

Abstract
Among the clusters of imprinted genes in humans, one of the most relevant regions involved in human growth is localised in 11p15. Opposite epigenetic and genomic disturbances in this chromosomal region contribute to two distinct imprinting disorders associated with disturbed growth, Silver–Russell and Beckwith–Wiedemann syndromes. Due to the complexity of the 11p15 imprinting regions and their interactions, the interpretation of the copy number variations in that region is complicated. The clinical outcome in case of microduplications or microdeletions is therefore influenced by the size, the breakpoint positions and the parental inheritance of the imbalance as well as by the imprinting status of the affected genes. Based on their own new cases and those from the literature, the authors give an overview on the genotype–phenotype correlation in chromosomal rearrangements in 11p15 as the basis for a directed genetic counselling. The detailed characterisation of patients and families helps to further delineate risk figures for syndromes associated with 11p15 disturbances. Furthermore, these cases provide us with profound insights in the complex regulation of the (imprinted) factors localised in 11p15.
Keywords: 11p15.5 imprinting region, Silver–Russell syndrome, Beckwith–Wiedemann syndrome, copy number variations
Introduction
Imprinted genes with a parent-of-origin specific expression are involved in various aspects of growth that are rooted in the prenatal period. As a result, disturbances of these genes result in aberrant growth as one key feature in nearly all known imprinting disorders (IDs). Imprinted genes tend to cluster, and thus the imprinting control is often not restricted to a single gene at an imprinted locus but affects the expression of several factors. Among these clusters of imprinted genes, one of the most relevant regions involved in human growth is localised on the short arm of chromosome 11 (11p15). Opposite epigenetic and genomic disturbances in this chromosomal region contribute to two distinct IDs associated with disturbed growth, Silver–Russell syndrome (SRS) and Beckwith–Wiedemann syndrome (BWS). Additionally, in nearly all patients with aberrant methylation at multiple loci (multilocus methylation defects), the imprinted 11p15 regions are affected (for review see Eggermann et al 1).
The 11p15 region contains a number of imprinted genes. Their expression is regulated by two different imprinting control regions (ICR1 and ICR2), also called H19 differentially methylated region (DMR) and KvDMR1 (figure 1). The telomeric ICR1 confers a differential chromatin architecture to the two parental alleles leading to reciprocal expression of H19 and IGF2 (insulin-like growth factor 2).
Figure 1.

UCSC genome browser custom tracks for the 11p15 region for the cases in this study and for the cases from the literature. (A) ICR1. (B) ICR2. (Blue: duplication; red: deletion; green: translocation; horizontal striping: paternal allele; vertical striping: maternal allele; BWS, Beckwith–Wiedemann syndrome phenotype; SRS, Silver–Russell syndrome phenotype; na, non-affected carrier; meso, mesodermal; endo, endodermal; skm, skeletal muscle enhancers.)
The ICR1 contains seven ccctc-binding factor (CTCF) target sites (CTCF1–CTCF7) in the DMR 2 kb upstream of H19 and shows allele-specific methylation. The zinc-finger binding factor CTCF binds to the maternal unmethylated ICR1 copy and thereby forms a chromatin boundary. This CTCF binding mechanism blocks IGF2 and promotes H19 transcription of the maternal 11p15 copy.
The centromeric ICR2 is maternally methylated and controls the monoallelic silencing of the non-coding antisense transcript KCNQ1OT1 as well as the monoallelic expression of cyclin-dependent kinase inhibitor 1c (CDKN1C) and potassium channel KQT-family member 1 (KCNQ1) in a not yet fully understood way.
As aforementioned, two IDs are known to be associated with molecular alterations in 11p15: SRS is mainly characterised by severe prenatal and postnatal growth restriction. The children are relatively macrocephalic and their faces are triangular-shaped with broad foreheads and pointed, small chins. In many cases, asymmetry of limbs and body and clinodactyly V are present. Growth failure is often accompanied by severe failure to thrive, and severe feeding difficulties in early childhood are frequent. However, the clinical diagnosis is difficult and requires an anamnestic investigation of an experienced clinician. The genetic basis of SRS is heterogeneous. In approximately 10% of SRS patients, a maternal uniparental disomy for chromosome 7 (upd(7)mat) can be observed (for review see Abu-Amero et al 2). More than 40% of SRS patients show a hypomethylation of the ICR1 in the imprinted region 11p15. However, upd(11)mat has been reported only once.3 Nearly 7% of SRS patients carrying an ICR1 hypomethylation show aberrant methylation at additional imprinted loci, and in all cases the ICR2 in 11p15 was affected (for review see Eggermann et al 1). Numerous (submicroscopic) chromosomal disturbances have been described in SRS patients; among them, 11p15 disturbances (tables 1 and 2).17 18
Table 1.
List of cases with rearrangements in 11p15 and their (putative) functional consequences
|
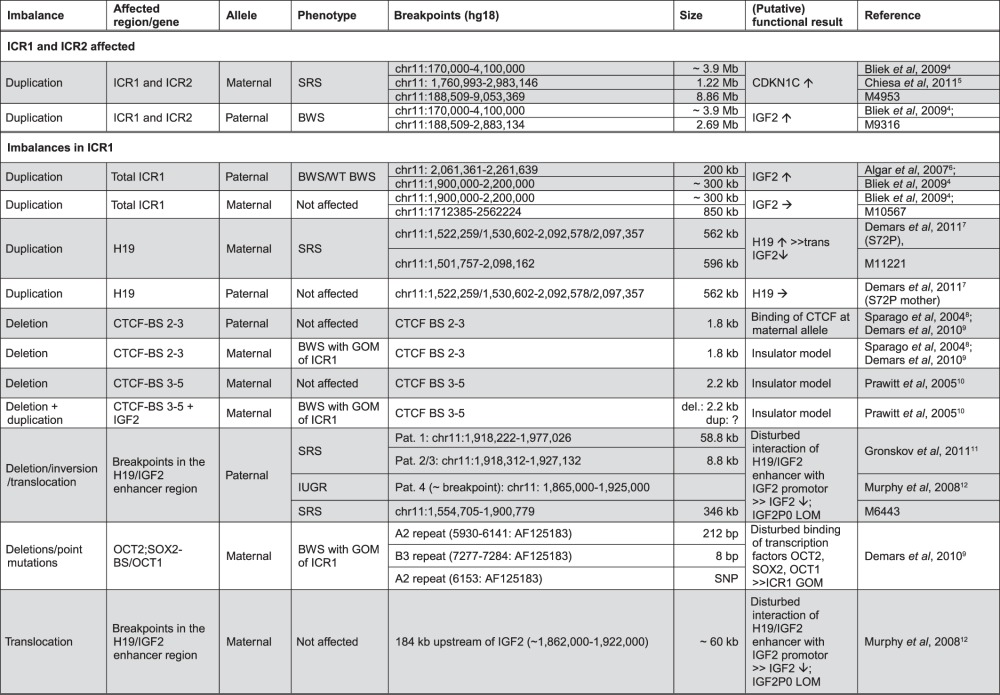

The functional result has either been reported or suggested in the specific reference, or has been delineated from the general knowledge about gene regulation and expression in 11p15. (↑ expression increased, ↓ expression decreased, → expression is not altered.)
BWS, Beckwith–Wiedemann syndrome phenotype; SRS, Silver–Russell syndrome phenotype; GOM, gain of mehylation; WT, wilms tumour; IUGR, intra uterine growth retardation.
Table 2.
Clinical outcomes in carriers of 11p15 imbalances as they can be delineated from some ‘key’ cases/families
|

The phenotype is associated with (a) origin—the parent contributing the variant, (b) type of imbalance (duplication/deletion), (c) the affected regions in both ICR1 and ICR2 and (d) the size of the aberration (grey: duplication, black: deletion; horizontal striping: this region is partially affected).
*Coordinates of the CDKN1C enhancer are from Algar et al 2011; the exact physical positions of the CDKN1C enhancer, the H19 DMR and the KCNQ1OT1 DMR are currently unclear.
BWS, Beckwith–Wiedemann syndrome phenotype; DMR, differentially methylated region; mat, maternal; pat, paternal; SRS, Silver–Russell syndrome phenotype.
Interestingly, the opposite 11p15 epigenetic and genetic findings can be observed in BWS. BWS was initially called EMG syndrome from its three main features of exomphalos, macroglossia and (neonatal) gigantism. Additional signs include neonatal hypoglycaemia, hemihypertrophy, organomegaly, earlobe creases, polyhydramnion, haemangioma and cardiomyopathy. In 5%–7% of children, embryonal tumours (most commonly Wilms' tumour) are diagnosed. The clinical diagnosis of BWS is often difficult due to its variable presentation and the phenotypic overlap with other overgrowth syndromes (for review see Enklaar et al 19). In nearly 70% of BWS patients, an altered expression or mutations of 11p15.5 encoded factors can be observed with a preponderance of an ICR2 hypomethylation accounting for nearly 50% of cases. Also, upd(11p15)pat is the second important alteration, while ICR1 hypermethylation is rare. In single cases, duplications or deletions in 11p15 have been reported (tables 1 and 2, figure 1). Most BWS cases are sporadic but familial inheritance is observed in 15% of all cases. In BWS families without aberrant 11p15 methylation, CDKN1C point mutations are frequent.
Whereas large duplications affecting both ICRs in 11p15 are known to be associated with growth retardation/SRS or overgrowth/BWS depending on the parental origin of the imbalance (table 1), the characterisation of carriers of rare small disturbances within either the ICR1 or the ICR2 allows the identification of key elements in 11p15.5 responsible for the clinical course. On the basis of three own cases and data from the literature, we review the role of submicroscopic imbalances in 11p15.5 in the aetiology of growth disturbances.
Patients
Two of the patients were initially referred as SRS for routine diagnostic testing (M11221, M6443), and the third patient (M10567) was molecularly karyotyped because of mental retardation but she did not show any aberrant growth phenotype. Appropriate informed consent was obtained from all participating patients or their legal representatives.
The 5.5-month-old girl M11221 was the third child of healthy non-consanguineous German parents (figure 2). The father's height was 180 cm (0.28 SD), and the mother's height of 160 cm was within the lower range (−0.78 SD). The maternal brother was of normal height (180 cm/0.28 SD), the maternal grandmother's height was within the lower range (157 cm/−1.29 SD), and the grandfather was of normal height (173 cm/−0.73 SD). The propositus' elder sisters were also small at birth, and persisted at the lower range (8 years old sister: 123 cm (−1.03 SD), 3.5 years old sister: 94 cm (−2.42 SD)). Further features were not observed. Family history was normal. One spontaneous abortion of the maternal grandmother was reported.
Figure 2.

Clinical features (age 7.5 months) and pedigree of patient M11221 with familial 598 kb duplication affecting 11p15.5 and uphd(6)mat.
During pregnancy low fetal movements were reported. The patient was born at 34 gestational weeks by primary caesarean section due to oligohydramnios, severe intrauterine growth retardation and pathological Doppler and cardiotocography. Birth length was 39 cm (−2.28 SD), weight 1030 g (−2.89 SD) and head circumference 28 cm (−2.18 SD). APGAR was 9-10-10. A small midface and a prominent forehead were reported. The placenta weight was remarkably reduced and corresponded to that of 20–25 weeks of gestation (P50 and P10, respectively).
At the age of 7.5 months, the girl was severely growth retarded (length 59 cm (−3.47 SD), weight 4.5 kg (−7.0 SD)). The head circumference of 42 cm (−0.68 SD) was in the normal range and thus corresponded to a relative macrocephaly. She showed a failure to thrive, had muscular hypotonia and motor development was slightly delayed. A mild asymmetry of the face was visible, whereas other parts of the body were not asymmetric. She had an umbilical hernia.
Patient M6443 is the second child of healthy German parents (father: 28 years old, 180 cm (0.28 SD); mother: 24 years old, 163 cm (−0.27 SD)). The elder sister was of normal growth. Pregnancy was uneventful but terminated at 38 weeks of gestation by caesarean section due to intrauterine growth retardation. Birth weight was 1390 g (−4.4 SD), length 40 cm (−4.83 SD) and head circumference 32 cm (−2.07 SD). Severe feeding difficulties led to gastral tube feeding at the age of 3 months and lasted for 5 weeks.
At the age of 1 1/12 years, the boy presented a short stature (height 65.5 cm/−4.42 SD; weight 5.3 kg/BMI: 12.35 SD) but with relative macrocephaly (head circumference: 44.5 cm/−2.04 SD). Further features suggestive of SRS were a triangular shaped face with a prominent forehead, a micrognathy and a clinodactyly V. Additionally, a cleft palate was reported.
Patient M10567 is the first child of healthy non-consanguineous German parents (father: 24 years old, 183 cm; mother: 20 years old, 158 cm). Family history was normal. Pregnancy was uneventful; however, the calculated due day was advanced by 4 weeks during pregnancy. She was born spontaneously at 40+3 weeks of gestation. Birth weight was reported as 2770 g (−1.67 SD), length 53 cm (0.59 SD) and head circumference 36 cm (0.85 SD). Motor development was delayed with free walking at 2 years of age, and language development was normal. At the age of 7 years, she attended a special school due to motor and mental delay. She was hypotonic, with no special neurological deficits. During the first years of life, growth and weight were slightly below the third centile, with catch-up growth at 7 5/12 years of age (weight 20.7 kg (P10), length 113.8 cm (0.5 cm, <P3), BMI 16.0 (P50), head circumference 50.9 cm (P25)). Average growth velocity between age 4 and 7 years was 6.2 cm/year (P54). IGF I was slightly low (53 ng/ml, <P5), and IGFBP3 was in the normal range (2958 ng/ml, >P5). She presented with discrete phenotypic features including a long narrow facial appearance, slightly upslanting palpebral fissures, a small mouth, a transverse groove of the chin, relatively large ears, a symmetric clinodactyly digit V with Dubois sign, simian crease on the right and bilateral syndactyly toes II/III. Early second dentition was observed. Asymmetry was not present. Fragile X-syndrome testing was negative.
Materials and methods
Genomic DNA was isolated from peripheral lymphocyte cells by a simple salting-out procedure.
Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) was performed using the SALSA MLPA kit ME030 from MRC Holland (Amsterdam, The Netherlands) according to the manufacturer's protocol.
For rapid screening of aberrant methylation of several DMRs which have been reported to be frequently affected in multilocus methylation defects patients (ie, PLAGL1 (6q24), IGF2R (6q25), GRB10 (7p13), MEST (7q32), H19 (11p15), KCNQ1OT1 (11p15), IGF2P0 (11p15), MEG3 (14q32)), we applied multilocus quantitative methylation-sensitive single-nucleotide primer extension (MS-SNuPE) tests.20
For the detection of submicroscopic genomic imbalances (<5 Mb), we typed genomic DNA of the patients by using the Affymetrix GeneChip®Genome-Wide Human SNP 6.0-Array (High Wycombe, UK) including 1.8M oligonucleotide markers. After PCR amplification and labelling of the patients' DNA the samples were hybridised to the arrays according to the manufacturer's instructions. Scanning was performed with an Affymetrix GeneChip®Scanner 3000 7G, and bioinformatics was done with the Affymetrix Genotyping Console 4.0 and the Chromosome Analysis Suite 1.1 software using annotation files version NA30 (hg18/NCBI build 36). For analysis and interpretation only copy number variations >100 kb with a mean marker distance <5 kb were considered.
To determine the inheritance of the 11p15 copy number variations in the families of patients M11221 and M10567 and to confirm the uniparental disomy of chromosome 6 in patient M11221, short tandem repeat typing with markers within 11p15 and on chromosome 6 was performed according to standard protocols.
Results
Three patients with rare submicroscopic imbalances in the telomeric ICR1 in 11p15 were recruited either because of their phenotype resembling to SRS (patients M11221 and M6443) or because of molecular karyotyping results (patient M10567).
In patient M11221 initial conventional cytogenetic analysis (500 bands) was normal. By MS-MLPA, increased hybridisation signals as well as aberrant methylation of the H19-specific probes could be determined, whereas the IGF2 probes as well as those for the ICR2 factors and the control probes showed normal copy numbers. The duplication of H19 in the ICR1 in 11p15.5 was then confirmed by molecular karyotyping and microsatellite analysis, indicating a 596 kb duplication in 11p15.5 (NCBI36/hg18: chr11:1.501.757-2.098.162) which affected the H19 but not the IGF2 gene (figure 1A). The duplication was inherited from the mother, and the two sisters also carried the disturbance. The maternal brother was not a carrier of the familial duplication.
Multilocus analysis by MS-SNuPE confirmed the 11p15 duplication; additionally, an increased methylation of both chromosome 6 loci PLAGL1 and IGF2R was observed. Further imprinted loci showed normal results except the H19 locus. Analyses of DNA samples of the mother and the two sisters revealed normal methylation patterns for the chromosome 6 loci. Microsatellite analysis of chromosome 6 markers confirmed a complete maternal uniparental heterodisomy of the whole chromosome 6 (uphd(6)mat) which was not detectable by SNP array typing.
In patient M6443, conventional karyotyping was performed after birth and showed a normal male karyotype. Further 11p15.5-specific MS-MLPA and testing for upd(7)mat did not reveal any irregularities. Subsequent SNP array analysis revealed a 346 kb deletion in 11p15.5 (NCBI36/hg18: chr11:1.554.705-1.900.779) which was inherited from the healthy father. Unfortunately, paternal grandparents were not available (figure 1A). This copy number variation affected the mesodermal enhancer of IGF2 but not the H19 DMR or other sequences analysed by the MS MLPA approach. By MS-SNuPE, a hypomethylation at the IGF2P0 DMR was visible.
Patient M10567 as well as her healthy mother carried a 849 kb duplication in 11p15.5 (NCBI36/hg18: chr11:1.712.385-2.562.224), affecting both H19 and IGF2 (figure 1A). In the mother, the duplication had originated de novo but affected the grandmaternal chromosome 11. We therefore assume that the patients' phenotype is not caused by the 11p15 duplication but another, so far unknown cause.
Discussion
The phenotype of carriers of duplications including both the telomeric ICR1 and the centromeric ICR2 in 11p15.5 is meanwhile well established: whereas duplication of maternal 11p15.5 material is generally associated with growth retardation and features suggestive for SRS, duplication of the paternal 11p15.5 copy results in overgrowth and BWS symptoms.
However, this strict association is at least in part derestricted in carriers of imbalances affecting only parts of the two ICRs. Here at a first glance unexpected clinical outcomes can be observed (figure 1, tables 1 and 2).
Duplications of the total paternal ICR1 including H19 and IGF2 result in overgrowth (and BWS), whereas duplication of the maternal ICR1 copy are not associated with an aberrant phenotype as our patient M10567 and his family as well as the family published by Bliek et al 4 show (figure 1A). The situation changes if the duplication is restricted to the H19 region (our patient M11221 and her family; patient S72P of Demars et al 7): carriers of microduplication affecting the maternal allele present a SRS phenotype whereas duplications of the paternal H19 copy are not associated with an unusual phenotype. In agreement with these observations, deletions within the maternal H19 DMR result in BWS7 8 10 whereas deletions within the paternal H19 gene/DMR do not cause a specific phenotype.7–10 In contrast, carriers of deletions of the paternal allele show a SRS phenotype if the deletion is localised telomeric from the H19DMR and affects the IGF2 telomeric enhancer (our patient M644311). In these patients with deletions within the telomeric IGF2 enhancers, a hypomethylation of the IGF2P0 DMR can be observed. This prompted Grønskov et al 11 to speculate that IGF2P0 methylation is established by the interaction with its telomeric enhancers and therefore reflects the IGF2 promoter activity.
Duplications of the total ICR2 region result in a SRS-like phenotype in case the maternal 11p15 copy is affected, whereas the duplication of paternal material is not associated with clinical symptoms (figure 1B).13 14 In contrast, a BWS phenotype can be observed in case the duplication of maternal material is restricted to the KCNQ1OT1 DMR or the putative CDKN1C enhancer region.5 7 Interestingly, the deletion of the maternal copy of both the KCNQ1OT1 DMR and the putative CDKN1C enhancer results in BWS, whereas the phenotype is normal if the paternal allele is deleted.6 15
The growing number of genomic imbalances within the two ICRs of paternal or maternal origin considerably helps us to understand the complex regulation mechanisms of the imprinting regions in 11p15 (table 1). These disturbances do not simply result in an increase or decrease of expression caused by an altered copy number, but they can also affect methylation patterns and chromatin organisation and thereby indirectly affect the regulation of imprinted genes.
In addition to the complex genotype–phenotype correlations in carriers of imbalances in 11p15.5, our patient M11221 and her family is furthermore remarkable as the index patients phenotype severely differs from that of her mother and sisters also carrying the ICR1 duplication: whereas her relatives are at the lower end of normal growth centiles, the patient is severely growth retarded. Further features were suggestive for the diagnosis of SRS. Indeed, we observed that the patient is a carrier of a uphd(6)mat, whereas the other 11p15.5 duplication carriers in her family were not. Homozygosity for a recessive mutation as the cause for the phenotype in our patient can nearly be excluded as SNP array typing did not reveal any evidence for isodisomic regions. One might rather speculate that the uphd(6)mat is responsible for the phenotype: indeed, at least two upd(6)mat patients have been reported to be associated with a severe IUGR, but generally upd(6)mat has not yet been reported to be associated with a specific clinical phenotype (for review see Salahshourifar et al 21).
From the diagnostic point of view, the advantages and limitations of the different applicable techniques for mutation detection have to be carefully considered.
For routine diagnostic of BWS and SRS, methylation-specific approaches are widely used. In particular, MS-MLPA is a well accepted testing procedure as it detects the majority of the currently known aberrations in the ICR1 and the ICR2 in 11p15 in both SRS and BWS patients. However, the currently available test discovers only aberrant methylation of the DMRs of H19 and KCNQ1OT1 as well as copy number variations of the coding sequences in IGF2, H19, CDKN1C and KCNQ1. As our cases and those from the literature show, patients might also carry copy number variations and methylation defects in other regulative regions within 11p15.5 (our patient M644311). In case of exclusion of the typical ICR1 and ICR2 alterations, further molecular characterisation is therefore needed to identify unusual mutations and epimutations. In carriers of copy number variations of 11p15, a precise breakpoint characterisation is essential to elucidate a genotype–phenotype correlation. Testing for aberrant methylation of further imprinted loci additional to the 11p15 DMRs should also be considered to identify patients with multilocus methylation defects, which might either carry genomic mutations like ZFP57 or genomic imbalances.22 23 Both findings can influence recurrence risks and have to be discussed in genetic counselling.
In summary, duplications or deletions affecting the ICR1 and/or ICR2 in 11p15 have been reported for both SRS and BWS as well as in clinically normal carriers. The clinical outcome in carriers of these microduplications or microdeletions is influenced by the size, the breakpoint positions and the parental inheritance of the imbalance reflecting the imprinting status of the affected genes (table 2). Due to the complexity of the 11p15 imprinting regions and their interactions, the interpretation of the copy number variations in that region is complicated and needs careful investigation.
Acknowledgments
We thank the families for participating in this study. The project was supported by the Bundesministerium für Bildung und Forschung (Network ‘Imprinting Diseases’, 01GM0884) and by an investigator-initiated, unrestricted research grant from Merck-Serono GmbH. MG had a scholarship of the German academic exchange service (DAAD).
Footnotes
Contributors: MB, SS, MG have carried out the analysis. MB, SS and TE have written the paper. UG, RB, MBo, AD and IS have described the patients and discussed the manuscript. MB and SS contributed equally.
Funding: The project was supported by the Bundesministerium für Bildung und Forschung (Network ‘Imprinting Diseases’, 01GM0884) and by an investigator-initiated, unrestricted research grant from Merck-Serono GmbH. MG had a scholarship of the German academic exchange service (DAAD).
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Eggermann T, Leisten I, Binder G, Begemann M, Spengler S. Disturbed methylation at multiple imprinted loci: an increasing observation in imprinting disorders. Epigenomics 2011;3:625–37 [DOI] [PubMed] [Google Scholar]
- 2. Abu-Amero S, Monk D, Frost J, Preece M, Stanier P, Moore GE. The genetic aetiology of Silver-Russell syndrome. J Med Genet 2008;45:193–9 [DOI] [PubMed] [Google Scholar]
- 3. Bullman H, Lever M, Robinson DO, Mackay DJ, Holder SE, Wakeling EL. Mosaic uniparental disomy of chromosome 11 in patient with Silver-Russell syndrome. J Med Genet 2008;45:396–9 [DOI] [PubMed] [Google Scholar]
- 4. Bliek J, Snijder S, Maas SM, Polstra A, van der Lip K, Alders M, Knegt AC, Mannens MM. Phenotypic discordance upon paternal or maternal transmission of duplications of the 11p15 imprinted regions. Eur J Med Genet 2009;52:404–8 [DOI] [PubMed] [Google Scholar]
- 5. Chiesa N, De Crescenzo A, Mishra K, Perone L, Carella M, Palumbo O, Mussa A, Sparago A, Cerrato F, Russo S, Lapi E, Cubellis MV, Kanduri C, Cirillo Silengo M, Riccio A, Ferrero GB. The KCNQ1OT1 imprinting control region and non-coding RNA: new properties derived from the study of Beckwith-Wiedemann syndrome and Silver-Russell syndrome cases. Hum Mol Genet 2011;21:10–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Algar EM, St Heaps L, Darmanian A, Dagar V, Prawitt D, Peters GB, Collins F. Paternally inherited submicroscopic duplication at 11p15.5 implicates insulin-like growth factor II in overgrowth and Wilms' tumorigenesis. Cancer Res 2007;67:2360–5 [DOI] [PubMed] [Google Scholar]
- 7. Demars J, Rossignol S, Netchine I, Lee KS, Shmela M, Faivre L, Weill J, Odent S, Azzi S, Callier P, Lucas J, Dubourg C, Andrieux J, Bouc YL, El-Osta A, Gicquel C. New insights into the pathogenesis of Beckwith-Wiedemann and Silver-Russell syndromes: contribution of small copy number variations to 11p15 imprinting defects. Hum Mutat 2011;32:1171–82 [DOI] [PubMed] [Google Scholar]
- 8. Sparago A, Cerrato F, Vernucci M, Ferrero GB, Silengo MC, Riccio A. Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith-Wiedemann syndrome. Nat Genet 2004;36:958–60 [DOI] [PubMed] [Google Scholar]
- 9. Demars J, Shmela ME, Rossignol S, Okabe J, Netchine I, Azzi S, Cabrol S, Le Caignec C, David A, Le Bouc Y, El-Osta A, Gicquel C. Analysis of the IGF2/H19 imprinting control region uncovers new genetic defects, including mutations of OCT-binding sequences, in patients with 11p15 fetal growth disorders. Hum Mol Genet 2010;19:803–14 [DOI] [PubMed] [Google Scholar]
- 10. Prawitt D, Enklaar T, Gärtner-Rupprecht B, Spangenberg C, Lausch E, Reutzel D, Fees S, Korzon M, Brozek I, Limon J, Housman DE, Pelletier J, Zabel B. Microdeletion and IGF2 loss of imprinting in a cascade causing Beckwith-Wiedemann syndrome with Wilms' tumor. Nat Genet 2005;37:785–6 [DOI] [PubMed] [Google Scholar]
- 11. Grønskov K, Poole RL, Hahnemann JM, Thomson J, Tümer Z, Brøndum-Nielsen K, Murphy R, Ravn K, Melchior L, Dedic A, Dolmer B, Temple IK, Boonen SE, Mackay DJ. Deletions and rearrangements of the H19/IGF2 enhancer region in patients with Silver-Russell syndrome and growth retardation. J Med Genet 2011;48:308–11 [DOI] [PubMed] [Google Scholar]
- 12. Murphy R, Baptista J, Holly J, Umpleby AA, Ellard S, Harries LW, Crolla J, Cundy T, Hatterlsey AT. Severe intrauterine growth retardation and atypical diabetes associated with a translocation breakpoint disrupting regulation of the insulin-like growth factor 2 gene. J Clin Endocrinol Metabol 2008;93:4373–80 [DOI] [PubMed] [Google Scholar]
- 13. Schönherr N, Meyer E, Roos A, Schmidt A, Wollmann HA, Eggermann T. The centromeric 11p15 imprinting centre is also involved in Silver-Russell syndrome. J Med Genet 2007;44:59–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bonaldi A, Mazzeu JF, Costa SS, Honjo RS, Bertola DR, Albano LM, Furquim IM, Kim CA, Vianna-Morgante AM. Microduplication of the ICR2 domain at chromosome 11p15 and familial Silver-Russell syndrome. Am J Med Genet A 2011;155:2479–83 [DOI] [PubMed] [Google Scholar]
- 15. Niemitz EL, DeBaun MR, Fallon J, Murakami K, Kugoh H, Oshimura M, Feinberg AP. Microdeletion of LIT1 in familial Beckwith-Wiedemann syndrome. Am J Hum Genet 2004;75:844–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Algar E, Dagar V, Sebaj M, Pachter N. An 11p15 imprinting centre region 2 deletion in a family with Beckwith Wiedemann syndrome provides insights into imprinting control at CDKN1C. PLoS One 2011;6:e29034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bruce S, Hannula-Jouppi K, Puoskari M, Fransson I, Simola KO, Lipsanen-Nyman M, Kere J. Submicroscopic genomic alterations in Silver-Russell syndrome and Silver-Russell-like patients. J Med Genet 2010;47:816–22 [DOI] [PubMed] [Google Scholar]
- 18. Spengler S, Schönherr N, Binder G, Wollmann HA, Fricke-Otto S, Mühlenberg R, Denecke B, Baudis M, Eggermann T. Submicroscopic chromosomal imbalances in idiopathic Silver-Russell syndrome (SRS): the SRS phenotype overlaps with the 12q14 microdeletion syndrome. J Med Genet 2010;47:356–60 [DOI] [PubMed] [Google Scholar]
- 19. Enklaar T, Zabel BU, Prawitt D. Beckwith-Wiedemann syndrome: multiple molecular mechanisms. Expert Rev Mol Med 2006;8:1–18 [DOI] [PubMed] [Google Scholar]
- 20. Begemann M, Leisten I, Soellner L, Zerres K, Eggermann T, Spengler S. Use of multilocus methylation-specific single nucleotide primer extension (MS-SNuPE) technology in diagnostic testing for human imprinted loci. Epigenetics 2012:7. [DOI] [PubMed] [Google Scholar]
- 21. Salahshourifar I, Halim AS, Sulaiman WA, Zilfalil BA. Maternal uniparental heterodisomy of chromosome 6 in a boy with an isolated cleft lip and palate. Am J Med Genet A 2010;152:1818–21 [DOI] [PubMed] [Google Scholar]
- 22. Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, Boonen SE, Dayanikli P, Firth HV, Goodship JA, Haemers AP, Hahnemann JM, Kordonouri O, Masoud AF, Oestergaard E, Storr J, Ellard S, Hattersley AT, Robinson DO, Temple IK. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet 2008;40:949–51 [DOI] [PubMed] [Google Scholar]
- 23. Begemann M, Spengler S, Kordaß U, Schröder C, Eggermann T. Segmental maternal uniparental disomy 7q associated with DLK1/GTL2 (14q32) hypomethylation. Am J Med Genet A 2012;158:423–8 [DOI] [PubMed] [Google Scholar]