


Abstract
Resolvase enzymes that cleave DNA four-way (Holliday) junctions are required for poxvirus replication, but clinically useful inhibitors have not been developed. Here, we report an assay for resolvase cleavage activity based on fluorescence polarization (FP) for high-throughput screening and mechanistic studies. Initial analysis showed that cleavage of a fluorescently labeled Holliday junction substrate did not yield an appreciable change in FP, probably because the cleavage product did not have sufficiently increased mobility to yield a strong FP signal. Iterative optimization yielded a substrate with an off-center DNA bulge, which after cleavage released a labeled short stand and yielded a greatly reduced FP signal. Using this assay, 133 000 compounds were screened, identifying 1-hydroxy-1,8-naphthyridin-2(1H)-one compounds as inhibitors. Structure-activity studies revealed functional parallels to Food and Drug Administration (FDA)-approved drugs targeting the related human immunodeficiency virus integrase enzyme. Some 1-hydroxy-1,8-naphthyridin-2(1H)-one compounds showed anti-poxvirus activity.
INTRODUCTION
Holliday junction resolvases cleave DNA four-way junctions (Holliday junctions) to yield two free duplex DNAs. For poxviruses, resolvase activity is required to complete synthesis of the viral genomic DNA (Figure 1A) (1–6). The early steps of poxvirus DNA replication result in the production of concatemeric arrays of viral DNA. The viral DNA at concatemer junctions can fold to make Holliday junction-like structures, which are cleaved to yield monomeric genomic DNAs. This reaction is required for poxvirus replication (1,2) and thus represents an attractive drug target for antiviral agents for biodefense applications.
Figure 1.

Holliday junction cleavage by poxvirus resolvase and DNA substrates studied. (A) The role of resolvase in poxvirus DNA replication. The initial product of DNA replication resembles a linear concatemer of many genomes, though in reality the structure is likely branched. The structure to the right emphasizes the Holliday junction formed by refolding of inverted repeats at the viral termini (DNA from the coordinate file 1FLO). (B) Cleavage of a Holliday junction substrate tracked by FP. (C) Cleavage of a Y-DNA substrate tracked by FP. (D) Lack of cleavage of a single DNA strand (3′ labeled). (E) Lack of cleavage of a single DNA strand (5′ labeled). (F) Lack of cleavage of a duplex DNA. (G) Efficient cleavage of an off-center DNA bulge substrate. Sequences of oligonucleotide substrates are in Supplementary Table S1.
Poxvirus resolvase is a member of the RNase H superfamily of enzymes, which also includes human immunodeficiency virus (HIV) integrase. Members of this enzyme family have a similar protein fold in their catalytic domains and likely share related catalytic mechanisms to carry out their respective nucleotidyl phosphotransfer reactions (7–9). Each enzyme is dependent on divalent metal ions for activity and contains three or four conserved acidic residues that bind metal ions in the active site. Structural studies of several family members reveal two metal binding sites at the active site, suggesting a conserved two metal-ion catalytic mechanism. We have previously reported biochemical evidence that poxvirus resolvase also has an active site that binds metal (6).
Small molecule inhibitors that target HIV integrase have been developed for the treatment of HIV infection and one such inhibitor, raltegravir, has been approved by the FDA (10,11). These inhibitors contain a metal chelating pharmacophore that is thought to disrupt catalysis by binding divalent metal-ion cofactors at the enzyme active site (12,13). These findings suggest that structurally related inhibitors may also inhibit poxvirus resolvase.
Initial studies of poxvirus resolvase have focused on the version of the enzyme encoded by vaccinia virus. This is of interest as an inhibitor target because it differs from the variola (smallpox) enzyme by only two amino acid substitutions, and variola virus is of concern as a bioweapon. However, we and others have found that the purified vaccinia enzyme is insoluble and is not suitable for high-throughput screening (1–6). Recently, we characterized the purified fowlpox virus resolvase and found it to be much more tractable in biochemical assays and well suited for high-throughput screening (5,6). The fowlpox resolvase shares only 43% amino acid identity with the vaccinia resolvase, though sequence alignments suggest the active sites are similar.
Here, we report an assay based on fluorescence polarization (FP) for monitoring DNA cleavage by fowlpox virus resolvase. FP relies on the anisotropic properties of the light emitted from fluorophores tumbling in solution after excitation with polarized light. To carry out the assay, a solution containing a fluorescently tagged molecule is exposed to a pulse of plane-polarized light and the polarization of the emitted light is measured in two orthogonal planes simultaneously. Large molecules rotate more slowly in solution and, when tagged with a fluorophore, the light emitted is more polarized than for a small molecule. Thus if the fluorescently tagged substrate and product differ significantly in size, the reaction progress can be monitored by FP.
We describe design of an optimal-bulged DNA substrate and its use to screen more than 133 000 small molecules for inhibitory activity. Mapping cleavage sites on the substrate suggested a new activity for the enzyme which may promote efficient DNA replication. The FP assay has excellent high-throughput screening parameters, which allowed us to identify a structural class of inhibitors, 1-hydroxy-1,8-naphthyridin-2(1H)-ones, with potent activity against fowlpox resolvase. Structure–activity relationship (SAR) analysis suggested a metal-chelating binding mode related to that of raltegravir. Several of these inhibitors also showed antiviral activity against vaccinia virus in cell culture assays.
MATERIALS AND METHODS
Purification of fowlpox virus resolvase
Fowlpox virus resolvase was purified as described (4,5). Briefly, resolvase protein was modified to contain a his-tag. The protein was over-expressed in Escherichia coli and purified by metal affinity chromatography. Protein concentrations were determined by Bradford assay. The fowlpox resolvase fraction was assessed to be >90% pure by SDS–PAGE.
Fluorescence polarization substrates
Oligonucleotides containing a 6-carboxyfluorescein end label (F) were purified by high performance liquid chromatography (HPLC) and all other oligonucleotides were purified by polyacrylamide gel electrophoresis (PAGE). DNA concentrations were determined by UV-spectrophotometry. The substrates were constructed by annealing together the indicated component oligonucleotides. Annealing reactions contained 10 µM labeled DNA and 2-fold excess unlabeled DNA and were carried out in the presence of 100 mM NaCl by heating to 95°C and allowing the solutions to cool slowly to room temperature over a period of 90 min.
Cleavage reactions
For reactions in 384-well plates, reagents were dispensed into wells using automated liquid handlers. Black polystyrene plates coated with a non-binding surface were used (Corning #3575).
For the National Screening Laboratory for the Regional Centers of Excellence in Biodefense and Emerging Infectious Disease (NSRB)-library screen, 20 µl of an enzyme solution or a buffer-only control (i.e. no enzyme) was dispensed into the plate wells. Next, 100 nl of each compound stock solution (5 mg/ml in DMSO) or DMSO was added to wells by robotic pin transfer. Then, 10 µl of a substrate solution was added to achieve a final volume of 30 µl and the plates were incubated at 37°C for 1 h. After incubation, FP values were measured using an Envision instrument (Perkin-Elmer). Final reagent concentrations were 2 nM fluorescein-labeled substrate and 10 nM fowlpox resolvase. Final solution conditions were 25 mM Tris–HCl (pH 8.0), 15 mM MgCl2, 100 mM NaCl and 1 mM DTT. Assuming a molecular weight of 500 g/mol, the final compounds concentration was 33 µM.
For the integrase-library screen, 20 µl of an enzyme solution or a buffer-only solution was dispensed into wells containing compound stock solution (in DMSO) or DMSO. Then, 10 µl of a substrate solution was added to achieve a final volume of 30 µl and the plates were incubated at 37°C for 1 h. After incubation, FP values were measured using an Analyst instrument (Molecular Devices). Final reagent concentrations and solution conditions were as above. The final concentration of each compound was 20 µM.
The screens were carried out in 384-well plates where each well contained a different compound from the library. The enzyme and compound were dispensed into wells first and then reactions were initiated by addition of the AB5 substrate and incubation at 37°C. After 1 h, FP measurements were obtained using a multilabel plate reader. Each plate contained 32 positive control wells (i.e. no enzyme, no inhibitor) and 32 negative control wells (i.e. enzyme, no inhibitor). Library 1 was screened at a compound concentration of 17 µg/ml (33 µM for a compound with molecular weight = 500 g/mol) and Library 2 was screened at a compound concentration of 20 µM. The average molecular weight of Library 2 was 416 g/mol (1 SD = 114), so for comparison with Library 1, the average concentration of Library 2 in µg/ml was 8.3 µg/ml (1 SD = 2.3), or ∼2-fold less.
Anisotropy of the emitted light is characterized by the polarization, P, and the anisotropy, r, so that P = (V − H)/(V + H) and r = (V − H)/(V + 2H), where V and H are the intensities of the emitted light in the vertical and horizontal planes, respectively. The total fluorescence intensity (TFI) is given by V + 2H. Both parameters are related to the rotational velocity of the tagged molecule in solution, which is proportional to its molecular volume.
We used the TFI measurements to identify compounds with either fluorescence enhancing or quenching properties so that we could remove these potential false positives and negatives from our analysis. We first normalized the TFI measurements from each compound-containing well by expressing it as a fraction of the mean TFI of the negative controls located on the same plate. Compound-containing wells with a TFI greater than or less than one standard deviation from the mean TFI of all compound-containing wells were excluded from subsequent analyses. For Library 1, we identified 2234 compounds (1.7%) that met these criteria, of which 98% were fluorescence enhancers. For Library 2, we identified 78 compounds (2.8%) that met these criteria, of which 46% were enhancers.
Percent inhibition values for each compound were calculated using the following equation: % inhibition = 100 * (P − x)/(P − N), where x is the FP value of the compound well, and P and N are the mean FP values of the positive and negative control wells within the same plate.
For IC50 determination, serial dilutions of the compounds were made in DMSO and the assay was carried out as above. For data analysis, FP values were transformed into anisotropy values using the following equation: r = 2P/(3 − P), where r is anisotropy and P is polarization. Percent inhibition values were calculated as above using the transformed data. Non-linear regression was used to fit the data against the logarithm of the compound concentrations using a sigmoidal dose-response model in Prism.
Kinetic analysis
Reactions for kinetic analysis were carried out in 100 µl buffer consisting of 25 mM Tris–Cl (pH 8.0), 100 mM NaCl, 15 mM MgCl2, and 1 mM DTT at 37°C. The concentration of fowlpox virus resolvase was 10 nM. Fluorescence labeled bulge substrate DNA at a final concentration of 2 nM was mixed with different amounts (0–1920 nM) of unlabeled bulge substrate DNA. Enzyme solutions were diluted in 2 µl of the reaction buffer and added immediately to the premixed substrate solution before analysis. Positive reference samples contained all components except the enzyme. Cleavage reactions were monitored by measuring change of FP using the Beacon 2000 Fluorescence polarization system (Invitrogen, CA) over a wide range of substrate concentrations (Supplementary Figure S1). Initial rates were calculated from non-linear regression using an exponential decay model in Prism. Km and Vmax were calculated using the Michaelis–Menten model in Prism.
Antiviral and cytotoxicity assays
Plaque assays were performed on confluent BSC1 monolayers in 12-well plates. The cells were overlaid with 1 ml of MEM (with 2% FBS) containing ∼60 PFU of vaccinia virus WR. Plates containing virus-inoculated cells were incubated for 1 h, the media was then removed and the cells were overlaid with 1 ml of MEM (with 2% FBS) containing serial 3-fold dilutions of the test compounds. Plaque formation was monitored at 48 h post-infection by staining monolayers with crystal violet. IC50 was calculated using a sigmoidal dose response model in Prism. DMSO carrier was found to be toxic above ∼5%.
Cytotoxicity was tested using the CellTiter-Glo Luminescent Cell Viability Assay kit from Promega (Madison, WI). Non-linear regression was used to fit the data against the logarithm of the compound concentrations as above.
Methods for synthesis of 1-hydroxy-1,8-naphthyridin-2(1H)-one derivatives can be found in the Supplementary data online.
RESULTS AND DISCUSSION
Developing a screening substrate for fluorescence polarization assays
Developing the resolvase substrate for high-throughput screening required an iterative series of synthesis and testing steps (Figure 1; oligonucleotide sequences are in Supplementary Table S1). Although Holliday junctions represent the physiologic substrate, cleavage of Holliday junctions into nicked duplex products yielded only a slight change in FP (Figure 1B, sub A), even though gel assays confirmed efficient cleavage. Evidently, the molecular weight change was not large enough to yield a significant difference in FP, or the fluorescein group bound to the double-stranded DNA end, which would also reduce its mobility and increase polarization. In an effort to increase the dynamic range, we tested a variety of splayed duplex substrates that each contained a single-to-double-strand transition (Figure 1C, sub B, and data not shown). These substrates were fluorescently tagged on the 5′-end single-stranded region, so cleavage would yield a short single strand (5 bases for sub B). After incubation with fowlpox resolvase, we were able to confirm complete conversion of the substrate to the expected products by native polyacrylamide gel electrophoresis (data not shown). However, sub B exhibited a relatively small change in polarization after incubation with resolvase (ΔP = −10 mP), resulting in a dynamic range that was not adequate for high-throughput screening.
For comparison, we also measured the polarization of single-strand DNAs labeled on the 5′ (Figure 1D, sub C) or 3′ (Figure 1E, sub D) ends. The observed polarization signals were close to the values observed for sub B (both in the presence and absence of resolvase). In contrast, we observed a higher polarization value with a duplex molecule containing complete sequence complimentary to the labeled strand (Figure 1F, sub E). This suggested mobility of the fluorescein-tagged single-stranded region prior to cleavage was reducing the polarization signal, likely due to rotational motion of single-stranded DNA or stacking of fluorescein on the double-stranded DNA end.
We thus reasoned that clamping down the single-stranded ends of a splayed duplex substrate by engineering base complementarity between the tips of the two single-stranded segments of sub B would result in a higher polarization signal and a greater difference following cleavage. We thus designed oligonucleotides that, after annealing, would introduce a 10 nt bulge region flanked on one side by a long region of duplex DNA and on the other side by a 5 bp duplex region, or stem (Figure 1G, sub F; ‘asymmetric bulge’). In a previous study, we showed that a substrate containing a central DNA bulge was efficiently cleaved at both sides of a bulge region (5). Sub F was fluorescein-labeled on the 3′-end of the stem region and tested with resolvase, demonstrating a 70 mP drop in FP. Evidently, the 5 bp of pairing is sufficient for the duplex be mostly paired in the starting substrate, but melt efficiently to allow release of the labeled strand after cleavage.
To characterize cleavage of this substrate more fully, we checked the structure of reaction products by gel electrophoresis (Figure 2A and B). Incubation with fowlpox resolvase yielded two cleavage products that appeared sequentially. Electrophoresis adjacent to synthetic oligonucleotides matching candidate cleavage products indicated sequential cleavage at the two single-to-double-strand transitions shown at the bottom of Figure 2B.
Figure 2.

Cleavage of the bulged DNA substrate by fowlpox resolvase. (A) Analysis of cleavage products by native gel electrophoresis. Products were characterized by co-migration with synthetic markers identical to each expected product. (B) Analysis of products of cleavage of the bulged DNA substrate analyzed by denaturing gel electrophoresis. The inferred sites of cleavage are shown at the bottom. ‘Mix’ indicates a mixture of the synthetic 15 nt expected cleavage product and an authentic reaction mixture, indicating co-migration with the indicated product. (C) Resolvase catalytic site mutants obstruct resolvase cleavage as measured in the FP assay. The bulged DNA substrate was mixed with D7A, D135A or wild-type resolvase, then cleavage tracked for the indicated times.
In the poxvirus replication cycle, this cleavage reaction may be important for resolving complex DNA structures generated during viral DNA replication. The poxvirus DNA polymerase is unusual for acting efficiently to fuse double strand broken ends containing terminal regions of homology, allowing use of the 3′-end at the double-strand break as a template for further elongation (14). Resolvase cleavage of an off-center-bulged DNA at the sites mapped in Figure 2 would thus generate a substrate for recombinational priming by the viral polymerase. The cleavage reaction described here would therefore allow restarting of DNA replication on molecules with complex unpaired regions.
A question that arises in any such study is whether the observed cleavage reaction is due to action of resolvase or cleavage by a low level contaminating protein in the resolvase preparation. To check this possibility, we compared cleavage by FP resolvase with two active site substitutions (D7A and D135A) previously shown to block cleavage by resolvase in vitro (5,6). These two mutants blocked all cleavage in the FP assay (Figure 2C) confirming that fowlpox resolvase was responsible for the observed cleavage activity.
Using the FP assay, we were able to show turn-over of fowlpox resolvase at high substrate concentrations, allowing an investigation of the cleavage reaction using Michaelis-Menton kinetics. We estimated Vmax to be 40 nM/min and KM to be 226 nM (Supplementary Figure S1). The apparent turn-over number kcat for the off-center bulge substrate was 4 per min, assuming that a dimer with two active sites cut a single double-stranded DNA on the two strands. The catalytic efficiency kcat/KM is thus 3 × 105 M−1 s−1. Cleavage of a Holliday junction by fowlpox resolvase in vitro was estimated at 13.8 per min in a single turn-over experiment, but only 0.24 per min in a multiple turn-over assay requiring completion of the catalytic cycle (5). Making the assumption that the rate limiting steps are the same for both substrates, the kinetic analysis suggests that cleavage is slower on the off-center bulge substrate, but this is associated with increased turn-over compared to the Holliday junction. Data in Figure 1G indicates that product is released efficiently after cleavage of the off-center bulge substrate, consistent with the idea that an increased rate of product release accounts for the acceleration of the reaction cycle.
To measure of the robustness of the assay, we calculated Z′- and Z-factor values, which provide a measure of both the dynamic range and variance. Z′- and Z-factor values range over −∞ < Z ≤ 1. Values greater than 0.5 indicate a large separation between the positive and negative controls with low variance suitable for high-throughput screening (15). Using data obtained from 5 days of consecutive screening, we calculated Z′- and Z-factor values of 0.78 and 0.68, respectively.
Screening compound libraries using the FP assays
We next used the off-center bulge substrate (sub F) to screen two small molecule libraries for inhibitors of resolvase DNA cleavage (Supplementary Table S2). Library 1, from the NSRB, contained a structurally diverse set of 130 540 compounds. Library 2 contained a structurally focused set of 2788 compounds from Merck designed to target enzymes of the RNAse H superfamily, the majority of which contain metal chelating pharmacophores, which have been reported to be important in HIV integrase and RNAase H inhibitors (10,11,16).
Percent inhibition (PI) was calculated for each compound from the FP measurements at a single concentration (see ‘Supplementary Methods’). For Library 1, we scored a compound as a hit if its PI was ≥2 SD from the mean PI of all compound-containing wells. This hit definition corresponded to compounds with PI values ≥15% in our assay. We identified 1936 compounds meeting this hit criteria, which corresponded to an overall hit rate of 1.5%. Using the same PI cut-off of 15% for Library 2, we obtained an overall hit rate of 45% for this library. Thus, we obtained a 30-fold higher hit rate for Library 2 (Supplementary Table S2). Since Library 2 contained primarily metal chelating compounds designed to target RNase H superfamily members, these data indicate that many of these metal chelating pharmacophores are inhibitors of poxvirus resolvase. The mechanism of inhibition of these metal chelators was not simple sequestration of the free Mg2+ cofactor away from the enzyme, because the Mg2+ concentration was 750-fold in excess of the inhibitor concentration.
Structure-activity analysis of 1-hydroxy-1,8-naphthyridin-2(1H)-one inhibitors
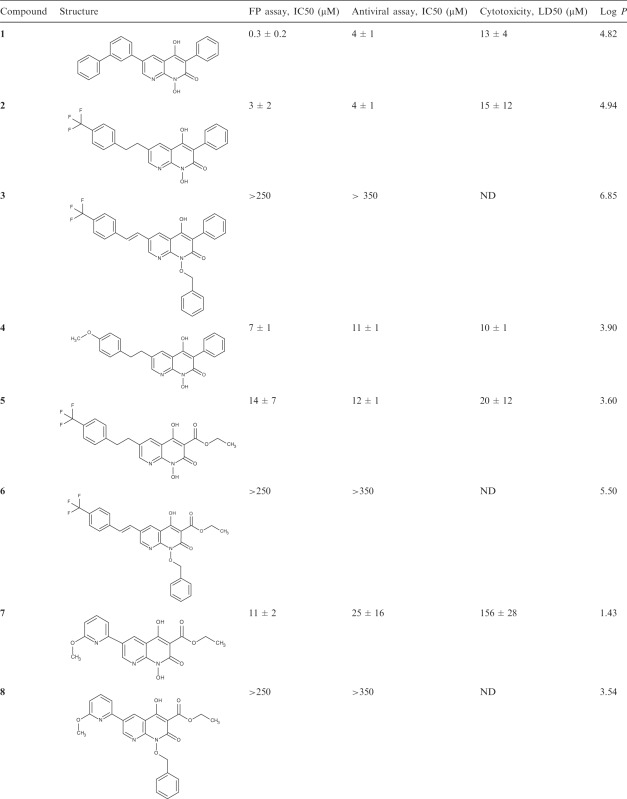
Based on initial results for resolvase inhibition in vitro, antiviral activity and toxicity, 1-hydroxy-1,8-naphthyridin-2(1H)-ones were selected as a chemical series for follow up SAR analysis (Figure 3 and Table 1). The 1-hydroxy-1,8-naphthyridin-2(1H)-one compounds have recently been reported to inhibit HIV RNase H, an enzyme structurally related to poxvirus resolvase (16). These compounds contain a potential metal chelating pharmacophore comprised of N8, the hydroxyl group at N1 and the carbonyl at position 2 in the naphthyridinone ring (Figure 3). Compound 1 (Table 1), the initial hit from the library screen, showed an IC50 value of 0.3 µM for resolvase inhibition in the FP assay. Resynthesis (Supplementary Data) and retesting of 1 confirmed that the activity was associated with the expected structure. To validate its activity, compound 1 was tested in resolvase cleavage reactions containing authentic Holliday junction substrates in vitro and found to show effective inhibition. Subsequent antiviral assays showed an IC50 value of 4 µM. Cytotoxicity however was high with LD50 value of 13 µM.
Figure 3.

The 1-hydroxynaphthrydinone backbone, showing the potential metal binding pharmacophore (shaded). For synthetic methods see Supplementary Figures S3 and 4.
Table 1.
SAR analysis of naphthyridone resolvase inhibitors
 |
ND indicates not done.
Compounds 2–8 were synthesized (Supplemental Methods in Supplementary Data) in an effort to increase potency, reduce cytotoxicity and begin to explore the SAR for this series of compounds. Since it is likely that the resolvase inhibitory activity of 1 is due to its ability to both bind to the active site as well as chelate an active site divalent metal, modifications to the structure of 1 were designed to explore these aspects of its activity. Modifications to the groups at the 3 and 6 positions around the naphthyridinone ring were made to determine if these structural components were essential for binding. Modifications to the N-hydroxy group were made to test whether metal chelation is essential for activity. Some of the substitutions were chosen based on the structures of other hits in the initial library, and for synthetic accessibility. Compounds 2 and 4 were synthesized to investigate the tolerance for substitutions at the 6 position of the naphthrydinone ring. Both were slightly less potent in the FP assay than compound 1 and showed no reduction in cytotoxicity. To test the importance of the potential metal chelating pharmacophore, an analog with the N1 position substituted with a benzyloxy group was tested and this eradicated activity, supporting the idea that metal binding by the pharmacophore in Figure 3 is essential for activity.
In an effort to improve the physiochemical properties of this series, the phenyl group at position 3 was substituted with an ethyl ester, yielding compounds 5–8. The ethyl ester was chosen based on its presence in active compounds the initial library (data not shown). This modification reduced the hydrophobicity (Table 1), which was higher in compounds 1–4 than is typical of inhibitors active in cells. Two pendant groups were compared at position 6 with the ethyl ester at position 3 (compounds 5 and 7). Compound 7 showed favorable characteristics. Initial tests of the IC50 in the FP assay showed a slight increase over compound 1. Inhibition of Holliday junction cleavage was monitored using gel-based assays and compound 7 was found to show a similar IC50 (Figure 4). As a test of specificity, activity of compound 7 was compared against the variola topoisomerase enzyme, and found to show no inhibitory activity (Supplementary Figure S2). Importantly, though the IC50 for viral infection was slightly increased, the LD50 was greatly increased, so that the differential between antiviral activity and LD50 was highest among the compounds studied (∼6-fold). Synthesis and testing of the N1 benzyloxy derivatives (6 and 8) confirmed for compounds 5 and 7 also that blocking the metal chelating pharmacophore abolished activity.
Figure 4.

Inhibition of cleavage of a Holliday junction substrate by fowlpox resolvase in the presence of compound 7. Holliday junction substrates were fluorescently labeled on one DNA strand. Reaction products were analyzed after separation by electrophoresis on a native polyacrylamide gel.
CONCLUSIONS
Variola virus is a category A agent due to concerns about its possible use as a biological weapon. A Holliday junction resolvase is required for poxvirus replication. Here, we report development of a bulged DNA substrate that reports resolvase cleavage activity efficiently using FP assays, making possible high-throughput screening. Besides the practical utility, the activity on this substrate suggests a new function for resolvase that may be important in linearizing branched DNA intermediates during poxvirus replication to restart DNA synthesis. We screened >100 000 compounds and found that 1-hydroxy-1,8-naphthyridin-2(1H)-one compounds constitute particularly active inhibitors. SAR analysis showed that potential metal chelating groups were important for inhibition, suggesting functional parallels with the FDA-approved inhibitors of HIV integrase. One 1-hydroxy-1,8-naphthyridin-2(1H)-one (compound 7) showed antiviral activity at concentrations below cytotoxic levels. Thus, these compounds represent starting points for developing inhibitors of variola virus for biodefense applications.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1 and 2, Supplementary Figures 1–4, Supplementary Data and Supplementary Methods.
FUNDING
National Institutes of Health NIAID Mid-Atlantic Regional Center of Excellence for Biodefense Research [Grant U54 AI057168 and UO1 U01 AI 082015]; training [T32 AI 07324 to M.J.C.]. Funding for open access charge: National Institutes of Health NIAID Mid-Atlantic Regional Center of Excellence for Biodefense Research [U54 AI057168 and UO1 U01 AI 082015].
Conflict of interest statement. Mike Miller and Jay Grobler are employees of Merck + Co., Inc.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to members of the Bushman laboratory and Rahul Kohli for help and suggestions.
REFERENCES
- 1.Garcia AD, Aravind L, Koonin EV, Moss B. Bacterial-type DNA holliday junction resolvases in eukaryotic viruses. Proc. Natl Acad. Sci. USA. 2000;97:8926–8931. doi: 10.1073/pnas.150238697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garcia AD, Moss B. Repression of vaccinia virus Holliday junction resolvase inhibits processing of viral DNA into unit-length genomes. J. Virol. 2001;75:6460–6471. doi: 10.1128/JVI.75.14.6460-6471.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Culyba MJ, Harrison JE, Hwang Y, Bushman FD. DNA cleavage by the A22R resolvase of vaccinia virus. Virology. 2006;352:466–476. doi: 10.1016/j.virol.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 4.Culyba MJ, Minkah N, Hwang Y, Benhamou OM, Bushman FD. DNA branch nuclease activity of vaccinia A22 resolvase. J. Biol. Chem. 2007;282:34644–34652. doi: 10.1074/jbc.M705322200. [DOI] [PubMed] [Google Scholar]
- 5.Culyba MJ, Hwang Y, Minkah N, Bushman FD. DNA binding and cleavage by the fowlpox virus resolvase. J. Biol. Chem. 2009;284:1190–1201. doi: 10.1074/jbc.M807864200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Culyba MJ, Hwang Y, Hu JY, Minkah N, Ocwieja KE, Bushman FD. Metal cofactors in the structure and activity of the fowlpox resolvase. J. Mol. Biol. 2010;28:182–195. doi: 10.1016/j.jmb.2010.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang W, Steitz TA. Recombining the structures of HIV integrase, RuvC, and RNase H. Structure. 1995;3:131–134. doi: 10.1016/s0969-2126(01)00142-3. [DOI] [PubMed] [Google Scholar]
- 8.Lovell S, Goryshin IY, Reznikoff WR, Rayment I. Two-metal active site binding of a Tn5 transposase synaptic complex. Nat. Struct. Biol. 2002;9:278–281. doi: 10.1038/nsb778. [DOI] [PubMed] [Google Scholar]
- 9.Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 2005;121:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 10.Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, et al. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science. 2000;287:646–650. doi: 10.1126/science.287.5453.646. [DOI] [PubMed] [Google Scholar]
- 11.Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Gonzalez Paz O, Hazuda DJ, et al. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J. Med. Chem. 2008;51:5843–5855. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- 12.Grobler JA, Stillmock K, Hu B, Witmer M, Felock P, Espeseth AS, Wolfe A, Egbertson M, Bourgeois M, Melamed J, et al. Diketo acid inhibitor mechanism and HIV-1 integrase: implications for metal binding in the active site of phosphotransferase enzymes. Proc. Natl Acad. Sci. USA. 2002;99:6661–6666. doi: 10.1073/pnas.092056199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature. 2010;464:232–236. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamilton MD, Nuara AA, Gammon DB, Buller RM, Evans DH. Duplex strand joining reactions catalyzed by vaccinia virus DNA polymerase. Nucleic Acids Res. 2007;35:143–151. doi: 10.1093/nar/gkl1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 16.Williams PD, Staas DD, Venkatraman S, Loughran HM, Ruzek RD, Booth TM, Lyle TA, Wai JS, Vacca JP, Feuston BP, et al. Potent and selective HIV-1 ribonuclease H inhibitors based on a 1-hydroxy-1,8-naphthyridin-2(1H)-one scaffold. Bioorg. Med. Chem. Lett. 2010;20:6754–6757. doi: 10.1016/j.bmcl.2010.08.135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.