.001). Low dose tramadol and hydromorphone were placebo-like for all items after both placebo and naltrexone pretreatment (data not shown). Relative to placebo, high dose tramadol and hydromorphone increased ratings of “Dry Mouth” after placebo, but not naltrexone, administration (data not shown). These doses generally produced no significant effects on the Withdrawal Scale or individual items from the adjective checklist after placebo pretreatment, but naltrexone enhanced the effects of 175 and 350 mg tramadol on those measures compared to placebo. Supplementary Figure 1 shows representative ratings for 175 and 350 mg tramadol and 16 mg hydromorphone for “Sick,” “Turning of Stomach” and “Heavy or Sluggish Feeling”. No significant dose-related main effects or interactions were observed for the subject- or observer-rated Agonist Scales.
Ocular and performance tasks
There were no significant effects on these measures.
Peak Effects
Table 2 presents the average nadir or maximum values for outcomes with statistically significant findings from peak analyses.
Table 2.
| Naltrexone | Tramadol | Tramadol with Naltrexone | Hydromorphone | Hydromorphone with Naltrexone | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Outcome Measure | F | Placebo | 50mg | 87.5mg | 175mg | 350mg | 87.5mg | 175mg | 350mg | 4mg | 16mg | 4mg | 16mg |
| Physiological | |||||||||||||
| End Tidal C02 | 3.3 | 3.2 (0.6) | 3.0 (0.7) | 4.8 (0.9) | 5.1 (0.7) | 7.1 (0.9) | 3.8 (1.0) | 4.7 (1.0) | 4.9 (0.9) | 4.7 (0.7) | 5.6 (1.0) | 3.1 (0.6) | 4.1 (1.1) |
| Oxygen Saturation * | 2.8 | −0.5 (0.2) | −0.3 (0.3) | −0.7 (0.2) | −1.1 (0.3) | −1.5 (0.3) | −0.7 (0.2) | −1.0 (0.2) | −0.7 (0.3) | −1.1 (0.2) | −1.7 (0.4) | −0.8 (0.2) | −1.0 (0.2) |
| Systolic Pressure | 2.6 | 6.6 (1.9) | 12.3 (2.4) | 10.7 (2.5) | 8.7 (2.0) | 13.9 (3.5) | 8.6 (2.7) | 11.6 (3.1) | 14.2 (2.2) | 6.4 (2.1) | 13.5 (2.7) | 5.2 (2.7) | 3.7 (1.6) |
| Pupil Diameter * | 10.3 | 4.3 (0.3) | 4.0 (0.3) | 4.2 (0.2) | 4.1 (0.3) | 3.8 (0.2) | 4.3 (0.3) | 4.1 (0.3) | 4.2 (0.3) | 3.6 (0.2) | 3.0 (0.2) | 4.0 (0.2) | 4.1 (0.3) |
| Pupil Diameter | 8.1 | 5.3 (0.3) | 4.9 (0.3) | 5.3 (0.3) | 5.2 (0.4) | 5.3 (0.3) | 5.4 (0.3) | 5.4 (0.3) | 5.7 (0.3) | 4.8 (0.3) | 4.9 (0.3) | 5.0 (0.3) | 5.0 (0.3) |
| Subject-rated | |||||||||||||
| Visual analogs | |||||||||||||
| High | 2.6 | 3.5 (3.2) | 1.1 (1.1) | 9.5 (6.9) | 6.7 (4.9) | 18.0 (10.8) | 13.1 (9.3) | 11.2 (9.9) | 24.2 (11.9) | 5.0 (3.2) | 32.9 (10.6) | 2.4 (1.2) | 1.1 (0.8) |
| Drug effect | 3.2 | 6.8 (4.9) | 6.8 (3.5) | 10.0 (6.2) | 8.8 (4.8) | 20.7 (10.3) | 15.0 (9.2) | 22.0 (10.1) | 32.4 (10.2) | 4.8 (2.4) | 34.0 (10.4) | 4.6 (1.7) | 3.5 (1.6) |
| Good effect | 2.5 | 5.3 (5.0) | 2.1 (1.4) | 9.7 (7.1) | 6.9 (4.9) | 18.2 (10.8) | 12.7 (9.4) | 11.9 (9.8) | 24.5 (11.9) | 5.1 (2.8) | 34.0 (10.6) | 2.8 (1.3) | 2.5 (1.1) |
| Bad effect | 2.8 | 1.6 (1.0) | 7.3 (5.4) | 1.4 (0.6) | 2.3 (1.5) | 19.9 (11.9) | 2.6 (1.2) | 21.4 (6.6) | 13.6 (5.2) | 0.6 (0.4) | 4.2 (2.1) | 1.9 (1.0) | 0.7 (0.7) |
| Like | 2.5 | 5.2 (5.0) | 1.3 (1.3) | 9.7 (7.2) | 6.4 (4.6) | 19.1 (10.8) | 13.1 (10.0) | 11.0 (9.9) | 20.6 (11.9) | 4.5 (2.5) | 34.7 (10.1) | 2.0 (1.0) | 1.7 (0.9) |
| Street Value | 2.8 | 1.1 (1.0) | 0.2 (0.2) | 2.9 (1.1) | 2.5 (1.1) | 6.6 (3.0) | 5.7 (4.0) | 6.3 (6.0) | 10.8 (5.8) | 4.2 (3.0) | 16.0 (3.8) | 1.7 (1.0) | 0.6 (0.5) |
| Adjectives | |||||||||||||
| Turning of Stomach | 4.5 | 0.1 (0.1) | 0.2 (0.1) | 0.6 (0.2) | 0.5 (0.2) | 1.3 (0.4) | 0.5 (0.2) | 1.0 (0.3) | 1.5 (0.3) | 0.3 (0.2) | 0.6 (0.2) | 0.2 (0.1) | 0.2 (0.1) |
| Dry Mouth | 2.8 | 0.2 (0.1) | 0.1 (0.1) | 0.1 (0.1) | 0.3 (0.2) | 1.0 (0.4) | 0.1 (0.1) | 0.4 (0.2) | 0.5 (0.2) | 0.2 (0.1) | 0.7 (0.3) | 0.1 (0.1) | 0.2 (0.1) |
| Flushing | 2.1 | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.2 (0.1) | 0.1 (0.1) | 0.0 (0.0) | 0.1 (0.1) | 0.3 (0.2) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
| Abdominal Pain | 2.0 | 0.0 (0.0) | 0.3 (0.2) | 0.0 (0.0) | 0.0 (0.0) | 0.5 (0.3) | 0.0 (0.0) | 0.1 (0.1) | 0.1 (0.1) | 0.2 (0.1) | 0.4 (0.2) | 0.1 (0.1) | 0.0 (0.0) |
| Bad | 2.2 | 0.2 (0.2) | 0.3 (0.2) | 0.6 (0.4) | 0.6 (0.4) | 0.6 (0.3) | 0.1 (0.1) | 1.2 (0.4) | 1.2 (0.4) | 0.0 (0.0) | 0.2 (0.1) | 0.3 (0.2) | 0.5 (0.3) |
| Sick | 2.8 | 0.0 (0.0) | 0.0 (0.0) | 0.4 (0.2) | 0.6 (0.4) | 0.9 (0.4) | 0.1 (0.1) | 0.9 (0.4) | 1.2 (0.4) | 0.0 (0.0) | 0.3 (0.2) | 0.1 (0.1) | 0.3 (0.2) |
| Relaxed | 1.9 | 1.0 (0.3) | 1.0 (0.3) | 1.4 (0.3) | 1.4 (0.2) | 2.0 (0.4) | 1.4 (0.3) | 1.7 (0.3) | 1.3 (0.3) | 1.2 (0.2) | 1.6 (0.3) | 1.2 (0.3) | 1.1 (0.2) |
| Opioid agonist scale | 3.2 | 7.6 (1.2) | 7.4 (1.3) | 8.6 (1.8) | 9.7 (1.3) | 12.1 (2.3) | 9.3 (1.5) | 11.3 (2.3) | 10.3 (1.6) | 8.4 (1.2) | 13.3 (2.2) | 9.7 (1.4) | 6.8 (1.1) |
| Opioid withdrawal scale | 2.2 | 3.5 (0.8) | 3.1 (0.9) | 3.5 (0.7) | 3.9 (1.0) | 5.8 (2.4) | 3.5 (0.9) | 7.5 (1.5) | 7.2 (1.6) | 3.1 (0.8) | 4.6 (1.2) | 3.2 (0.8) | 3.6 (1.4) |
| Performance | |||||||||||||
| CPT (hits) * | 2.2 | 85.5 (7.4) | 79.1 (7.4) | 70.5 (10.3) | 90.9 (8.6) | 69.9 (10.8) | 77.6 (11.6) | 83.3 (9.2) | 53.1 (11.6) | 90.2 (8.6) | 80.5 (7.6) | 81.2 (8.4) | 83.1 (11.6) |
| CPT (misses) * | 2.4 | 12.9 (6.1) | 16.5 (6.0) | 16.3 (6.6) | 11.6 (5.9) | 17.3 (7.4) | 14.5 (7.1) | 16.6 (7.3) | 22.2 (7.7) | 13.9 (6.5) | 18.2 (6.5) | 15.5 (6.8) | 12.6 (6.4) |
| Observer-rated | |||||||||||||
| Opioid agonist scale | 2.2 | 8.5 (0.3) | 8.4 (0.6) | 9.2 (0.4) | 9.3 (0.9) | 9.3 (0.7) | 9.0 (0.5) | 9.4 (0.8) | 9.1 (0.5) | 9.3 (0.5) | 10.4 (0.5) | 8.1 (0.5) | 8.7 (0.5) |
Measures were analyzed as peak maximum score unless nadir is indicated (*). Values are mean (SEM) scores (n = 10) for placebo, naltrexone, tramadol and hydromorphone.
Physiological Measures
Compared to placebo, tramadol (350 mg) and both hydromorphone doses decreased nadir pupil diameter after placebo but not naltrexone pretreatment. Both hydromorphone doses decreased maximum pupil diameter relative to placebo, which was blocked by naltrexone pretreatment. Significant mydriasis was observed when the high tramadol dose was combined with naltrexone compared to placebo. High dose tramadol increased maximum expired CO2 and both high dose tramadol and hydromorphone decreased nadir oxygen saturation (all change from baseline scores), compared to placebo, following placebo but not naltrexone pretreatment. An effect also was observed on systolic blood pressure (change from baseline), but post hoc testing revealed no active versus placebo differences. No significant effects were observed for other physiological measures.
Subject- and observer-rated measures
Significant dose effects were observed on several subject- and observer-rated measures (Table 2). Compared to placebo, hydromorphone (16 mg) increased ratings on VAS ratings (i.e., “High,” “Drug Effect,” “Good Effect” and “Liking,” Opioid Agonist Scale) and Street Value; naltrexone blocked these effects. Tramadol (350 mg) increased peak Opioid Agonist Scale ratings (mean peak: 12.1) following placebo pretreatment that were similar in magnitude to hydromorphone 16 mg (mean peak: 13.3); this test condition also resulted in increased ratings of “Bad Effect,” “Turning of Stomach,” “Dry Mouth” and “Relaxed” that were not observed after hydromorphone 16 mg. Tramadol (350 mg) with naltrexone pretreatment increased “Drug Effect” ratings and significantly increased unpleasant feelings that, with the exception of “Turning of Stomach,” were different than those observed when this dose was combined with placebo (i.e., “Flushing” and “Sick”). Compared to placebo, the intermediate tramadol dose produced no significant effects following placebo pretreatment but increased ratings of “Bad Effects” and “Turning of Stomach” after naltrexone pretreatment. A significant effect also was observed on the Opioid Withdrawal Scale, but post hoc testing revealed no active versus placebo differences.
Ocular and Performance Tasks
A significant effect of dose was observed for nadir hits and misses on the CPT (Table 2) task only, whereby tramadol 350 mg with naltrexone decreased hits and increased misses.
Drug Identification
Only high dose tramadol and hydromorphone given with placebo pretreatment were identified as an opioid. The mode drugs identified following tramadol were tramadol and hydromorphone, whereas hydromorphone was identified most commonly as hydromorphone. All other dose conditions, including doses combined with naltrexone, were identified as placebo.
Discussion
The primary findings of this experiment were 1) hydromorphone produced prototypic mu opioid agonist-like effects (e.g., miosis, increased ratings of “Liking”) that were dose-dependent and were blocked by naltrexone, 2) tramadol produced some effects consistent with mu opioid receptor activation (e.g., miosis, increased ratings of “Liking”), which were of reliably lower magnitude compared to hydromorphone despite the testing of supratherapeutic doses, but also increased ratings of “Bad Effects,” 3) naltrexone blocked tramadol-induced physiological effects (e.g., miosis; reduced oxygen saturation) and unveiled pharmacodynamic effects consistent with tramadol’s monoaminergic activity (i.e., mydriasis, increased ratings of “Flushing” and “Sick,” increased vomiting) and 4) naltrexone pretreatment attenuated, but did not completely block, the positive subjective effects of tramadol observed over time (e.g., “Liking,” “Good Effects”), which is in contrast to the complete blockade of the positive subjective effects of hydromorphone by naltrexone.
Hydromorphone (16 mg) alone, the positive opioid control condition, reliably produced prototypic opioid effects, which were completely blocked by naltrexone, consistent with previous work (Preston and Bigelow, 1993; Schuh et al., 1999; Walsh et al., 1996). These results demonstrate that there was pronounced mu opioid agonist blockade produced by the dose of naltrexone employed here (50 mg, p.o.), validating the experimental method.
The onset of subjective effects of tramadol occurred within approximately 1 hr of dosing, consistent with the pharmacokinetic profile of the parent drug, whereas the miotic effects of tramadol appeared later (i.e., within approximately 2.5 h), which is more consistent with the time action profile of M1 (i.e., the Tmax of M1 is approximately 2 hr after dosing; Liao et al., 1992; Lintz et al., 1986; Rouini et al., 2006). These findings may suggest that the parent drug is responsible for producing the early reported subjective profile, while M1 is responsible for producing miosis as previously suggested (Lofwall et al 2007). Other outcomes observed herein with tramadol under the condition of naltrexone blockade further support this suggestion. For example, for 350 mg tramadol, naltrexone blocked miosis and mydriasis occurred (see Table 2), which on visual inspection, appeared to occur within 1 hr after tramadol administration in contrast to the later emergence of miosis with tramadol alone (see Figure 1).
Naltrexone only partially attenuated the time course and magnitude of most of the positive subjective effects of tramadol (e.g., “Liking,” “Good Effects,” in Figure 2). This finding, in combination with the early onset of tramadol subjective effects (consistent with the parent drug kinetics), suggest that these effects may not be mediated entirely by action at the mu opioid receptor. There is a striking similarity of the time course and magnitude of subjective effects produced by high dose tramadol following placebo and naltrexone, which is in contrast to the time course results for high dose hydromorphone following placebo and naltrexone (see Figure 2). These results support the idea that there may be a non-mu mechanism of action, in addition to mu opioid activity, which contributes to the overall constellation of positive subjective effects of this tramadol dose.
Interestingly, while neither tramadol nor naltrexone alone significantly increased scores on the Opioid Withdrawal Scale or items sensitive to norepinephrine or serotonin reuptake inhibition (e.g., “Flushing”), the naltrexone-tramadol combination did increase scores on these measures. Increases on the Withdrawal Scale are not likely due to actual opioid withdrawal as these were not physically dependent individuals, but rather are attributable to symptoms of opioid withdrawal that also overlap with monoamine reuptake blockade effects, such as “Turning of Stomach” and “Flushing” (e.g., flushing is similar to hot flashes that can be part of opioid withdrawal). Vomiting occurred most commonly in naltrexone-tramadol sessions; gastrointestinal side effects such as nausea and vomiting are not uncommon side effects of serotonin reuptake inhibitors, particularly if started at a high dose without an initial dose run-up (e.g., fluoxetine). Also, norepinephrine reuptake inhibitors cause mydriasis, which was only observed for tramadol after naltrexone administration (Matouskova et al., 2011; see also Laeng et al., 2012). Taken together, these outcomes suggest that naltrexone blockade allowed the serotonergic and noradrenergic effects of tramadol to emerge or to become more evident and/or enhanced.
These data suggest that high doses of tramadol produce a signal of abuse potential, with increases on some prototypic measures such as “Liking” and increased Street Value ratings, but that this effect is seen only at supratherapeutic doses for which the profile also includes negative subjective effects (i.e., increased ratings of “Bad Effects”). Moreover, tramadol was distinguishable from hydromorphone by its lower potency (and likely lower intrinsic activity; Volpe et al., 2011), its slower time to reach maximum physiological effect and the participants’ ability to distinguish correctly between the two drugs. These findings are generally concordant with conclusions in the literature that tramadol has reduced abuse potential relative to prototypic opioid analgesics (e.g., Epstein et al., 2006; Raffa, 2008).
Naltrexone pretreatment resulted in an emergence of negative subjective effects for the intermediate tramadol dose (e.g., peaks for “Bad Effects,” “Turning of Stomach”) and changed the negative subjective effects observed after the high tramadol dose (e.g., peaks for “Flushing,” “Sick”) relative to those observed following placebo pretreatment (e.g., peaks for “Bad Effect,” “Dry Mouth”), which indicate that the relative degree and type of bad effects of tramadol were altered by administration of an opioid antagonist. This outcome is supportive of previous research implicating the mu opioid receptor in several of the behavioral effects of tramadol (Filip et al., 2004; Lanier et al., 2010; Lofwall et al., 2007; Ren and Zheng, 2000; Tzschentke et al., 2002). For example, in one human laboratory study, naloxone administration precipitated withdrawal in tramadol-maintained subjects (Lanier et al., 2010). In another study, while tramadol produced negative effects comparable to those observed here, it also suppressed withdrawal among morphine-maintained subjects in a time-dependent manner consistent with the pharmacokinetic profile of M1 (Lofwall et al., 2007). These data indicate that some effects of tramadol/M1, particularly those related to opioid physical dependence and withdrawal suppression, are likely mediated by opioid receptors. However, in contrast to prevailing opinion, because naltrexone pretreatment did not completely block the positive, abuse-related subjective effects of 350 mg tramadol, unlike the complete attenuation that was observed in the positive control condition, it is possible that its subjective profile of effects is not wholly attributable to mu opioid receptor activation. This conclusion is inconsistent with the results of preclinical studies implicating the mu opioid receptor in the rewarding effects of tramadol (Ide et al., 2006; Tzschentke et al., 2002). Those studies suggest that the conditioned place preference effects of tramadol are 1) reduced in mu opioid receptor knockout mice relative to wildtype mice (Ide et al., 2006) and 2) attenuated by naloxone administration (Tzschentke et al., 2002). The finding also is inconsistent with reports that neither serotonin nor norepinephrine reuptake inhibitors have abuse potential (Heil et al., 2002; Jasinski et al., 2008; Yen and Fuller, 1992). It is possible that tramadol produces positive subjective effects through some other pharmacological mechanism. For example, drugs that block reuptake of serotonin or norepinephrine also often interact with the dopamine transporter (e.g., Daws, 2009) and dopamine reuptake blockade is associated with abuse-related subjective effects (Volkow et al., 1997); however, tramadol has not been shown to block dopamine reuptake (Raffa et al., 1992). Overall, the present findings and extant literature lend support to the conclusion that tramadol produces its effects, including those related to abuse liability, through mixed activation of mu opioid and monoamine receptors.
In this study, there were some discrepancies between time course and peak effect statistical outcomes, particularly for positive subjective effects of tramadol (e.g., tramadol significantly increased ratings of “Good Effects” after placebo and naltrexone in the time course, but not the peak effect, analysis). Following placebo and naltrexone pretreatment, tramadol did increase peak effect ratings for positive subjective effects, with higher magnitude ratings observed in the tramadol/naltrexone conditions, but these outcomes failed to reach statistical significance. This failure to reach significance relative to placebo could be due to the reduced degrees of freedom in the peak effect analyses, which in turn reduced statistical power to detect an effect with post hoc testing, or it could be due to increased variability observed after tramadol dosing relative to what was observed following hydromorphone adminstration. This individual variability in response, indicated by the relatively large error bars in the time course figures, could be related to difference in genotypes (which were not tested here) as polymorphisms in CYP2D6 have been shown to differentially alter the response to tramadol (Matouskova et al., 2011). However, the impact of individual variability is partially obviated by the use of a within-subject design.
There are several other aspects of the study that warrant further consideration. First, this experiment only used opioid receptor blockade to study the neuropharmacology of tramadol. Future research could test noradrenergic or serotonergic antagonists combined with tramadol to demonstrate more definitively whether these neurotransmitter systems are critical to the positive subjective effects observed here. Second, this study used only one oral naltrexone dose (50 mg). This dose results in near complete blockade of mu opioid receptors (Lee et al., 1988) and was sufficient to block the effects of hydromorphone and attenuate the physiological effects of tramadol. However, it is possible that an even higher dose may be needed to produce complete blockade of tramadol’s mu opioid effects, as previous studies have shown that higher antagonist doses are needed to block the effects of drugs with lower intrinsic activity relative to full agonists (e.g., Eissenberg et al., 1996; Lanier et al., 2010; Martin et al., 1976; Schuh et al., 1996). Because a higher opioid antagonist dose was not tested in this study, the receptor systems mediating the positive subjective effects of tramadol cannot be completely determined from our results. Moroever, although tramadol and naltrexone are selective for the mu opioid receptor, they also have affinity for kappa and delta opioid receptors so the specific receptor mediating the opioid-like effects or blockade of tramadol cannot be completely elucidated (Ananthan et al., 1999; Lai et al., 1996; Raffa et al., 1992; Takemori et al., 1988; Volpe et al., 2011; Walsh et al., 2008b). Third, the drug identification data are confounded by the disclosure in the consent document of the drugs under study, a University of Kentucky IRB requirement, so subjects may have been biased to select tramadol or hydromorphone when they identified the drug they received as an opioid. Even with this confound in place, subjects were able to identify hydromorphone correctly, despite having a range of other options (e.g., methadone, morphine, tramadol, oxycodone), but were less able to identify tramadol accurately.
Taken together, these findings show that naltrexone can be used to disentangle the mixed neuropharmacological actions of tramadol. Naltrexone blocked the mu opioid-related physiological effects of tramadol (i.e., miosis, reduced oxygen saturation). It modestly attenuated the magnitude and time course of tramadol’s positive subjective effects and revealed pharmacodynamic effects consistent with tramadol’s serotonergic and noradrenergic activity. These data suggest that the non-mu opioid actions of tramadol play a role in its subjective profile and may possibly contribute to its abuse potential.
Supplementary Material
Acknowledgments
This research was supported by grant number R01DA025649 from the National Institute on Drug Abuse (PI: William W. Stoops) and by grant number UL1RR033173 from the National Center for Research Resources (PI: Philip A. Kern). The content is solely the responsibility of the authors and does not necessarily represent the official views of NIDA, NCRR or NIH.
The authors wish to thank the staff at the University of Kentucky Center on Drug and Alcohol Research for technical and medical assistance.
Footnotes
The authors declare no conflicts of interest relevant to this project.
References
- Ananthan S, Kezar HS, III, Carter RL, et al. Synthesis, opioid receptor binding and biological activities of naltrexone-derived pyrido- and pyrimidomorphinans. J Med Chem. 1999;42:3527–3538. doi: 10.1021/jm990039i. [DOI] [PubMed] [Google Scholar]
- Carroll CP, Walsh SL, Bigelow GE, Strain EC, Preston KL. Assessment of agonist and antagonist effects of tramadol in opioid-dependent humans. Exp Clin Psychopharmacol. 2006;14:109–120. doi: 10.1037/1064-1297.14.2.109. [DOI] [PubMed] [Google Scholar]
- Cicero TJ, Inciardi JA, Adams EH, Geller A, Senay EC, Woody GE, Muñoz A. Rates of abuse of tramadol remain unchanged with the introduction of new branded and generic products: Results of an abuse monitoring system, 1994–2004. Pharmacoepidemiol Drug Saf. 2005;14:851–859. doi: 10.1002/pds.1113. [DOI] [PubMed] [Google Scholar]
- Daws LC. Unfaithful neurotransmitter transporters: Focus on serotonin uptake and implications for antidepressant efficacy. Pharmacol Ther. 2009;121:89–99. doi: 10.1016/j.pharmthera.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driessen B, Reimann W, Giertz H. Effects of the central analgesic tramadol on the uptake and release of noradrenaline and dopamine in vitro. Br J Pharmacol. 1993;108:806–811. doi: 10.1111/j.1476-5381.1993.tb12882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duke AN, Bigelow GE, Lanier RK, Strain EC. Discriminative stimulus effects of tramadol in humans. J Pharmacol Exp Ther. 2011;338:255–262. doi: 10.1124/jpet.111.181131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissenberg T, Greenwald MK, Johnson RE, Liebson IA, Bigelow GE, Stitzer ML. Buprenorphine’s physical dependence potential: Antagonist-precipitated withdrawal in humans. J Pharmacol Exp Ther. 1996;276:449–459. [PubMed] [Google Scholar]
- Epstein DH, Preston KL, Jasinski DR. Abuse liability, behavioral pharmacology and physical-dependence potential of opioids in humans and laboratory animals: Lessons from tramadol. Biol Psychol. 2006;73:90–99. doi: 10.1016/j.biopsycho.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filip M, Wydra K, Inan SY, Dziedzicka-Wasylewska M, Przegalinski E. Opioid and monoamine systems mediate the discriminative stimulus of tramadol in rats. Eur J Pharmacol. 2004;498:143–151. doi: 10.1016/j.ejphar.2004.07.090. [DOI] [PubMed] [Google Scholar]
- Frink MC, Hennies HH, Englberger W, Haurand M, Wilffert B. Influence of tramadol on neurotransmitter systems of the rat brain. Arzneimittelforschung. 1996;46:1029–1036. [PubMed] [Google Scholar]
- Gillen C, Haurand M, Kobelt DJ, Wnendt S. Affinity, potency and efficacy of tramadol and its metabolites at the cloned human mu-opioid receptor. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:116–121. doi: 10.1007/s002100000266. [DOI] [PubMed] [Google Scholar]
- Grond S, Sablotzki A. Clinical pharmacology of tramadol. Clin Pharmacokinet. 2004;43:879–923. doi: 10.2165/00003088-200443130-00004. [DOI] [PubMed] [Google Scholar]
- Heil SH, Holmes HW, Bickel WK, Higgins ST, Badger GJ, Laws HF, Faries DE. Comparison of the subjective, physiological and psychomotor effects of atomoxetine and methylphenidate in light drug users. Drug Alcohol Depend. 2002;67:149–156. doi: 10.1016/s0376-8716(02)00053-4. [DOI] [PubMed] [Google Scholar]
- Ide S, Minami M, Ishihara K, Uhl GR, Sora I, Ikeda K. Mu opioid receptor-dependent and independent components in effects of tramadol. Neuropharmacology. 2006;51:651–658. doi: 10.1016/j.neuropharm.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Jasinski DR, Faries DE, Moore RJ, Schuh LM, Allen AJ. Abuse liability assessment of atomoxetine in a drug-abusing population. Drug Alcohol Depend. 2008;95:140–146. doi: 10.1016/j.drugalcdep.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Jasinski DR, Preston K, Sullivan JT, Testa MP. Abuse potential of oral tramadol. NIDA Res Monogr. 1993;132:103. [Google Scholar]
- Laeng B, Sirois S, Gredebäck G. Pupillometry: A window into the preconscious? Perspect Psychol Sci. 2012;7:18–27. doi: 10.1177/1745691611427305. [DOI] [PubMed] [Google Scholar]
- Lai J, Ma S-w, Porreca F, Raffa Tramadol, M1 metabolite and enantiomer affinities for cloned human opioid receptors expressed in transfected HN9.10 neuroblastoma cells. Eur J Pharmacol. 1996:369–372. doi: 10.1016/s0014-2999(96)00770-4. [DOI] [PubMed] [Google Scholar]
- Lanier RK, Lofwall MR, Mintzer MZ, Bigelow GE, Strain EC. Physical dependence potential of daily tramadol dosing in humans. Psychopharmacology (Berl) 2010;211:457–466. doi: 10.1007/s00213-010-1919-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Wagner HN, Jr, Tanada S, Frost JJ, Bice AN, Dannals RF. Duration of occupancy of opiate receptors by naltrexone. J Nucl Med. 1988;29:1207–1211. [PubMed] [Google Scholar]
- Liao S, Hill JF, Nayak RK. Pharmacokinetics of tramadol following single and multiple oral doses in man. Pharm Res. 1992;9(Suppl):308. [Google Scholar]
- Lintz W, Barth H, Osterloh G, Schmidt-Bothelt E. Bioavailability of enteral tramadol formulations. 1st communication: Capsules. Arzneimittelforschung. 1986;36:1278–1283. [PubMed] [Google Scholar]
- Lofwall MR, Walsh SL, Bigelow GE, Strain EC. Modest opioid withdrawal suppression efficacy of oral tramadol in humans. Psychopharmacology. 2007;194:381–393. doi: 10.1007/s00213-007-0847-3. [DOI] [PubMed] [Google Scholar]
- Manchikanti L, Fellows B, Ailinani H, Pampati V. Therapeutic use, abuse and nonmedical use of opioids: A ten-year perspective. Pain Physician. 2010;13:401–435. [PubMed] [Google Scholar]
- Martin CA, Guenthner G, Bingcang C, Rayens MK, Kelly TH. Measurement of the subjective effects of methylphenidate in 11- to 15-year-old children with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2007;17:63–73. doi: 10.1089/cap.2006.0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin WR, Eades CG, Thompson JA, Huppler RE, Gilbert PE. The effects of morphine- and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. J Pharmacol Exp Ther. 1976;197:517–532. [PubMed] [Google Scholar]
- Matouskova O, Slanar O, Chytil L, Perlik F. Pupillometry in healthy volunteers as a biomarker of tramadol efficacy. J Clin Pharm Ther. 2011;36:513–517. doi: 10.1111/j.1365-2710.2010.01203.x. [DOI] [PubMed] [Google Scholar]
- Matthiesen T, Wohrmann T, Coogan TP, Uragg H. The experimental toxicology of tramadol: An overview. Toxicol Lett. 1998;95:63–71. doi: 10.1016/s0378-4274(98)00023-x. [DOI] [PubMed] [Google Scholar]
- McLeod D, Griffiths RR, Bigelow GE, Yingling J. An automated version of the digit symbol substitution test (DSST) Behav Res Meth Ins. 1982;14:433–436. [Google Scholar]
- Mello NK, Mendelson JH, Bree MP. Naltrexone effects on morphine and food self-administration in morphine-dependent rhesus monkeys. J Pharmacol Exp Ther. 1981;218:550–557. [PubMed] [Google Scholar]
- O'Connor EC, Mead AN. Tramadol acts as a weak reinforcer in the rat self-administration model, consistent with its low abuse liability in humans. Pharmacol Biochem Behav. 2010;96:279–286. doi: 10.1016/j.pbb.2010.05.018. [DOI] [PubMed] [Google Scholar]
- Preston KL, Bigelow GE. Differential naltrexone antagonism of hydromorphone and pentazocine effects in human volunteers. J Pharmacol Exp Ther. 1993;264:813–823. [PubMed] [Google Scholar]
- Preston KL, Jasinski DR, Testa M. Abuse potential and pharmacological comparison of tramadol and morphine. Drug Alcohol Depend. 1991;27:7–17. doi: 10.1016/0376-8716(91)90081-9. [DOI] [PubMed] [Google Scholar]
- Radbruch L, Grond S, Lehmann KA. A risk-benefit assessment of tramadol in the management of pain. Drug Saf. 1996;15:8–29. doi: 10.2165/00002018-199615010-00002. [DOI] [PubMed] [Google Scholar]
- Raffa RB. Basic pharmacology relevant to drug abuse assessment: Tramadol as example. J Clin Pharm Ther. 2008;33:101–108. doi: 10.1111/j.1365-2710.2008.00897.x. [DOI] [PubMed] [Google Scholar]
- Raffa RB, Friderichs E, Reimann W, Shank RP, Codd EE, Vaught JL. Opioid and nonopioid components independently contribute to the mechanism of action of tramadol, an 'atypical' opioid analgesic. J Pharmacol Exp Ther. 1992;260:275–285. [PubMed] [Google Scholar]
- Ren YH, Zheng JW. Influence of tramadol on morphine discriminative behavior in rats. Acta Pharmacol Sin. 2000;21:924–926. [PubMed] [Google Scholar]
- Rouini MR, Ardakani YH, Soltani F, Aboul-Enein HY, Foroumadi A. Development and validation of a rapid HPLC method for simultaneous determination of tramadol and its two main metabolites in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;830:207–211. doi: 10.1016/j.jchromb.2005.10.039. [DOI] [PubMed] [Google Scholar]
- Schneider MF, Bailey JE, Cicero TJ, Dart RC, Inciardi JA, Parrino M, Muñoz A. Integrating nine prescription opioid analgesics and/or four signal detection systems to summarize statewide prescription drug abuse in the United States in 2007. Pharmacoepidemiol Drug Saf. 2009;18:778–790. doi: 10.1002/pds.1780. [DOI] [PubMed] [Google Scholar]
- Schuh KJ, Walsh SL, Bigelow GE, Preston KL, Stitzer ML. Buprenorphine, morphine and naloxone effects during ascending morphine maintenance in humans. J Pharmacol Exp Ther. 1996;278:836–846. [PubMed] [Google Scholar]
- Schuh KJ, Walsh SL, Stitzer ML. Onset, magnitude and duration of opioid blockade produced by buprenorphine and naltrexone in humans. Psychopharmacology (Berl) 1999;145:162–174. doi: 10.1007/s002130051045. [DOI] [PubMed] [Google Scholar]
- Sprague JE, Leifheit M, Selken J, Milks MM, Kinder DH, Nichols DE. In vivo microdialysis and conditioned place preference studies in rats are consistent with abuse potential of tramadol. Synapse. 2002;43:118–121. doi: 10.1002/syn.10025. [DOI] [PubMed] [Google Scholar]
- Stamer UM, Musshoff F, Kobilay M, Madea B, Hoeft A, Stuber F. Concentrations of tramadol and O-desmethyltramadol enantiomers in different CYP2D6 genotypes. Clin Pharmacol Ther. 2007;82:41–47. doi: 10.1038/sj.clpt.6100152. [DOI] [PubMed] [Google Scholar]
- Stoops WW, Hatton KW, Lofwall MR, Nuzzo PA, Walsh SL. Intravenous oxycodone, hydrocodone and morphine in recreational opioid users: Abuse potential and relative potencies. Psychopharmacology (Berl) 2010;212:193–203. doi: 10.1007/s00213-010-1942-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Substance Abuse and Mental Health Services Administration. Results from the 2010 national survey on drug use and health: Summary of national findings, NSDUH Series H-41, HHS Publication No (SMA) Rockville, MD: 2011. pp. 11–4658. [Google Scholar]
- Substance Abuse and Mental Health Services Administration, Office of Applied Studies. The NSDUH report: Trends in nonmedical use of prescription pain relievers: 2002 to 2007. Rockville, MD: 2009. [Google Scholar]
- Substance Abuse and Mental Health Services Administration, Office of Applied Studies. The NSDUH report: Patterns and trends in nonmedical prescription pain reliever use: 2002 to 2005. Rockville, MD: 2007. [Google Scholar]
- Sullivan MA, Vosburg SK, Comer SD. Depot naltrexone: Antagonism of the reinforcing, subjective and physiological effects of heroin. Psychopharmacology. 2006;189:37–46. doi: 10.1007/s00213-006-0509-x. [DOI] [PubMed] [Google Scholar]
- Swedberg MD, Shannon HE, Nickel B, Goldberg SR. Pharmacological mechanisms of action of flupirtine: A novel, centrally acting, nonopioid analgesic evaluated by its discriminative effects in the rat. J Pharmacol Exp Ther. 1988;246:1067–1074. [PubMed] [Google Scholar]
- Swedberg MD, Shannon HE, Nickel B, Goldberg SR. D-16949 (anpirtoline): A novel serotonergic (5-HT1B) psychotherapeutic agent assessed by its discriminative effects in the rat. J Pharmacol Exp Ther. 1992;263:1015–1022. [PubMed] [Google Scholar]
- Takemori AE, Ho BY, Naeseth JS, Portoghese PS. Nor-Binaltorphimine, a highly selective kappa-opioid antagonist in analgesic and receptor binding assays. J Pharmacol Exp Ther. 1988;246:255–258. [PubMed] [Google Scholar]
- Tzschentke TM, Bruckmann W, Friderichs E. Lack of sensitization during place conditioning in rats is consistent with the low abuse potential of tramadol. Neurosci Lett. 2002;329:25–28. doi: 10.1016/s0304-3940(02)00571-2. [DOI] [PubMed] [Google Scholar]
- Vanderkooy JD, Kennedy SH, Bagby RM. Antidepressant side effects in depression patients treated in a naturalistic setting: A study of bupropion, moclobemide, paroxetine, sertraline and venlafaxine. Can J Psychiatry. 2002;47:174–180. doi: 10.1177/070674370204700208. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fischman MW, Foltin RW, Fowler JS, Abumrad NN, Vitkun S, Logan J, Gatley SJ, Pappas N, Hitzemann R, Shea CE. Relationship between subjective effects of cocaine and dopamine transporter occupancy. Nature. 1997;386:827–830. doi: 10.1038/386827a0. [DOI] [PubMed] [Google Scholar]
- Walsh SL, Chausmer AE, Strain EC, Bigelow GE. Evaluation of the mu and kappa opioid actions of butorphanol in humans through differential naltrexone blockade. Psychopharmacology (Berl) 2008b;196:143–155. doi: 10.1007/s00213-007-0948-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh SL, Nuzzo PA, Lofwall MR, Holtman JR., Jr The relative abuse liability of oral oxycodone, hydrocodone and hydromorphone assessed in prescription opioid abusers. Drug Alcohol Depend. 2008a;98:191–202. doi: 10.1016/j.drugalcdep.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh SL, Preston KL, Bigelow GE, Stitzer ML. Acute administration of buprenorphine in humans: Partial agonist and blockade effects. J Pharmacol Exp Ther. 1995;274:361–372. [PubMed] [Google Scholar]
- Walsh SL, Sullivan JT, Preston KL, Garner JE, Bigelow GE. Effects of naltrexone on response to intravenous cocaine, hydromorphone and their combination in humans. J Pharmacol Exp Ther. 1996;279:524–538. [PubMed] [Google Scholar]
- Yanagita T. Drug dependence potential of 1-(m-methoxyphenyl)-2-dimethylaminomethyl)-cyclohexan-1-ol hydrochloride (tramadol) tested in monkeys. Arzneimittelforschung. 1978;28:158–163. [PubMed] [Google Scholar]
- Yen TT, Fuller RW. Preclinical pharmacology of fluoxetine, a serotonergic drug for weight loss. Am J Clin Nutr. 1992;55(1 Suppl):177S–180S. doi: 10.1093/ajcn/55.1.177s. [DOI] [PubMed] [Google Scholar]
- Volpe DA, McMahon Tobin GA, Mellon RD, Katki AG, Parker RJ, Colatsky T, Kropp TJ, Verbois SL. Uniform assessment and ranking of Mu receptor binding constants for selected opioid drugs. Regul Toxicol Pharmacol. 2011;59:385–390. doi: 10.1016/j.yrtph.2010.12.007. [DOI] [PubMed] [Google Scholar]
- Zacny JP. Profiling the subjective, psychomotor and physiological effects of tramadol in recreational drug users. Drug Alcohol Depend. 2005;80:273–278. doi: 10.1016/j.drugalcdep.2005.05.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.