


Abstract
Accurate estimation of expression levels from RNA-Seq data entails precise mapping of the sequence reads to a reference genome. Because the standard reference genome contains only one allele at any given locus, reads overlapping polymorphic loci that carry a non-reference allele are at least one mismatch away from the reference and, hence, are less likely to be mapped. This bias in read mapping leads to inaccurate estimates of allele-specific expression (ASE). To address this read-mapping bias, we propose the construction of an enhanced reference genome that includes the alternative alleles at known polymorphic loci. We show that mapping to this enhanced reference reduced the read-mapping biases, leading to more reliable estimates of ASE. Experiments on simulated data show that the proposed strategy reduced the number of loci with mapping bias by ≥63% when compared with a previous approach that relies on masking the polymorphic loci and by ≥18% when compared with the standard approach that uses an unaltered reference. When we applied our strategy to actual RNA-Seq data, we found that it mapped up to 15% more reads than the previous approaches and identified many seemingly incorrect inferences made by them.
INTRODUCTION
Advances in high-throughput sequencing have enabled the development of sequence-based approaches for transcriptome quantification. RNA sequencing (RNA-Seq) makes use of next-generation sequencing to estimate the expression levels of individual genes. This is achieved by first converting messenger RNA to complementary DNA, then sequencing using deep-sequencing technologies (1,2) and finally quantifying expression levels based on the relative numbers of reads obtained from individual transcripts. However, multiple sources of bias inherent to these technologies need to be accounted for to more accurately quantify gene expression levels (3).
The vast amount of information captured by sequence reads allows us to go beyond expression-level quantification and answer more specific questions, such as the identification of loci with allele-specific expression (ASE). ASE refers to instances where one of the two alleles at a heterozygous locus in an individual is expressed more than the other (4–8). In RNA-Seq data, a heterozygous position in the genome with statistically significant difference in the number of reads carrying the two alleles indicates ASE.
Reliable estimation of ASE depends on the ability to accurately map the reads to their correct positions in the genome. The reads are usually mapped to a reference genome that contains only one of the possible alleles at any polymorphic locus. However, this method is inherently biased. Reads with the reference alleles, i.e. the alleles contained in the reference genome, exactly match the reference genome, whereas reads that contain non-reference alleles differ from the reference genome in at least one position. Hence, the reads with the reference allele are more likely to be mapped than the reads with non-reference alleles. Degner et al. (9) have shown that these read-mapping biases have a significant effect on the estimation of ASE. To correct this bias, they modified the reference genome, so that each known single-nucleotide polymorphism (SNP) locus is masked with a third base that is neither the reference allele nor non-reference allele. Although this method eliminated the systematic bias toward the reference allele, they also observed that it did not eliminate biases at individual loci. In one experiment, they simulated data with an equal number of reads carrying the reference and non-reference alleles at each polymorphic locus. Then, they mapped these reads to the original, unmodified reference genome and found that the proportion of mapped reads with the reference allele was significantly higher than 50%, indicating a systematic bias toward the reference allele. When they mapped the reads to a modified reference in which polymorphic loci were masked, they found that, on average, the proportion of mapped reads carrying the reference allele was closer to 50%. However, there were many loci at which ≥75% of the mapped reads contained one of the two alleles. This suggests that although the systematic bias toward the reference allele may have been eliminated, the bias was probably just redistributed, with some loci biased toward the reference allele and others toward the non-reference allele. This led them to conclude that some individual SNPs were inherently biased due to problems in read mapping. A locus can be considered inherently biased if reads carrying one of the two alleles have an equally good or better match at some other location in the genome, thereby resulting in a read-mapping bias at the current location.
In this article, we argue that the fundamental source of read-mapping bias at most loci is the absence of non-reference alleles in the reference genome. Therefore, we propose an alternative strategy that is based on the construction of an enhanced reference genome that, in addition to the reference alleles, contains all alternate alleles at each known SNP locus. Using simulated reads, we show that the mapping biases at most of these loci can be eliminated by using such an enhanced reference, strongly suggesting that many of these loci are not inherently biased. Also, by applying our approach to actual RNA-Seq data set provided by Degner et al. (9), we show that some loci previously reported to exhibit ASE, in fact, do not show significant ASE. In addition, we identify new loci that are statistically significant for ASE.
The fundamental idea of incorporating SNP variants into the reference has been previously implemented in the GSNAP program (10) for detecting variants. However, the solution presented by them is specific to their hash-based mapping algorithm. In contrast, the solution we propose builds a generalized enhanced reference that can be used with any mapping algorithm. Recently, Rozowsky et al. (11) proposed an alternative strategy that constructs the personal diploid genome of a subject. This approach requires the availability of genotype data for the specific individual, along with genotype data for a pedigree of related individuals, for accurate phasing of the genotype into the constituent haplotypes. Although such an approach was feasible due to the availability of all the necessary genotype data for the specific HapMap (12) individual selected in their study, their approach is not generalizable to any arbitrary individual. Turro et al. (13) proposed a similar approach that is based on phasing the haplotypes from genotype calls made by an initial mapping. However, in addition to intrinsic difficulties in phasing accurately, the genotype calls based on the initial mapping can themselves be incorrect at many loci due to the inability to map any of the reads with the alternate allele. In contrast, the approach we present in this study only requires knowledge of the polymorphic loci in the human genome and hence can be used to map the RNA-Seq data of any individual. Other approaches to this problem include the GenomeMapper software (14) that attempts to map the reads to multiple genomes. However, their approach, in addition to being computationally intensive, can only map the reads to one of the previously sequenced genomes, whereas our approach has the potential to map reads to novel haplotypes that need not be present in any previously sequenced genome.
MATERIALS AND METHODS
The basis for our approach is the observation that most read-mapping biases are caused by the absence of alternate alleles (i.e. the non-reference alleles) in the reference genome. The absence of these alleles implies that a read with a non-reference allele differs from the reference genome in at least one position. Furthermore, sequencing errors or multiple SNP loci within a single read can result in more than one mismatch between the read and the reference genome. This leads to the read-mapping software failing to map the read, or worse, mapping the read to an incorrect locus. This, in turn, results in the underestimation of the expression levels of non-reference alleles, causing both false-positive and false-negative inferences of ASE.
Construction of an enhanced reference genome
Our method attempts to correct the biases in read mapping by constructing an enhanced reference genome that incorporates alternate alleles at each known SNP position. Assuming a fixed read length r, we add sequence fragments to the reference genome to create an enhanced reference, so that every possible length-r segment that overlaps a non-reference allele is part of an added sequence fragment. Figure 1A shows the added fragments for two SNPs that are at least r − 1 bases apart. In cases where there are multiple SNPs within any r-window, we need to ensure that every possible length-r segment that overlaps any of the possible haplotypes is represented by an added sequence fragment. Figure 1B shows an example with two SNPs within a single r-window. In this case, three separate sequence fragments, each representing a haplotype not present in the reference genome, need to be added. The left and right boundaries of the added fragments are selected, so that each r-window of the added segment is unique with respect to both the reference genome and the other added segments that overlap it.
Figure 1.

Schematic representation of the enhanced segments added for two SNPs, S1 and S2. The read length is indicated by r. (A) Enhanced segments added when the distance between two adjacent SNPs S1 and S2 is ≥r. No read can overlap both SNPs in this scenario. A single enhanced segment with the non-reference allele is added for each SNP. The enhanced segment extends r − 1 bases on either side of the SNP to ensure an exact match with any read carrying the non-reference allele. (B) Scenario when the distance between S1 and S2 is <r − 1. Because there can be reads that overlap both SNPs, we need to add three segments to cover all possible haplotypes formed by S1 and S2. It is also necessary that none of the added segments is identical to another enhanced segment (or the reference) in any window of length ≥r. This ensures that the read uniquely maps to the reference or one of the enhanced segments. Multiple solutions satisfying these conditions are possible. The figure shows one such possible solution.
There are two fundamental objectives in constructing the enhanced reference: (i) the enhanced reference should contain all the possible haplotypes within every r-window in the genome and (ii) none of the added segments in the enhanced reference should be identical to another added segment (or to the original reference) in an r-window. We designed a greedy algorithm to construct an enhanced reference that conforms to both these objectives for a fixed read length r. The algorithm has been implemented in C++ using SeqAn libraries (15). Details about the algorithm are provided in the Supplementary Data.
Simulated data
To evaluate the effectiveness of the enhanced reference in correcting read-mapping biases, we simulated reads overlapping exonic SNP loci in the human chromosome 1 (build 36.3). We used the HapMap Yoruba SNPs from release r22 (12,16) and exon positions from the National Center for Biotechnology Information MapView (ftp://ftp.ncbi.nih.gov/genomes/MapView/Homo_sapiens/sequence/ (7 May 2012, date last accessed)). Using a procedure identical to that used by Degner et al. (9), we generated simulated reads with lengths 35, 70 and 100. At each SNP position, we generated equal numbers of reads with the reference allele and non-reference allele, obtaining one read for each possible position overlapping the SNP in both strands. We arbitrarily assigned a high-quality score of 66 to each base of the simulated reads. For each read length, we also generated two data sets with random errors added, such that each base in the read had a Bernouli probability of 0.01 or 0.02 of being randomly changed to any other base.
RNA-Seq data
RNA-Seq data set with accession number GSE18156 provided by Degner et al. (9) was downloaded from Gene Expression Omnibus. The data set contains 35-bp Illumina reads generated from two HapMap Yoruba lymphoblastoid cell lines (GM19238 and GM19239). Details about the RNA-Seq process are given in Degner et al. (9). The data set contains 15 579 717 reads from the individual GM19238 and 16 780 153 reads from the individual GM19239.
Mapping the reads
Both simulated and actual RNA-Seq reads were mapped against the unaltered reference genome, SNP-masked reference genome and enhanced reference genome using MAQ version 0.7.1 (17) and BWA version 0.5.0 (18) software. For the actual RNA-Seq data, both programs were run with default parameters. For mapping the simulated data with MAQ, we used the default parameters for the 35-bp reads. For mapping the 70- and 100-bp reads, one of the parameters, the maximum sum of quality scores at mismatch bases (the command-line parameter e) was set to 300 and 500, respectively. We used the default settings to map the simulated reads with the BWA program. We decided not to filter the mapped reads based on mapping quality, as, by design, the enhanced reference contains duplicated segments, which can result in low mapping quality scores because the mapping software is not aware that these are artificially induced duplications. Downstream analysis of the mapped reads was performed with the help of the SAMtools package (19).
RESULTS
The mappings generated by the BWA and MAQ programs produced very similar ASE profiles when mapping qualities were ignored. Thus, in what follows, we only present results from the MAQ mappings. The results from the BWA mappings and results when the reads were filtered based on mapping quality are provided in the Supplementary Data.
Construction of the masked reference and enhanced reference
To allow for a comparison with Degner et al. (9), we used the human genome build 36.3. The combined length of all chromosomes in this version of the human genome is 3.08 Gbp. Both the masked and enhanced references were constructed using the Yoruba SNPs from HapMap release r22. This data set consists of 3.7 million SNPs. We limited the maximum number of SNPs within an r-window to 5 in constructing the enhanced reference. Because of this limit, if there were more than five SNPs within an r-window, the enhanced segments were generated only for the first five SNPs within the window, and the remaining were ignored. The combined length of the entire set of generated enhanced segments was 275 Mbp, which represented an ∼10% increase in the length of the original, unaltered reference.
Results on simulated reads
We generated simulated reads for both alleles at each SNP, with one read from each strand for all positions overlapping the SNP, as discussed in the ‘Materials and Methods’ section. Figure 2 shows the mapping statistics for the three mapping methods when we mapped 35-bp reads with different error rates using the MAQ program. Figure 2A shows that the enhanced reference method consistently mapped a higher percentage of the input reads for all error rates. The difference between the masked approach and the enhanced reference approach widened as the error rate increased. Although the masked approach could only map 83% of the reads at an error rate of 0.02, the enhanced reference approach still mapped ≥97% of the reads. Similar trends held for longer read lengths: Supplementary Figure S3 shows that the enhanced reference approach consistently outperformed the other two approaches for simulated reads of length 70 and 100.
Figure 2.

Mapping results of simulated 35-bp reads for the three approaches. (A) The enhanced reference approach was able to map a much higher percentage of the input reads, especially for higher error rates. (B) Approximately 50% of the mapped reads carried the reference allele, both for the masked reference and the enhanced reference approaches.
When we mapped 35-bp reads with no errors against the unaltered reference, 50.20% of the mapped reads carried the reference allele. When we increased the error rate to 0.01 and 0.02 mutations per base, however, the proportion of mapped reads with the reference allele increased to 51.22 and 53.32%, respectively. Mapping the same 35-bp reads against the masked reference seemed to almost completely eliminate this systematic bias: 49.93, 49.93 and 49.95% of the mapped reads, on average, carried the reference allele for error rates of 0.00, 0.01 and 0.02, respectively (Figure 2B). Mapping against the enhanced reference resulted in similar numbers, with 49.96, 49.96 and 49.99% of the mapped reads, on average, carrying the reference allele for error rates of 0.00, 0.01 and 0.02, respectively. The results were similar for longer read lengths. Based on these results, one would be tempted to conclude that the enhanced reference approach offers no significant improvements over the masked reference approach in eliminating the mapping bias.
Analysis of the bias at individual loci, however, presented a drastically different picture. Figure 3 shows histograms of the fraction of times the mapped reads contained the reference allele for each of the three mapping methods. For mapping against the unaltered reference, Figure 3A shows that a considerable percentage of loci were biased toward the reference allele. The number of biased loci increased with the error rate. There were almost no loci that showed a bias toward the non-reference allele. Figure 3B shows that mapping against the masked reference only re-distributed this bias: significant numbers of loci were still biased toward the reference allele, with an equally large number of loci biased toward the non-reference allele. For error rates of 0.00 and 0.01, the overall proportion of unbiased loci was virtually the same as mapping against the unaltered reference, whereas there was a significant improvement for error rate of 0.02. Mapping against the enhanced reference, as shown in Figure 3C, resulted in drastic improvements for all error rates: a very small proportion of loci showed a bias toward the reference allele or the non-reference allele. The loci that showed bias were only slightly biased: there were almost no loci with ≥70% of the mapped reads carrying the reference allele or non-reference allele. Biases at individual loci for the simulated 70- and 100-bp reads showed similar trends, with the enhanced reference method consistently outperforming the masked reference method. However, the overall number of loci with read-mapping bias decreased with increasing read lengths. This was expected, as longer reads were more likely to be mapped correctly despite mismatches. Supplementary Figures S4 and S5 show the histograms for 70-bp reads and 100-bp reads, respectively.
Figure 3.

Histograms of the proportions of mapped reads for the different mapping approaches. (A) Mapping against the unaltered reference showed a clear bias toward the reference allele. (B) Mapping against the masked reference showed that there was no systematic bias, but a significant percentage of the loci were still biased. (C) Mapping against the enhanced reference eliminated the bias at the majority of the loci.
For each mapping, we computed the number of biased loci, i.e. the number of loci that deviated from the expected 50:50 distribution of the reference and the non-reference alleles among the mapped reads. We computed the biased loci for various levels of bias, where one of the alleles represents ≥55, ≥52.5 or ≥51% of the mapped reads. For the error-free 35-bp reads, at the ≥55% bias level, the numbers of biased loci were 141, 319 and 116, respectively, for mapping against the unaltered, masked and enhanced reference. This indicates that the masked reference approach significantly increased the number of strongly biased loci when compared with the unaltered reference approach. In comparison with the masked reference approach, the number of biased loci decreased by ∼63% in the enhanced reference approach. Similar trends were observed for both longer reads and larger error rates, with the enhanced reference approach showing a significant reduction in the number of biased loci. Detailed numbers for various read lengths, error rates and significance levels are given in Supplementary Table S1.
A locus can appear to be biased either due to shortcomings in the used mapping approach or due to some characteristic of the sequence around the locus that renders it impossible to map reads with one of the alleles. It is intriguing, however, why some loci appear to be biased even in the enhanced reference approach. Because the enhanced reference always contains an exact match for error-free reads, one might expect that the reads would always map to the correct location. Additional analysis of the mappings revealed that these biased loci were always from repeated regions. The reads carrying one of the alleles from these loci had an exact match with some other location in the genome. Thus, the mapping algorithm arbitrarily assigned the read to one of the exact matches, which led to the mapping of some of these reads to an alternate location. The reads carrying the alternate allele, however, may not necessarily have an exact match somewhere else, and all of them mapped to the intended location, thereby resulting in an imbalance between the numbers of mapped reads from the two alleles. One such SNP locus is shown in Supplementary Figure S6. The likelihood of having an exact match somewhere else in the genome decreases with increasing read length. Hence, we noticed that the number of biased loci was smaller for longer read lengths as shown in Supplementary Table S1.
We obtained similar results with BWA when mapping qualities were ignored. Supplementary Figures S7–S10 provide details about these mappings. When we limited the analysis to reads with non-zero mapping quality, however, we observed significant differences between MAQ and BWA mappings. Most of these differences, however, were due to different methodologies used to compute the mapping quality. Additional information about these differences and detailed results of read-mapping biases for reads with mapping quality >0 are presented in the Supplementary Data (Supplementary Figures S11–S16 and Supplementary Table S2).
Results on RNA-Seq data
We mapped the 35-bp reads from two Yoruba HapMap individuals (GM19238 and GM19239) provided by Degner et al. (9) using the MAQ program with default parameters. We mapped these reads to the standard unaltered reference genome, the reference genome in which all the Yoruba HapMap SNPs were masked with a third allele, and the enhanced reference genome, which was constructed as explained at the beginning of this Section. From the mapped reads, we isolated the reads overlapping the HapMap Yoruba SNP loci within exons and untranslated regions (UTRs). There were ∼95 000 such SNPs in total. Table 1 lists the number of reads carrying the reference allele, the known non-reference allele or some other allele. The appearance of alleles that were neither the reference allele nor the non-reference allele was, in most cases, due to sequencing errors or mis-mapped reads. Results presented in Table 1 suggest that the enhanced reference approach was successful in mapping the maximum number of reads in both individuals. For GM19238, the enhanced reference approach mapped 6% more reads than the unaltered reference approach and 15% more reads than the masked reference approach. Similarly, for GM19239, the enhanced reference approach mapped 5% more reads than the unaltered reference approach and 13% more reads than the masked reference approach. These results strongly suggest that the enhanced reference approach possesses a superior ability to map reads that the other two methods failed to map.
Table 1.
Reads mapped to HapMap heterozygous exonic and UTR Yoruba SNP loci in each method
| Individual | GM19238 |
GM19239 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Method | Ref | Non-ref | Other | Total | Ref % | Ref | Non-ref | Other | Total | Ref % |
| Unaltered | 43 647 | 35 672 | 644 | 79 963 | 55.0 | 38 727 | 32 299 | 655 | 71 681 | 54.5 |
| Masked | 37 167 | 35 447 | 692 | 73 306 | 51.2 | 33 575 | 32 218 | 716 | 66 509 | 51.0 |
| Enhanced | 43 840 | 40 211 | 654 | 84 705 | 52.2 | 38 847 | 35 490 | 679 | 75 016 | 52.3 |
The columns labeled ‘other’ provide the number of reads carrying an allele that is neither the reference allele nor the non-reference allele.
Although 54–55% of the reads mapped to the unaltered reference carried the reference allele, only ∼52% of the reads mapped to the enhanced reference carried the reference allele. Given that the actual number of mapped reads carrying the reference allele was higher in the enhanced reference approach than the other two methods, this result indicates the ability of the enhanced reference approach to successfully map many more reads carrying the non-reference allele, rather than inefficiency in mapping reads carrying the reference allele. In the masked reference approach, only 51% of the mapped reads carried the reference allele. Although this number is the closest to 50%, it is important to note that the number of reads carrying the reference allele in an actual data set may not necessarily be 50%. One or two highly expressed loci that are allele specific can tilt the numbers one way or the other. Comparing the number of loci that exhibit higher expression of the reference allele with the number of loci that exhibit higher expression of the non-reference allele is a more appropriate way to analyze the effectiveness of each method in eliminating biases.
Accordingly, to obtain a more accurate picture, we first filtered the results to retain only the loci with ≥20 mapped reads. Table 2 lists the numbers of loci and numbers of mapped reads when the results were filtered with this criterion. The results show that the enhanced reference approach had the largest number of loci with ≥20 mapped reads, as it was able to map the largest number of reads overall. In both individuals, the masked reference approach was the closest to 50% in the proportion of reads mapping to the reference. Altogether, there were 716 distinct loci in GM19238 with ≥20 mapped reads using any one of the three methods. There was only one locus to which the enhanced reference approach mapped <20 reads, but the unaltered reference approach mapped ≥20 reads. In case of GM19239, there were no loci to which the enhanced approach mapped <20 reads, but any of the other methods mapped ≥20 reads.
Table 2.
Numbers of exonic and UTR loci with ≥20 mapped reads and the numbers of reads mapping to these loci
| Individual | GM19238 |
GM19239 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Method | Loci | Ref | Non-ref | Other | Total | Ref % | Loci | Ref | Non-ref | Other | Total | Ref % |
| Unaltered | 685 | 29 084 | 22 230 | 425 | 51 739 | 56.7 | 772 | 23 882 | 18 358 | 463 | 42 703 | 56.5 |
| Masked | 645 | 23 268 | 21 868 | 408 | 45 544 | 51.6 | 727 | 19 390 | 18 038 | 459 | 37 887 | 51.8 |
| Enhanced | 715 | 29 445 | 26 150 | 432 | 56 027 | 53.0 | 802 | 24 171 | 20 902 | 483 | 45 556 | 53.6 |
The enhanced reference approach was able to map the largest number of reads.
Next, we computed the loci with ≥20 reads that showed significant ASE in each method. To determine the number of loci with significant ASE, we used the statistical testing procedure described by Degner et al. (9). We compared the observed distribution of the proportion of mapped reads coming from the reference allele to the expected distribution from symmetric binomial sampling. At each SNP, we used two one-sided binomial tests to evaluate the complementary hypotheses that expression of the reference allele was greater than or less than 0.5. We analyzed each mapping separately and applied a different P-value threshold in each mapping, corresponding to a false discovery rate (FDR) of 1% in that mapping. Table 3 lists the number of loci with significant ASE in each method. The table shows that, for all three methods, the number of loci with ASE specific to the reference allele was higher than that for the non-reference allele. In particular, the unaltered reference approach was heavily biased toward the reference allele: very few loci were specific to the non-reference allele in both individuals under this mapping. The proportions of the loci specific to the reference alleles in the masked reference approach and the enhanced reference approach were similar to each other Figure 4 shows the number of overlapping loci reported by each method. The enhanced reference approach identified many loci that were missed by the other two approaches. The details of the loci with significant ASE based on the mapping to the enhanced reference are shown in Supplementary Table S5 for GM19238 and Supplementary Table S6 for GM19239, and the histograms of reference bias for all loci with ≥20 mapped reads are shown in Supplementary Figure S19. We did not filter out pseudoautosomal regions in the sex chromosomes, and, hence, we observed both alleles of the locus rs7205 in chrX being expressed in GM19239, even though this cell line was from a male individual. There were many more loci from chrX in the data from GM19238, who was female.
Table 3.
Number of loci with significant ASE for each method (at 1% FDR)
| Individual | GM19238 |
GM19239 |
||||
|---|---|---|---|---|---|---|
| Method | Ref | Non-ref | Total | Ref | Non-ref | Total |
| Unaltered | 55 | 6 | 61 | 43 | 12 | 54 |
| Masked | 24 | 13 | 37 | 21 | 18 | 39 |
| Enhanced | 40 | 21 | 61 | 38 | 28 | 66 |
Figure 4.
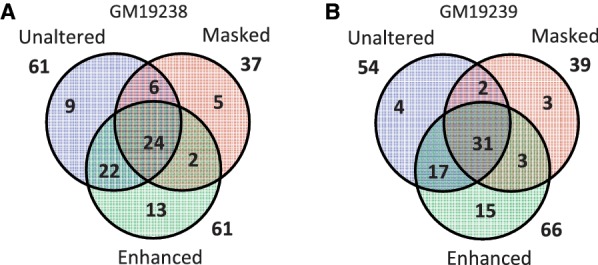
Overlaps between loci reported to be allele specific in different methods. Venn diagrams show the overlaps between the number of loci that were positive for ASE at 1% FDR in (A) GM19238 and (B) GM19239. The masked reference approach missed many loci reported by the other two methods. Loci unique to the masked approach were the result of its inability to map reads with one of the alleles and, hence, were false positives. The enhanced reference approach identified many loci that were missed by the other two approaches.
Supplementary Tables S7 and S8 show the reads mapped to each allele in all the three methods. There were many loci that were significant for ASE in the mapping to the enhanced reference that were not significant using the other two methods. In each such instance, the difference was due to the enhanced reference approach being able to map many reads carrying the reference allele or the non-reference allele that the other two approaches failed to map. For instance, SNP rs11619791 in GM19238 (Supplementary Table S7) had only a few reads mapping to both alleles in both the unaltered reference and masked reference approaches. However, mapping to the enhanced reference showed that there were 140 reads that carried the non-reference allele, whereas only four reads carried the reference allele, clearly indicating a strong signal of ASE.
Many loci indicate that the masked reference approach was unable to map the reads carrying both the reference and non-reference alleles. For instance, the masked reference approach was able to map only six reads to SNP rs2814966 in GM19238 (Supplementary Table S7), which did not provide a strong signal for ASE even though all six reads carried the reference allele. The enhanced reference approach mapped as many as 130 reads to this locus, all of them carrying the reference allele, which provided strong support for the hypothesis that this locus exhibits ASE. In this particular case, 121 of these reads were also mapped to the unaltered reference. However, there were many cases in which mapping to the unaltered reference produced incorrect results: in case of SNP rs1042448 in GM19239 (Supplementary Table S8), both the unaltered reference and masked reference approaches failed to map a large number of reads carrying the non-reference allele, which led to the incorrect identification of this locus as positive for ASE. In the mapping to the enhanced reference, we observed that 61 reads carried the non-reference allele in addition to 57 reads that carried the reference allele, which indicated that there was no ASE at this locus.
Interestingly, even some highly expressed loci were affected by mapping bias. The SNP rs1803621 in GM19238 (Supplementary Table S7) showed significant ASE in mapping to both the unaltered reference (2694 reads carried the reference allele and 1690 reads carried the non-reference allele) and the masked reference (2269 reads carried the reference allele and 1695 reads carried the non-reference allele). However, mapping to the enhanced reference indicated that the numbers were almost evenly balanced (2705 reads carried the reference allele and 2541 reads carried the non-reference allele), which did not indicate any significant ASE. Altogether, the results from the actual RNA-Seq data showed that the enhanced reference approach was effective in correcting read-mapping biases and in eliminating both false positives and false negatives in the identification of loci with ASE.
DISCUSSION
Herein, we presented a simple, practical and generalizable approach to reduce read-mapping biases for accurate identification of ASE. Results on simulated and actual RNA-Seq data show that our approach is superior to alternative approaches that use the standard reference or a SNP-masked reference. In addition to reducing mapping biases, our approach is also effective in mapping a significant percentage of the reads that cannot be mapped by other methods. The ability to map these reads results in a more accurate estimation of expression levels. We observed, however, that among the loci that showed statistically significant ASE in the actual RNA-Seq data, there were more loci specific to the reference allele than to the non-reference allele. We conjecture that this might be due to either unresolved biases in read mapping or artifacts in the RNA-Seq data.
Similar to masking, the enhanced reference approach can only correct mapping biases at known SNP loci. In practice, it is reasonable to expect that every individual has at least some variations not listed in dbSNP or other SNP databases. Correcting mapping biases at these loci might require a two-stage approach in which the reads are first mapped to an enhanced reference constructed from a standard set of SNPs. Novel, putative SNPs should be identified from this mapping, so that an individualized enhanced reference could then be constructed to accommodate these novel SNPs. Mapping against such an individualized enhanced reference would eliminate biases at these novel SNP loci.
The approach we present herein works best with short- to moderate-sized read lengths, where the variability in read lengths is small. When the variability in read lengths is too large, the shorter reads might match too many enhanced segments in the enhanced reference, which might trigger the mapping algorithms to assume that the read is coming from a repeat region and ignore all the alignments. In theory, too many SNPs within an r-window make it impractical to incorporate all possible haplotypes into the enhanced segments. However, in practice, we found that this situation is extremely rare in actual data sets, and, even when it occurs, it has minimal impact on the performance of the enhanced reference approach. Details about the frequency of these high-SNP-density regions and the mapping biases in these regions are provided in the Supplementary Data (Supplementary Tables S3 and S4; Supplementary Figures S17 and S18).
Our results strongly suggest that many reads carrying a non-reference allele fail to map to the standard reference. This might significantly affect the overall expression levels of the genes with a high density of polymorphic loci. Table 4 shows a few loci in our results from actual RNA-Seq data that support this hypothesis: in each instance, our approach was able to map >100 reads, whereas the other methods mapped ≤20 reads. It is reasonable to expect that these differences can significantly affect the expression levels of the genes containing these loci. In our analysis of actual RNA-Seq data, we considered only heterozygous loci. There could be many more instances such as these if homozygous loci are also taken into consideration, thus underscoring the importance of the enhanced reference approach.
Table 4.
A few instances where our approach is able to map >100 reads where the other methods map very few reads
| SNP ID | Chromosome | Position (hg 18) | Individual | Unaltered |
Masked |
Enhanced |
|||
|---|---|---|---|---|---|---|---|---|---|
| Ref | Non-ref | Ref | Non-ref | Ref | Non-ref | ||||
| rs1807676 | chr22 | 25 613 521 | GM19238 | 0 | 0 | 0 | 0 | 0 | 101 |
| rs11619791 | chr13 | 24 568 984 | GM19238 | 5 | 3 | 2 | 1 | 4 | 140 |
| rs12610462 | chr19 | 22 343 302 | GM19238 | 0 | 13 | 0 | 8 | 0 | 259 |
| rs1807676 | chr22 | 25 613 521 | GM19239 | 0 | 1 | 0 | 1 | 0 | 101 |
| rs7662013 | chr4 | 174 791 881 | GM19239 | 0 | 18 | 0 | 14 | 0 | 171 |
AVAILABILITY
The executables to construct the enhanced reference genome and the Perl scripts to analyze the mapped reads are available for download from http://www.bhsai.org/downloads/ase/ (7 May 2012, date last accessed).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–8 and Supplementary Figures 1–19.
FUNDING
Funding for open access charge: United States (U.S.) Department of Defense (DoD) High Performance Computing Modernization Program, under the High Performance Computing Software Applications Institutes Initiative.
Conflict of interest statement. None declared. The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the U.S. Army or the U.S. DoD. This article has been approved for public release with unlimited distribution.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Degner et al. (9) for making their RNA-Seq data available and Jacob Degner for providing us some of the R scripts for data analysis.
REFERENCES
- 1.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 2.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwartz S, Oren R, Ast G. Detection and removal of biases in the analysis of next-generation sequencing reads. PLoS One. 2011;6:e16685. doi: 10.1371/journal.pone.0016685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heap GA, Yang JH, Downes K, Healy BC, Hunt KA, Bockett N, Franke L, Dubois PC, Mein CA, Dobson RJ, et al. Genome-wide analysis of allelic expression imbalance in human primary cells by high-throughput transcriptome resequencing. Hum. Mol. Genet. 2010;19:122–134. doi: 10.1093/hmg/ddp473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan H, Yuan W, Velculescu VE, Vogelstein B, Kinzler KW. Allelic variation in human gene expression. Science. 2002;297:1143. doi: 10.1126/science.1072545. [DOI] [PubMed] [Google Scholar]
- 6.Knight JC. Allele-specific gene expression uncovered. Trends Genet. 2004;20:113–116. doi: 10.1016/j.tig.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Milani L, Lundmark A, Nordlund J, Kiialainen A, Flaegstad T, Jonmundsson G, Kanerva J, Schmiegelow K, Gunderson KL, Lonnerholm G, et al. Allele-specific gene expression patterns in primary leukemic cells reveal regulation of gene expression by CpG site methylation. Genome Res. 2009;19:1–11. doi: 10.1101/gr.083931.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ronald J, Akey JM, Whittle J, Smith EN, Yvert G, Kruglyak L. Simultaneous genotyping, gene-expression measurement, and detection of allele-specific expression with oligonucleotide arrays. Genome Res. 2005;15:284–291. doi: 10.1101/gr.2850605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Degner JF, Marioni JC, Pai AA, Pickrell JK, Nkadori E, Gilad Y, Pritchard JK. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics. 2009;25:3207–3212. doi: 10.1093/bioinformatics/btp579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rozowsky J, Abyzov A, Wang J, Alves P, Raha D, Harmanci A, Leng J, Bjornson R, Kong Y, Kitabayashi N, et al. AlleleSeq: analysis of allele-specific expression and binding in a network framework. Mol. Syst. Biol. 2011;7:522. doi: 10.1038/msb.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turro E, Su SY, Goncalves A, Coin LJ, Richardson S, Lewin A. Haplotype and isoform specific expression estimation using multi-mapping RNA-seq reads. Genome Biol. 2011;12:R13. doi: 10.1186/gb-2011-12-2-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneeberger K, Hagmann J, Ossowski S, Warthmann N, Gesing S, Kohlbacher O, Weigel D. Simultaneous alignment of short reads against multiple genomes. Genome Biol. 2009;10:R98. doi: 10.1186/gb-2009-10-9-r98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doring A, Weese D, Rausch T, Reinert K. SeqAn an efficient, generic C++ library for sequence analysis. BMC Bioinformatics. 2008;9:11. doi: 10.1186/1471-2105-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAM tools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.