


Abstract
DNA mimic proteins have evolved to control DNA-binding proteins by competing with the target DNA for binding to the protein. The Ocr protein of bacteriophage T7 is the most studied DNA mimic and functions to block the DNA-binding groove of Type I DNA restriction/modification enzymes. This binding prevents the enzyme from cleaving invading phage DNA. Each 116 amino acid monomer of the Ocr dimer has an unusual amino acid composition with 34 negatively charged side chains but only 6 positively charged side chains. Extensive mutagenesis of the charges of Ocr revealed a regression of Ocr activity from wild-type activity to partial activity then to variants inactive in antirestriction but deleterious for cell viability and lastly to totally inactive variants with no deleterious effect on cell viability. Throughout the mutagenesis the Ocr mutant proteins retained their folding. Our results show that the extreme bias in charged amino acids is not necessary for antirestriction activity but that less charged variants can affect cell viability by leading to restriction proficient but modification deficient cell phenotypes.
INTRODUCTION
DNA mimic proteins have evolved to control DNA-binding proteins by competing with the DNA for binding. Their mimicry of the structure and electrostatics of DNA means they can slot into the nucleic acid binding groove on their target protein and turn the DNA-binding protein ‘off’ (1–3). The two most striking examples of DNA mimics are Ocr from the lytic bacteriophage T7 (4) and ArdA (5) from conjugative plasmids and transposons that mediate horizontal gene transfer. Both of these proteins enhance the propagation of the phage or mobile elements by overcoming the host restriction/modification (RM) system which would normally destroy the invading DNA (6,7).
The first protein to be produced during infection of Escherichia coli by bacteriophage T7 is overcome classical restriction (Ocr), the product of gene 0.3 (8,9). The Ocr protein is the best characterized example of an anti-RM protein and of a structural mimic of DNA (10–20). Ocr is a highly negatively charged protein with a similar shape to that of a bent, double-stranded B-form DNA molecule ∼24 base pairs in length.
This molecular mimicry accounts for the ability of Ocr to inhibit, virtually irreversibly (4,9,13) archetypal versions of all Type I RM enzymes found in many diverse eubacteria (21). Ocr binds to and completely occupies the DNA-binding site on the enzyme thereby inhibiting the restriction endonuclease activity and protecting the phage genome as it enters the bacterium. Thus, Ocr greatly assists the spread of phage infection in the bacterial population. The fact that Ocr mimics the general shape and charge of the DNA segment bound by the RM enzyme, rather than any specific base pair sequence, means that the protein can act against all Type I RM enzymes, each of which recognizes a different defined base pair sequence. The Type I RM enzymes (2,22–25) are complex oligomeric multifunctional enzymes comprising an S subunit for DNA sequence recognition, two M subunits for DNA methyltransferase (MTase) activity and two R subunits for ATP-hydrolysis-dependent DNA translocation and restriction endonuclease activities. The M2S1 complex can function as a sequence-specific MTase and the R2M2S1 complex can switch between MTase and restriction endonuclease activities. The structure of complete Type I RM enzymes has recently been determined (26). The restriction endonuclease only operates on DNA containing unmethylated recognition sequences typically found in invading foreign phage DNA. The structure of the MTase core, referred to as M.EcoKI, has been determined for the archetypal EcoKI Type I RM system from E. coli K12 (27). The target sequence recognized by EcoKI is AACNNNNNNGTGC with methylation occurring on the adenines at the underlined positions. The bipartite nature of the target sequence is characteristic of the Type I RM enzymes. The S subunit contains two target recognition domains (TRDs) each responsible for recognizing one part of the bipartite target sequence.
The structure of Ocr is an elongated, curved dimer. Each 116 amino acid monomer is decorated with 34 surface-exposed Asp and Glu residues but only 2 Lys and 4 Arg residues (4,10). Many of the negatively charged residues of Ocr can be superimposed upon the equivalent phosphate groups on the DNA molecule recognized by the RM enzyme (1,4). In addition to mimicking the charge distribution, Ocr mimics the bend of ∼46° in the DNA helical axis induced in DNA when it binds to the RM enzyme (4). The introduction of the bend in the DNA by the RM enzyme is energetically costly. This cost is ‘saved’ when the RM enzyme binds to Ocr because Ocr is already ‘pre-bent’ (28). By combining electrostatic mimicry of DNA and mimicry of the bent shape, binding of the RM enzyme to Ocr is energetically more favourable than binding to DNA. Thus, the overall binding affinity of the MTase core of the RM enzyme for Ocr is KA of ∼2 × 1010 M−1, which is 50-fold greater than the affinity for DNA (13,18–20).
Mutational and chemical modification studies of Ocr revealed the robustness of the DNA mimicry activity. A large number of single or double mutations in the Ocr monomer produced no observable effect in vivo or in vitro (17). Only deletion of the C terminus of Ocr with concomitant structural destabilization (10,14) or disruption of the dimer interface (19) led to a loss of activity. Chemical modification to remove the negative charges indicated that ∼16 Asp or Glu side chains per Ocr monomer needed to be neutralized before the binding affinity of Ocr for M.EcoKI fell to approximately the same level as the DNA-M.EcoKI affinity (16). Further chemical modifications were required to completely inactivate the DNA mimicry. Importantly, the extensive chemical modification of over 15% of the amino acid sequence had no deleterious effect on the fold of the Ocr protein.
In the current work, we have mutated groups of five or six Asp or Glu located together in the primary structure of Ocr and then combined these groups to produce Ocr molecules containing up to 17 mutations per monomer. The effect of these mutations on the ability of Ocr to act as an anti-RM DNA mimic against the archetypal EcoKI Type I RM system was examined. We found that ∼12 Asp and Glu located in the central portion of the Ocr dimer were important for DNA mimicry. The clustering of these amino acids and the decrease in abundance of positively charged amino acids would be the minimal evolutionary requirements for the formation of the DNA mimic. However, the evolutionary path to the Ocr protein appears to have to surmount a barrier. The Ocr variants can be grouped into classes: fully active, inactive but showing some toxicity for cell viability and inactive with no deleterious effect on cell viability. Our data seem to indicate an evolutionary progression from an inactive precursor protein via a toxic protein to the fully active anti-RM protein which has lost the toxicity.
MATERIALS AND METHODS
Chemicals, bacterial strains and phage
All chemicals were purchased from Sigma-Aldrich unless otherwise stated. Escherichia coli XL1 Blue supercompetent cells were obtained from Stratagene (La Jolla, CA) and E. coli BL21(DE3) was from Invitrogen Life Technologies (Carlsbad, CA). Escherichia coli NM1261 (r−m−; no RM system) and E. coli NM1049 (r+m+; EcoKI Type IA RM system) were a kind gift of Professor Noreen Murray (School of Biology, University of Edinburgh, UK) (29). These two NM strains were converted to DE lysogens as described by McMahon et al. (5). Escherichia coli NM1041 (E. coli MG1655 r+m+; EcoKI Type IA RM, system clpX ::kan) (29) and E. coli NM1057 (E. coli MG1655 r−m+; no RM system, ΔhsdREcoKI, clpX ::kan) were kind gifts from Dr Angela Dawson (School of Physics and Astronomy, University of Edinburgh). Virulent unmodified bacteriophage lambda λv.o, or modified lambda λv.k were provided by Professor Noreen Murray.
Plasmids
Plasmid pAR2993, based upon the vector pET1, harbours the gene encoding wild-type Ocr located just downstream of an isopropyl β-d-1-thiogalactopyranoside (IPTG) inducible T7 promoter (14). Plasmids pAR2993 and pAR3786 and pAR3790 encoding truncated forms (deletions of the C-terminal 7 or 17 amino acid residues, termed Ocr109 and Ocr99, respectively) of wild-type Ocr were a kind gift from Dr Alan Rosenberg and Professor William Studier (Brookhaven National Laboratory, USA). Plasmids pOcrD12N/D26N, pOcrD25C and pOcrD73C were derivatives of pAR2993 encoding genes to produce Ocr containing a double substitution D12N/D26N or single substitutions D25C and D73C (13,17). Plasmid pET24a was purchased from Novagen (Madison, WI). pBRsk1 is an engineered version of pBR322 (4361 bp) in which one of the two EcoKI sites (4024–4036) has been removed by site-directed mutagenesis (30). The unmethylated form of pBRsk1 used in the nuclease assays was prepared from E. coli NM1261 (r−m−). The plasmid pTrc99A (31) was a gift from Dr Angela Dawson.
Generation of mutants of Ocr
Standard DNA manipulation procedures were used throughout (32). All constructs were sequenced to ensure no errors had been introduced during amplification. For clarity, references to the amino acid sequence of the Ocr correspond to the numbering system for the mature full-length protein that lacks the N-terminal methionine, which is subject to post-translational cleavage. We generated a series of Ocr mutants, each containing multiple (between 5 and 17) amino acid substitutions that targeted acidic residues on the surface of the protein. The majority of mutations were generated using the QuikChange II Site-Directed Mutagenesis kit from Stratagene (La Jolla, CA) and plasmid pAR2993 and derivatives. The primer pairs for the polymerase chain reaction (PCR) along with the corresponding DNA template used to generate each set of mutations are given in Supplementary Table S1. The PCR comprised an initial denaturation step of 95°C for 30 s followed by 18 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 1 min and elongation at 68°C for 6 min 30 s. The PCR mix (total volume 50 µl) included PfuUltra HF DNA polymerase (2.5 U), 10 mM Tris-HCl (pH 9.0 at room temperature), 50 mM KCl, 1.5 mM MgCl2, 200 μM of each deoxynucleoside triphosphate, 40 pM of each primer and ∼5 ng of template DNA. DpnI (10 U) was then added directly to the amplification reaction and incubated at 37°C for 60 min to remove the original template. After transformation of E. coli XL1-Blue supercompetent cells, recombinant clones were selected by growth on LB-agar plates containing 100 µg/ml carbenicillin. Plasmid DNA was prepared from individual colonies and sequenced to verify whether the anticipated mutations had been introduced.
The strategy adopted for the creation of pOcr/POcr involved using PCR to amplify and fuse together two different fragments of DNA encoding Ocr (using pAR2993) and a synthetic version of the gene encoding Ocr, known as POcr (plasmid pPOcr created by GeneArt, Regensberg, Germany), in which all the acidic acid residues had been mutated (Asp and Glu to Asn and Gln, respectively). Two PCRs (PCR1 and PCR2) were conducted separately using the following primer pairs: primer A 5′-AGCTCATATGGCTATGTCTAACATGACTTACAACAACGTTTTCGAC-3′ with primer B 5′-CTGCAGAATACGGATTACGTTCTTGGTGTCAGGCATCAGACC-3′ and primer C 5′-GGTCTGATGCCTGACACCAAGAACGTAATCCGTATTCTGCAG-3′ with primer D 5′-GAATTCAAGCTTTTACTGTTGGTTCTGCTGATATTGCTG-3′ to amplify specific sections from pAR2993 and pPOcr, respectively. The PCR cycle parameters comprised an initial denaturation step of 95°C for 30 s followed by 25 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 1 min and elongation at 72°C for 1 min. The composition of the reaction mixture was the same as described earlier. Both PCRs generated amplified products of the anticipated size, which were gel purified and then used as template for a third PCR using primers A and D (PCR3). Note that primers B and C of PCR1 and PCR2 were designed to generate a partial overlap, which facilitated the generation of full length template during the first extension cycle of PCR3. This full length product then allowed amplification of Ocr/POcr using primers A and D. The amplified product was then digested with NdeI and HindIII and ligated into the corresponding sites of pET24a. The ligation mix was used to transform E. coli XL1-Blue supercompetent cells and recombinant clones were then selected by growth on LB-agar plates supplemented with 50 µg/ml kanamycin. Plasmid DNA was prepared from individual colonies and sequenced to verify whether the anticipated mutations had been introduced.
For the purposes of performing the in vivo restriction alleviation (RA) assay described below, the gene encoding wild-type Ocr or mutant Ocr had to be transferred to an expression system not based upon the T7 promoter. Each plasmid was amplified by PCR using primers that included an NdeI restriction recognition site (CATATG), which overlapped the ATG start codon of the gene, and a HindIII site immediately 3′ of the stop codon. The PCR products were then ligated into the corresponding NdeI/HindIII sites of pTrc99a. The resulting plasmids were sequenced to ensure no mistakes had been introduced during the amplification.
In vivo phage restriction assay
The activity of the mutated forms of Ocr was assessed using an in vivo restriction assay with virulent unmodified bacteriophage lambda λv.o or EcoKI-modified lambda λv.k as described previously (33). The efficiency of plating of the phage was determined on E. coli NM1261 (DE3) (r−m−) transformed with plasmids expressing Ocr or its mutants compared to E. coli NM1049 (DE3) (r+m+) transformed with the same plasmids. Cells containing a non-functional Ocr would not be able to inhibit EcoKI and phage propagation would be restricted whereas those containing a functional Ocr would be able to inhibit EcoKI and phage propagation would not be restricted. No IPTG was present in the BBL agar because the promoter is sufficiently leaky to produce sufficient Ocr protein (4).
In vivo phage modification assay
The activity of the mutated forms of Ocr was assessed using an in vivo modification assay. Virulent unmodified bacteriophage lambda λv.o was plated on a r−m+ strain (E. coli NM1057) after the strain had been transformed with a vector control or the Ocr mutant overexpression plasmids (33). IPTG (50 µg/ml) was included in the BBL agar. Plaques found after overnight incubation were expected to be modified if Ocr activity was compromised. If Ocr activity was functional, then the phage would not be modified. The recovered phage were assessed for modification using the in vivo restriction assay comparing efficiency of plating on E. coli NM1041 (r+m+, clpX−) versus E. coli NM1057(r−m+, clpX−).
In vivo restriction alleviation assay
The RA assay exploits the ClpXP protease found in E. coli K12 cells and the observation that the addition of the adenine analogue and mutagen, 2-aminopurine (2AP), leads to the production of unmethylated targets for EcoKI on the chromosome. ClpXP, which cleaves the HsdR subunit of EcoKI, is absent in the strain E. coli NM1041(r+m+). In the absence of ClpXP but presence of 2AP, the EcoKI enzyme cleaves the chromosome and the cell dies. However, if an active Ocr protein is also present, the EcoKI is inactivated and the cell survives even when growing on a 2AP medium without ClpXP. Therefore, the anti-restriction activity of Ocr can be measured by determining cell survival, as previously described by Makovets et al. (34).
The RA activity was assayed with 100 μl cultures of E. coli NM1041 (r+m+) transformed with plasmids expressing each of the Ocr mutants. Cultures were diluted using a solution of L Broth and glycerol (5:2 ratio) to provide equal starting cell concentrations. Each of the cultures were diluted in 10-fold increments from 10−1 to 10−8 and spotted in triplicate onto LB-agar plates containing different concentrations of 2AP: 0, 5, 10, 20 or 40 μg/ml. The plates were supplemented with kanamycin (30 μg/ml), ampicillin (50 μg/ml) and IPTG (50 μg/ml). In addition, as a control and to determine the total cell concentration, the cultures were plated onto a plate supplemented only with kanamycin (30 μg/ml) and ampicillin (50 μg/ml). The plates were inverted and sealed in an air-tight container and incubated at 37°C for 18 h after which colony numbers were counted and the number of viable cells per ml calculated.
Bacterial growth curves
Cultures of the E. coli NM1041 (r+m+, clpX−) transformed with each of the plasmids expressing Ocr or its mutants were incubated at 37°C. About 250 μl samples were taken from each culture and used to inoculate two lots of 5 ml L Broth, one with and one without IPTG (50 µg/ml). These were incubated for 15 min to obtain a similar population density for each culture. All of the cultures were in the lag phase. Sterile covered microplates (96-well plates) and lids were obtained from Greiner. About 200 μl samples from the prepared cultures were pipetted, in triplicate, with and without 50 µg/ml IPTG, into the 96-well plates. Bacterial growth curves were obtained by measuring the optical density of the cultures as a function of time. The optical density measurements were carried out by a Fluorostar OPTIMA plate reader (BMG Labtech). The reader was programmed to measure the samples on the plate every 15 min for 10 h at a wavelength of 600 nm. The reader maintained a temperature of 37°C throughout the experiment and a medium shaking speed of 200 rpm between readings.
Cell growth on solid media in the presence of Ocr variants
Determining the loss of viability for E. coli NM1041 (r+m+, ClpX−) and E. coli NM1057 (r−m+, HsdR−, ClpX−) transformed with various plasmids expressing Ocr or its mutants was performed as follows. Cultures of the bacterial strains were grown after transformation with the plasmid expressing Ocr or its mutants. The cells were grown in L Broth with aeration at 37°C, shaking at 200 rpm for 18 h. Serial dilutions of the cultures were spotted onto LB-agar plates, supplemented with Kanamycin (30 µg/ml), Ampicillin (50 µg/ml) and IPTG (50 µg/ml). The plates were incubated for 18 h at 37°C. Viable counts for each strain transformed with each mutant were determined. Controls were cells transformed with either the plasmid expressing wild-type Ocr protein or the pTrc99A expression vector.
Microscopy
Overnight cultures of E. coli NM1041 (r+m+, ClpX−) cells, transformed with wild-type Ocr or the mutant derivatives, were used to inoculate 10 ml of L Broth (50 µl). The cultures were grown to an optical density of 0.2 at 600 nm and subsequently induced with IPTG (50 µg/ml) and incubated for a further 2.5 h (the OD600 was measured to ensure each culture was still in the logarithmic phase of growth). To condense the nucleoid, chloramphenicol (10 µl) was added to each culture and incubation at 37°C continued for 30 min. The bacterial cells were fixed with the addition of methanol at a 2:1 ratio and a further incubation on ice for 10 min. The cells were collected by centrifugation and resuspended in buffer; 10 mM Tris–HCl (pH 7.5) 10 mM MgSO4. Prior to microscopy analysis, the cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) (0.5 µl, 5 mg/ml) and observed under a fluorescence microscope.
Protein production and purification
Escherichia coli BL21(DE3) was transformed with each of the recombinant plasmids encoding the mutated forms of Ocr. Cells were grown at 37°C in two 2 -l conical flasks, each containing 1 l L Broth supplemented with the appropriate antibiotic for selection (Kanamycin 30 µg/ml or Ampicillin 50 µg/ml) to an optical density at 600 nM of ∼0.5. Heterologous gene expression was then induced by adding IPTG to a final concentration of 1 mM and growth was then continued for a further 2.5 h before harvesting the cells by centrifugation (8000g, 15 min, 4°C). Analysis of the cell-free extracts by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie-blue staining showed that most of the mutated forms of Ocr were overexpressed at comparable levels to that observed for wild-type protein. Furthermore, all the overexpressed proteins were produced in a soluble form and could be purified to homogeneity using the protocol described previously (17). M.EcoKI and EcoKI were prepared as previously described (35,36). All Ocr protein concentrations refer to the dimeric form of Ocr or its derivatives.
Glutaraldehyde crosslinking
Chemical crosslinking was used to assess whether each of the Ocr mutant proteins exist as a homodimer in solution. A 60 μg aliquot of each protein was diluted with 10 ml of 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (pH 7.5) i.e. final protein concentration of ∼430 nM. Glutaraldehyde (25% w/v) was added to the solution to give a final concentration of 1% (w/v). The mixture was incubated at room temperature for 2 min and then the reaction was quenched by adding 0.25 ml of freshly prepared 2 M sodium borohydride in 0.1 M NaOH. After incubating for a further 20 min at room temperature, a 10 μl aliquot of a 10% (w/v) sodium deoxycholate solution was added. The protein was then precipitated by addition of trichloroacetic acid (TCA) and pelleted by centrifugation. Each pellet was dissolved in a small volume of SDS-PAGE loading buffer. A small amount of 1 M Tris HCl pH 8.0 was added to the samples to neutralize the acid. Samples were then analysed by SDS-PAGE on a 4–12% polyacrylamide gradient gel. Each sample was run alongside non-treated protein for comparison. A wild-type Ocr sample was used as a positive control.
Circular dichroism for assessment of Ocr folding
Far UV (190–260 nm) circular dichroism (CD) analysis was performed on a Jasco model J-180 spectropolorimeter (Jasco Corp., Tokyo, Japan) at 25°C in 10 mM Tris HCl pH 8.0, 50 mM NaF, 7 mM mercaptoethanol buffer at a protein concentration of 30 µM using a 0.2 mm pathlength cell. Spectra were corrected for buffer contribution. Each spectrum was an accumulation of four individual scans.
Isothermal titration calorimetry for assessment of binding of Ocr to M.EcoKI MTase
All isothermal titration calorimetry (ITC) experiments were conducted using a VP-ITC instrument (Microcal, Northampton, MA). Protein samples were exchanged into 20 mM Tris–HCl pH 8.0, 6 mM MgCl2, 7 mM 2-mercaptoethanol using a PD-10 column (GE Healthcare, Piscataway, NJ) for each experiment. The protein concentration was adjusted and then S-adenosyl-l-methionine (SAM, New England Biolabs) added to a final concentration of 100 µM. All solutions were thoroughly degassed prior to use. Typically, Ocr at a concentration of 30 µM was titrated into M.EcoKI at a concentration of 3 µM (1.4 ml active volume). All titrations were performed at 25°C. The data were then fitted to a single-site binding model using the Microcal LLC Origin software package.
In vitro DNA cleavage assay for assessment of inhibition of the endonuclease activity of EcoKI
The in vitro endonuclease assay monitored the linearization of unmethylated plasmid pBRsk1 by EcoKI RM enzyme in the presence or absence of Ocr or its variants. Typically, the reaction was performed in a 50 µl reaction volume containing 10 mM Mg acetate, 10 mM Tris-acetate, 7 mM 2-mercaptoethanol, 50 µg/ml BSA, 2 mM ATP, 0.1 mM SAM, 3 nM pBRsk1 and initiated by adding EcoKI to 30 nM. Where required, each Ocr protein was preincubated with the EcoKI at a molar ratio of 10:1 or 20:1 (Ocr:EcoKI), for 2 min at room temperature prior to adding to the other reaction components. Digests were performed for 8 min at 37°C before stopping by incubation at 68°C for 10 min. The extent of reaction was then analysed by agarose gel electrophoresis on a 0.8% 1× TAE gel. DNA bands were visualized under UV illumination after staining with ethidium bromide.
Structure visualization
PDB files (Ocr: 1S7Z and M.EcoKI: 2Y7C (with Ocr bound) 2Y7H (with DNA bound)) were viewed using UCSF Chimera (37).
RESULTS
Location of mutations on the atomic structure of Ocr
Mutant forms of Ocr containing multiple amino acid substitutions (5–17 per monomer) were generated to help define specific structural features of the molecule that are key to its interaction with the Type I RM system. The atomic structure of Ocr facilitated the rational design of a series of mutant forms of the molecule in which patches of negative charge were systematically removed from the protein surface. Specifically, a number of acidic residues were conservatively mutated to neutral amino acids (i.e. Asp to Asn and Glu to Gln). However, for convenience, some existing mutated forms of Ocr (14) were employed as templates for the mutagenic PCR. In such instances (i.e. Mut2, Mut3, Mut7, Mut10 and Mut11), one of the acidic residues was substituted with Cys. Our strategy was to generate a succession of clustered mutations to target defined structural features of the Ocr protein, such as exposed loops or helices, which together covered the entire surface of the protein. The multiple mutations of Ocr are shown in Table 1 and their locations on the Ocr structure in Figure 1.
Table 1.
The Ocr variants examined in this work
| Mutant name | Changes to wild-type amino acid sequence |
|---|---|
| Mut1 | D12N, E16Q, E20Q, D25N, D26N |
| Mut2 | D25C, D26N, D29N, D31N, D32N, D35N |
| Mut3 | E59Q, D62N, E64Q, E66Q, D67N, D73C |
| Mut4 | D92N, E95Q, D96N, E98Q, D99N |
| Mut12 | E59Q, D62N, E64Q, E66Q, D67N |
| Mut7 | Mut2 + Mut4 |
| Mut10 | Mut2 + Mut4 + Mut12 |
| Mut11 | Mut2 + Mut12 |
| Mut13 | Mut4 + Mut12 |
| Mut16 | Mut1 + Mut12 |
| Ocr99 | Deletion of C-terminal amino acids 99–116 |
| Ocr109 | Deletion of C-terminal amino acids 109–116 |
| Ocr/POcr | D76N, E87Q, D92N, E95Q, D96N, E98Q, D99N, E103Q, E106Q, E107Q, E109Q, E110Q, E112Q, E113Q, D114N, E115Q, E116Q |
| POcr | All 34 Asp and Glu changed to Asn and Gln, respectively. |
The Ocr99 and Ocr109 variants have been described previously (14).
Figure 1.

The location of the Ocr mutants on the dimeric atomic structure (PDB 1S7Z, 4). The residues targeted for mutagenesis are highlighted as coloured sticks. (A) The locations of mutations for Mut2 (magenta), Mut4 (blue) and Mut12 (red) are shown in left-hand monomer and Mut1 (yellow) and Ocr/POcr (orange) in the right-hand monomer. (B) A view rotated 90° from that in panel (A) showing the bottom of the structure.
In vivo restriction assay
The activity of each mutated form of Ocr was assessed in vivo using a restriction assay. Infection with unmodified virulent λ phage (λv.o) of two different E. coli strains, transformed with pET24a or the plasmids expressing Ocr or its variants, was carried out. Escherichia coli NM1049(DE3) contains the EcoKI Type IA RM system (r+m+) while E. coli NM1261(DE3) lacks the RM system (r−m−). As anticipated, the efficiency of plating (eop) of λv.o on E. coli NM1049(DE3) containing pET24a was four orders of magnitude lower than the eop on E. coli NM1261(DE3) with the vector (Table 2). The methylated form of λ (λv.k) showed an eop of ∼1 as expected (data not shown). Comparing the two strains when transformed with the plasmid expressing the wild-type Ocr gave essentially identical numbers of plaques and an eop of 1.45, demonstrating that Ocr had inhibited the Type I RM system. Assaying the mutated forms of Ocr in the same way showed that most of the mutants were fully active with an eop ranging from 0.54 to 1.5. Cells expressing Mut10, Mut11 or Mut13 showed low eop similar to cells containing the vector alone. These results suggest that Mut10, Mut11 and Mut13 have no significant antirestriction activity in vivo. The synthetic gene encoding POcr in which all 34 Asp or Glu residues in Ocr were substituted by Asn and Gln was also inactive in this assay (data not shown) and was not investigated further.
Table 2.
In vivo restriction and modification assays
| Ocr variant expressed | Efficiency of plating of λv.o [Phage titre on E. coli NM1049(DE3) (r+m+)]/[Phage titre on E. coli NM1261(DE3) (r−m−)] | Efficiency of plating of λv.o [Phage titre on E. coli NM1041 (r+m+, clpX−)]/[Phage titre on E. coli NM1057(r−m+, clpX−)] | Active versus whole RM system in vivo | Active versus M.EcoKI alone in vivo |
|---|---|---|---|---|
| pET24a | 3.80 × 10−4 | 0.96 | N | N |
| Wild type | 1.45 | <10−4 | Y | Y |
| Mut1 | 0.94 | <10−4 | Y | Y |
| Mut2 | 0.77 | <10−4 | Y | Y |
| Mut3 | 0.82 | 0.79 | Y | N |
| Mut4 | 1.33 | <10−4 | Y | Y |
| Mut7 | 1.5 | <10−4 | Y | Y |
| Mut10 | 2.30 × 10−4 | 1 | N | N |
| Mut11 | 1.15 × 10−4 | 1.31 | N | N |
| Mut12 | 1.29 | 0.16 | Y | Y/N |
| Mut13 | 5.00 × 10−4 | 0.67 | N | N |
| Mut16 | 0.54 | 0.47 | Y | N |
| Ocr/POcr | 0.78 | 1 | Y | N |
| Ocr99 | 0.30a | 3.80 × 10−4 | Y | Y |
| Ocr109 | 0.32a | 0.55 | Y | N |
Note that an efficiency of plating of ∼1 indicates an active Ocr protein in the restriction assay whereas an eop of ∼0 (i.e. <10−4) in the modification assay indicates an active Ocr protein. Errors in the number of plaques where ±20% from full plate assays performed in triplicate.
In vivo phage modification assay
To assess the methylation activity of each of the mutants, a phage modification assay was carried out. Unmethylated phage, λv.o, was used to infect the modifying host E. coli NM1057 (r−m+, clpX-) transformed with either a control vector or with plasmids expressing Ocr or its variants (IPTG was added 50 µg/ml). This strain lacks the ClpXP protease and the gene expressing the R subunit of EcoKI. The strain still expresses the M.EcoKI MTase and, therefore, phages recovered from a single plaque should have been methylated by the action of M.EcoKI unless the Ocr protein was active. The activity of Ocr was assessed by determining the eop of the phages recovered from a single plaque on E. coli NM1057 (r−m+, clpX−). The eop was determined on the restrictive host E. coli NM1041 (r+m+, clpX−) relative to the modifying host E. coli NM1057 (r−m+, clpX−). If the Ocr variant was active then the titre of recovered phages per ml after plating λv.o on E. coli NM1057 would be lower than the titre of phages per ml on E.coli NM1041, and an eop reflecting a fully restrictive condition would be observed.
The results showed that wild-type Ocr and mutants Mut1, Mut2, Mut4, Mut7 and Ocr99 were active in preventing DNA modification by M.EcoKI as their eop was very low (e.g. Ocr99) or ∼0 (i.e <10−4), Table 2. Ocr mutant Mut12 showed a greatly reduced effectiveness (eop of 0.16) while the others showed no activity (eop of ∼0.5–1). Arbitrarily, we define that an eop of 0.5 or greater indicates an Ocr that is inactive in inhibiting M.EcoKI in E. coli NM1057. It is noteworthy that some Ocr variants active against restriction in vivo were not able to prevent modification in vivo (Mut3, Mut12, Mut16 and Ocr/POcr). Also of note is that addition of one further substitution (D73C) to change Mut12 into Mut3 converts the Ocr protein from being able to partially inhibit modification to being unable to inhibit modification.
In vivo restriction alleviation assay
For E. coli NM1041 (r+m+, clpX), the mutagen 2-aminopurine is toxic. Toxicity results from cleavage of the chromosome in the absence of ClpXP control or Ocr inhbition, so that cell growth is reduced or prevented as EcoKI destroys the chromosome. The presence of an active Ocr protein in the cell will then rescue the viability by blocking the action of EcoKI, a phenomenon known as RA. Thus colonies will grow on media containing 2AP in the presence of an active Ocr antirestriction protein.
The effect on the viable cell count for E. coli NM1041 when transformed with plasmid vector alone or with plasmids encoding the Ocr mutants is shown in Figure 2 as a function of 2AP concentration. In the absence of 2AP and IPTG, the strain grew equally well when transformed with various Ocr plasmids or the vector alone (∼108 cells per ml). The addition of IPTG caused an unanticipated reduction in cell viability for three of the mutants, Mut12, Mut16 and Ocr/POcr even when no 2AP was present (∼103 to ∼104 cells per ml). The addition of 2AP to 5 μg/ml had no further effect on any of the transformed strains. A 2AP concentration of 10 μg/ml led to a complete loss of viability for cells expressing Mut3. Concentrations of greater than 10 μg/ml also killed cells expressing Mut7, Mut10, Mut11, Mut12, Mut13, Mut16, Ocr/POcr and those containing only the vector. Those cells expressing the wild-type Ocr, Mut1, Mut2, Mut4, Ocr99 or Ocr109 were viable even at the highest concentrations of 2AP used (40 μg/ml).
Figure 2.

The RA assay measuring cell survival of E. coli NM1041 (clpX−, r+m+) when transformed with various plasmids expressing Ocr or its mutant forms. The graph shows growth on LB-agar plates supplemented with antibiotics (cyan) only, antibiotics and ITPG (green) and, in the presence of antibiotics, IPTG plus increasing concentrations of 2AP (5, 10, 20 and 40 µg/ml are red, magenta, blue and olive, respectively).
The results show four distinct phenotypes, namely active antirestriction giving full protection against all concentrations of 2AP (Wild type Ocr, Mut1, Mut2, Mut4, Ocr99, Ocr109), no antirestriction effect and indistinguishable from the vector (Mut7, Mut10, Mut11, Mut13), no antirestriction coupled with a deleterious effect on cell growth when induced with IPTG (Mut12, Mut16 and Ocr/POcr) and lastly no IPTG effect on cell growth but also no antirestriction and an enhanced sensitivity to 2AP (Mut3). Again the single amino difference between Mut12 and Mut3 causes a noticeable difference in behaviour.
Bacterial growth
The RA assay revealed that even in the absence of 2AP to induce RA, cell viability could be compromised by some Ocr variants (Mut12, Mut16 and Ocr/POcr). To further explore this phenomenon, growth properties of plasmid-containing E. coli NM1041 (clpX−, r+m+) were examined without 2AP but with IPTG and selective antibiotics.
Although the growth curves (Figure 3) generally highlighted the same deleterious Ocr mutants, Mut3, Mut12, Mut16 and Ocr/POcr, as the RA assay, there were some minor differences in growth behaviour between these mutants. The presence of Ocr or mutants Mut1, Mut2, Mut4, Mut7, Mut10, Mut11, Mut13, Ocr99 and Ocr109 showed normal exponential growth with a doubling time of ∼130 min after a lag time of ∼150 min whether ITPG was present or not. Mut3 which produced a strong effect in the RA assay allowed normal growth in the absence of IPTG and slow growth with a doubling time of ∼150 min in the presence of IPTG. The presence of Mut12 in the absence of ITPG caused a long lag time of over 300 min before exponential growth started with a doubling time of ∼150 min. The addition of ITPG stopped growth altogether. The presence of Mut16 strongly slowed growth in the presence or absence of IPTG as suggested by the RA assay. The cells started to grow exponentially but then halted growth at a low optical density. Ocr/POcr, which also reduced cell viability in the RA assay, allowed normal growth in the absence of IPTG and limited growth with a doubling time of ∼200 min and halting at a low optical density in the presence of IPTG. Thus Ocr/POcr would appear to be not as deleterious as Mut12 or Mut16.
Figure 3.
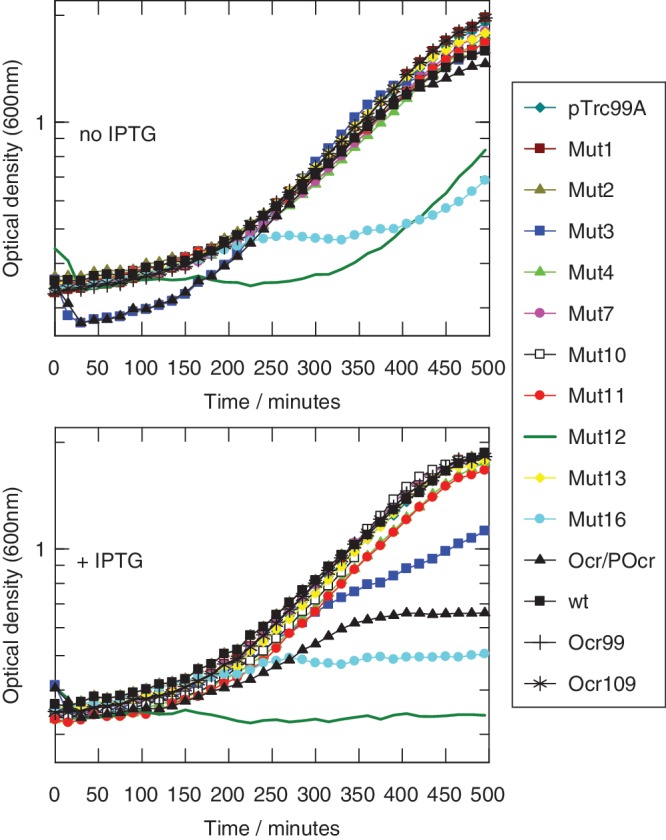
Cell growth with the Ocr mutants expressed in E. coli, NM1041 (clpX−, r+m+) in the absence of 2AP. It is clear that cells expressing different Ocr mutants grow at different rates. pTrc99A is the vector alone, all other data are for cells expressing Ocr wild-type (wt), Ocr99, Ocr109, Ocr/POcr or mutants Mut1 to Mut16. OD600 measurements were recorded every 15 min for 500 min. Errors are ±7% for triplicate measurements (error bars not shown for clarity).
Cell growth on solid media in the presence of Ocr variants
The deleterious effect of Mut12, Mut16 and Ocr/POcr on viability even in the absence of RA induced by 2AP and the strong effect of Mut3 in the presence of 2AP was examined on agar plates using the two strains, E. coli NM1041 (r+m+, clpX) and E. coli NM1057 (r−m+, clpX) (Figure 4). 2AP was not added so the chromosome would be modified by EcoKI. Spotting serial dilutions of the cultures onto the plates showed that cells expressing an active EcoKI system (E. coli NM1041) and Ocr/POcr, Mut12 or Mut16 grew poorly. Cells expressing EcoKI and the Mut3 Ocr mutant were slightly affected compared to the controls. However, E. coli NM1057 cells also lacking the R subunit of EcoKI, and thus an active RM system, grew normally or almost normally (Mut16) in the presence of all the Ocr variants tested. These results are comparable to the changes in growth rate measured in liquid culture. The results indicate that the deleterious effect is specific to the RM pathway and suggest that some of the Ocr mutants are creating a situation where EcoKI is unable to modify the target sites on the chromosome leading to restriction of the chromosome (i.e. an r+m+/− phenotype).
Figure 4.

Loss of cell growth of E. coli NM1041 (clpX−, r+m+) and NM1057 (clpX−, r−m+) expressing the mutants Mut3 (m3), Ocr/POcr, Mut16 (m16) and Mut12 (m12) compared to cells expressing wild-type Ocr (wtOcr) or transformed with vector alone (pTrc99A). Serial dilutions of cultures were spotted onto LB-Agar plates supplemented with appropriate antibiotics. The dilutions of 10−2, 10−3 and 10−4 fold of the liquid culture are spotted in triplicate clockwise for E. coli NM1041 and anticlockwise for E. coli NM1057 from the top of the plate.
Microscopy
Escherichia coli NM1041 cells expressing Ocr or its variants were induced with IPTG, stained with DAPI and examined by fluorescence microscopy (Figure 5). Most cultures contained normal cells with intact nucleoids similar to those expressing wild-type Ocr or transformed with the vector alone. Ocr variants Mut3, Mut12, Mut16 and Ocr/Pocr, however, showed extensive filamentation indicating induction of the SOS response and inhibition of cell division. This effect is consistent with DNA damage caused by endonucleolytic cleavage. These microscopy data suggest that EcoKI restriction function is active in the presence of mutants Mut3, Mut12, Mut16 and Ocr/POcr, but that modification is compromised leading to unmodified target sites on the chromosome that are then cleaved.
Figure 5.

Fluorescence microscopy images of cell morphology of bacterial cultures of E. coli NM1041 (clpX−, r+m+) transformed with plasmids expressing wild-type Ocr (wtOcr) or its variants. Control cells were transformed with the vector pTrc99A. Cell nucleoids were revealed with DAPI staining. Scale bar is 10 microns.
Protein expression and purification
Each mutated version of Ocr gave levels of heterologous gene expression similar to that of the wild-type protein except for the synthetic gene expressing the POcr variant which showed no protein product on SDS-PAGE of cell extracts. The other mutated recombinant proteins were all soluble and could be purified to near homogeneity using the same protocol as for wild-type Ocr (17). The far UV CD analysis indicated that the introduction of the various mutations had no significant effect on the secondary structure of the mutated Ocr proteins, a selection of which are shown in Figure 6. Spectra were analysed using the Dichroweb online secondary structure deconvolution program (38). Analysis using the CDSSTR method gave an α-helical content of 60% for wild-type Ocr, which is in good agreement with the crystal structure. Similar values (±2%) were obtained for the mutant proteins. It was therefore concluded that all the mutant proteins retained the overall fold of wild-type Ocr.
Figure 6.

Far-UV CD spectra of wild-type Ocr (black) and several variants: Mut1 (olive), Mut3 (orange), Mut7 (magenta), Mut10 (red) and Ocr/POcr (green). The protein concentration was ∼30 µM in each case.
The wild-type Ocr protein is known to begin to dissociate into monomers below ∼300 nM (14).Glutaraldehyde crosslinking was used to determine whether dimers existed for each of the mutant Ocr proteins. It was found that Ocr and its mutants showed the presence of dimers (and some higher order species) even at concentrations of 400 nM (Supplementary Figure S1) and thus we conclude that the extensive mutagenesis of Ocr produces no changes in its quaternary structure behaviour.
Isothermal titration calorimetry for measurement of binding of Ocr to M.EcoKI MTase
ITC was used to analyse the interaction between M.EcoKI and the wild-type or mutated forms of Ocr in vitro. Each injection peak was integrated and the heat of dilution subtracted. A plot of the heat of interaction versus the molar ratio of Ocr dimer to M.EcoKI was then generated. As found previously (13,17), the interaction between wild-type Ocr and M.EcoKI was highly exothermic and exhibited essentially stoichiometric binding (one Ocr dimer per M.EcoKI) within the concentration range used in these experiments (Table 3). The binding was too strong to allow determination of the dissociation constant which has been estimated as ∼50pM by other methods (13). Such tight binding is outside the range accessible by ITC. Except for Mut10 and Mut11, which displayed no apparent binding (ΔH = 0), Figure 7 and Table 3, the ITC data showed a highly exothermic interaction between the Ocr mutants and M.EcoKI under the experimental conditions employed. Furthermore, a stoichiometry consistent with one Ocr dimer per M.EcoKI was evident for all of the Ocr mutants (Table 3). However, several of the Ocr variants showed sigmoidal titration curves, Figure 7, indicating binding intermediate between the tight binding of the wild-type Ocr and the absence of binding by Mut10 and Mut11. For these sigmoidal transitions obtained with Mut3, Mut12, Mut13 and Mut16, estimates of the dissociation constant, Kd, lay in the range of 20–80 nM. This range of Kd indicates that these mutant proteins bind less well than DNA to M.EcoKI (Kd ∼1 nM under these conditions as determined using a fluorescence anisotropy assay (28,39)). The Kd for the interaction of Mut10 or Mut11 with M.EcoKI must be greater than ∼1 µM. Fluorescence spectroscopy measurements using a GFP-tagged version of M.EcoKI (40) suggest that the Kd for Mut10 binding to M.EcoKI is ∼1.5 µM (Stephanou,A.S., unpublished data).
Table 3.
ITC analyses for the interaction of the M.EcoKI MTase with Ocr or its variants
| Ocr variant | Stoichiometry Ocr dimers per M.EcoKI | Enthalpy (kcal mol−1) | Dissociation constant (Kd) (nM) |
|---|---|---|---|
| Wild typea | 0.87 ± 0.01 | −20.5 ± 0.3 | Too tight |
| Mut1 | 0.98 ± 0.01 | −13.8 ± 0.3 | Too tight |
| Mut2 | 1.04 ± 0.01 | −14.5 ± 0.2 | Too tight |
| Mut3 | 0.93 ± 0.02 | −14.7 ± 0.5 | 79 ± 21 |
| Mut4 | 0.83 ± 0.01 | −19.4 ± 0.2 | Too tight |
| Mut7 | 0.99 ± 0.01 | −12.5 ± 0.3 | Too tight |
| Mut10 | ∼0 | ∼0 | ∼no binding |
| Mut11 | ∼0 | ∼0 | ∼no binding |
| Mut12 | 0.90 ± 0.01 | −15.4 ± 0.4 | 20 ± 9 |
| Mut13 | 1.18 ± 0.03 | −11.0 ± 0.4 | 41 ± 16 |
| Mut16 | 0.98 ± 0.03 | −8.2 ± 0.4 | 71 ± 30 |
| Ocr/POcr | 0.89 ± 0.01 | −15.3 ± 0.2 | Too tight |
| Ocr99a | 0.71 ± 0.01 | −22.4 ± 0.7 | Too tight |
| Ocr109a | 0.74 ± 0.01 | −25.0 ± 0.6 | Too tight |
aData from (17).
Figure 7.

Isothermal titration calorimetry of the interaction between M.EcoKI and several variants of Ocr. (A) Titration of M.EcoKI with Mut4 protein showing tight binding as observed previously with wild-type Ocr (13). (B) Titration of M.EcoKI with Mut3 protein showing a sigmoidal transition characteristic of weaker binding than wild-type Ocr. (C) Titration of M.EcoKI with Mut10 protein showing no binding interaction at all.
In vitro DNA cleavage assay for assessment of inhibition of the endonuclease activity of EcoKI
We performed in vitro nuclease assays to analyse the level of inhibition exerted by the mutated forms of Ocr. A 10-fold excess of Ocr was mixed with EcoKI prior to addition to a plasmid substrate, pBRsk1, containing a unique restriction recognition site, and the cofactors. It has been shown previously that Ocr must be mixed with the Type I RM enzyme prior to introducing DNA as once ATP hydrolysis and translocation have commenced, the Ocr can no longer bind to the translocating enzyme (12). The mixture was then incubated at 37°C for 8 min before heat quenching the reaction. Linearization of the plasmid DNA was then assessed by subjecting the reaction mixture to agarose gel electrophoresis (Figure 8). As can be seen, a 10-fold excess of EcoKI over DNA in the absence of Ocr was sufficient to fully digest the DNA in the time available. However, preincubation of the nuclease with a 10-fold excess of wild-type Ocr over EcoKI completely inhibited digestion of the plasmid DNA. Similar levels of inhibition were also observed for the Mut1, Mut2, Mut4 Ocr mutants and the two truncated versions of Ocr99 and Ocr109. However, no observable inhibition was evident for Mut3, Mut10, Mut11, Mut12, Mut13 and Mut16 under the same experimental conditions. Mut7 displayed partial inhibition of DNA digestion. However, when the experiment was conducted at a molar ratio of 1:20, EcoKI:Mut7 the inhibition of nuclease activity was complete (Roberts, G.A., unpublished data).
Figure 8.

The effect of Ocr and variants on DNA cleavage by EcoKI. The end lanes show 1 kbp DNA size markers increasing from 2 kb to 7 kb. The pBRsk1 lanes show the uncut plasmid predominantly in the supercoiled form with a small amount of open circular form running more slowly. The EcoKI lanes show linearization of the DNA by EcoKI. The other lanes show the effect of Ocr and its variants on EcoKI activity. The enzyme was mixed with the Ocr proteins at a ratio of 1 enzyme per 10 Ocr prior to addition to the DNA sample.
DISCUSSION
Structural implications of the mutational analysis of Ocr
Our data allow four groups of Ocr mutants to be defined (Table 4). These groups are: Ocr mutants with normal activity indistinguishable from the wild-type Ocr; mutants with nearly normal activity; mutants with some activity but with further deleterious effects on cell viability and lastly Ocr mutants with no activity. As expected from previous chemical modification experiments (16), the structural fold of Ocr proved to be very robust to mutagenesis. Introducing extensive changes to its surface with up to 17 amino acid substitutions per monomer were tolerated. Thus it would be reasonable to suppose that the structure would be maintained in vivo even if the activity of mutants had been compromised.
Table 4.
Summary of results with Y indicating yes, N indicating no and X nM indicating the dissociation constant for binding
| Ocr variant expressed | Active versus whole RM system in vivo | Active versus M.EcoKI alone in vivo | Protection against restriction of chromosome in presence of 2AP | Normal growth in liquid culture without IPTG | Normal growth in liquid culture +IPTG | Normal cell morphology | Normal growth on solid media without IPTG | Binding to M.EcoKI in vitro | Inhibition of nuclease in vitro | Summary |
|---|---|---|---|---|---|---|---|---|---|---|
| Wild type | Y | Y | Y | Y | Y | Y | Y | Y | Y | Normal Ocr |
| Mut1 | Y | Y | Y | Y | Y | Y | Y | Y | ||
| Mut2 | Y | Y | Y | Y | Y | Y | Y | Y | ||
| Ocr99 | Y | Y | Y | Y | Y | Y | Y | Y | ||
| Mut4 | Y | Y | Y | Y | Y | Y | Y | Y | ||
| Ocr109 | Y | N | Y | Y | Y | Y | Y | Y | ∼Normal but some odd effects | |
| Mut7 | Y | Y | N | Y | Y | Y | Y | Y | ||
| Ocr/pOcr | Y | N | N | ∼Y | N | N | N | Y | N | Weakening binding, some loss of activity. Cells sick due to lack of RA. |
| Mut3 | Y | N | N | Y | ∼N | N | ∼Y | 79 nM | N | |
| Mut12 | Y | ∼N | N | N | N | N | N | 20 nM | N | |
| Mut16 | Y | N | N | N | N | N | N | 71 nM | N | |
| Mut13 | N | N | N | Y | Y | Y | 41 nM | N | Total loss of activity and cells are well. | |
| Mut10 | N | N | N | Y | Y | Y | N | N | ||
| Mut11 | N | N | N | Y | Y | Y | N | N | ||
We find that our mutational analysis indicates that mimicry of the electrostatics of the bend at the centre of the EcoKI DNA target sequence is a major contributor to the activity of Ocr as an anti-RM protein. This complements the conclusions reached by our chemical modification study (16) where it was found that mimicry of the bend in the DNA target was important but not sufficient to make Ocr a strong inhibitor of Type I RM enzymes. In other words, mimicry by Ocr of the charge and the shape of the bend in the DNA target add together.
Wild type activity is observed when the aspartate and glutamate residues in the sequence ranges aa12–26, aa25–35 or 92–99 (Mut1, 2 and 4) are mutated. Adding together the mutations of Mut2 and Mut4 to create Mut7 has a mild effect with the loss of the ability of Ocr to protect a clp− strain, in the presence of 2AP, from EcoKI.
The section of mutated amino acids, aa59–73 (Mut3 and 12), has a severe effect on activity and cell viability particularly when the amino acid substitution D73C is made in Mut3. Combining this deleterious section with aa12–26 in Mut1 has a slight effect on binding affinity in vitro in addition to the effects observed with Mut3 or Mut12. However combining this deleterious section (Mut3 or Mut12) with Mut2 or Mut4 abolishes Ocr activity and restores cell viability. This section is clearly important for activity.
The effects of deleting or mutating the C-terminal tail of Ocr are varied. Truncation of Ocr has either no effect (Ocr99) or is unable to prevent methylation in vivo but can prevent restriction in vivo and in vitro (Ocr109). The inability of Ocr109 to prevent methylation in vivo is difficult to explain given that it can bind tightly to the EcoKI MTase in vitro. The Ocr/POcr mutant with extensive replacement of aspartate and glutamates from aa76 onwards, although it can bind tightly to the Mtase, otherwise behaves as the Mut3 and Mut12 mutants with severe effects on cell viability. Since Ocr/POcr contains the aa changes made to construct Mut4, which have no effect on Ocr activity, and the tail region which can be deleted (Ocr99 and Ocr109) with little effect, it would appear that the section from aa76 to 91 are crucial for the effects observed with Ocr/POcr (Ocr/POcr substitutions minus those in Ocr99 and Mut4).
Combining this region of Ocr/POcr with the region identified in Mut3, aa59–73, then indicates that a block of charge found between aa59 and 91 on the surface of Ocr is crucial for Ocr activity. The region between aa59 and 91 comprises the bend in the centre of the convex surface of the Ocr protein (Figure 9). Recalling the bipartite nature of the DNA target sequence recognized by a Type I RM enzyme, e.g. EcoKI recognizes AACNNNNNNGTGC and bends the non-specific spacer sequence, then our data indicate that the central region of Ocr mimics the six base pairs comprising the bent non-specific spacer. The majority of Asp and Glu residues in this region are not making significant contacts with the TRDs of the S subunit as they lie in the region between the TRDs over the helical spacer which links the TRDs. The one exception to this is amino acid D73 which is close to the region of the TRDs predicted to be in contact with DNA in the M.EcoKI–DNA complex. Since the TRDs contain a significant number of Arg and Lys residues for DNA recognition then the major effect of removing the charge of D73 by substituting it with Cys can be rationalized. Its removal would upset the electrostatics of this Ocr–TRD interface and account for the enhanced effect of Mut3 over Mut12, which lacks the D73C substitution. The aa59–91 region extends up the face of Ocr and would make additional contacts to the M subunit.
Figure 9.
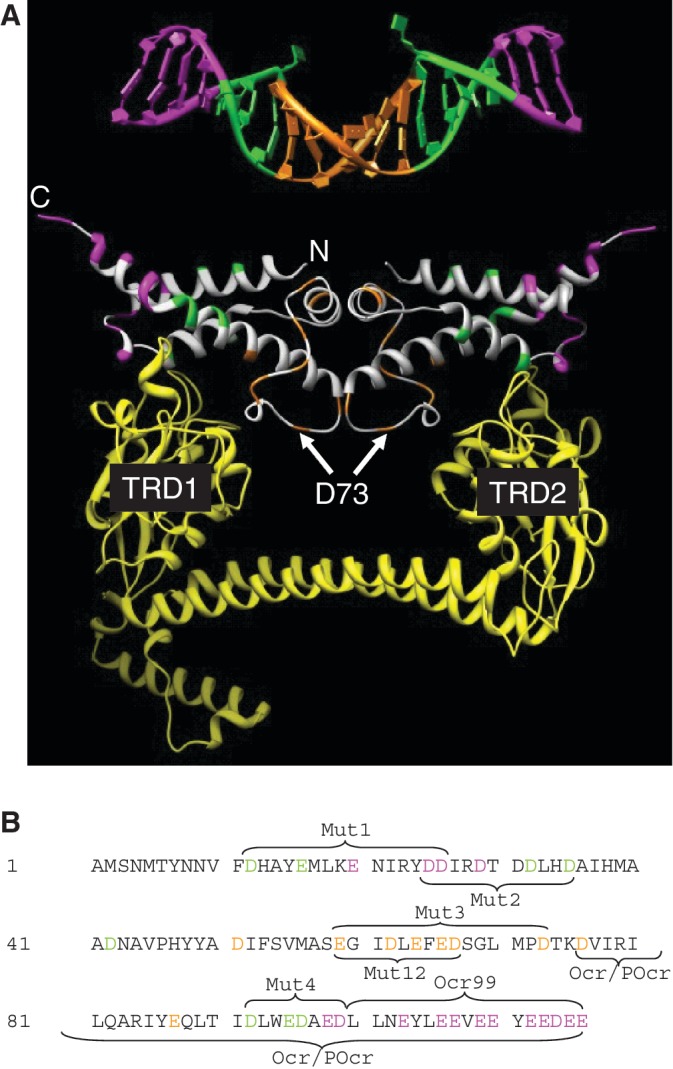
A comparison of the location of the amino acid substitutions made in Ocr with the bent DNA target bound by M.EcoKI (DNA coordinates from 27; pdb 2Y7H). (A) The DNA is coloured magenta for base pairs lying outside of the target sequence, specific base pairs recognized by M.EcoKI (AAC and GTGC) are coloured green and the non-specific spacer of six base pairs is coloured orange. Asp and Glu residues in Ocr equivalent to these regions in DNA are coloured in the same way. Residue D73 found to be important in the Mut3 variant of Ocr is highlighted. The yellow ribbon structure is the S subunit of EcoKI bound to Ocr (27; pdb 2Y7C) showing that the TRDs regions contact the green region in Ocr and the DNA while the alpha helical linkers between the TRDs are close to the orange region of Ocr and the DNA. (B) The sequence of Ocr with the residues equivalent to the magenta, green and orange regions of the DNA in panel A coloured similarly. The regions of the mutations are indicated.
The other mutations outside of aa59–91 identify regions which are not absolutely essential for activity but changes in them enhance the deleterious effects of mutations in the aa59–91 region. The amino acids in Mut1, Mut2, Mut4 and after aa99 in Ocr/POcr can be seen to occupy positions in the Ocr structure equivalent to the AAC and GTGC segments of the target sequence plus non-specific DNA sequence extending beyond the target site, Figure 9, in agreement with regions identified previously (13). The regions of Ocr mimicking the AAC and GTGC elements of the target sequence do not seem to be major contributors to the effectiveness of Ocr as an inhibitor of Type I RM enzymes. As previously noted (4,9), this would be expected as Ocr is a non-specific inhibitor of all Type I RM enzymes irrespective of the target sequence recognized by the RM enzyme. The regions of Ocr mimicking the non-specific DNA flanking the target sequence similarly appear to make minor contributions to the effectiveness of Ocr as an inhibitor.
Effects of Ocr mutations on cell viability
The effect of these central Ocr mutants on cell viability was unexpected as wild-type Ocr has no such effect nor do the mutants which have completely lost anti-RM activity. The results for the mutants affecting cell viability appear contradictory in that the mutant Ocr proteins which retain the ability in vivo to halt restriction of phage DNA do not prevent restriction of unmodified host DNA induced by the presence 2AP. In vitro, they do not halt restriction despite the fact that they bind the MTase reasonably well. This difference in behaviour towards unmodified sites on phage and the chromosome has been observed previously and attributed to the different abilities of the Type I RM enzyme to translocate on naked phage DNA versus DNA contained in the nucleoid (29,41). It may also indicate that the larger RM enzyme is more excluded from the nucleoid than the smaller MTase.
However it has also been noted that the R restriction subunit is in short supply in vivo compared to the MTase core in the EcoKI Type I RM system (42). Thus an excess of MTase is usually present in vivo. Normally, wild-type Ocr removes all of the MTase and the RM enzyme activity in the cell thus resulting in unmodified chromosomal DNA. A totally inactive Ocr mutant does not remove the RM enzyme thus the chromosome remains modified by EcoKI. However, if the Ocr activity is only partially diminished then both the MTase and the RM enzyme will be diminished in quantity. The MTase would be unable to maintain chromosomal methylation thus allowing even a single unmodified target site to stimulate cleavage of the chromosome by even a single copy of the RM enzyme. However the presence of minimal amounts of the RM enzyme would appear to be insufficient to give effective restriction of invading phage during the relatively short phage life cycle and thus the Ocr mutant appears to be active in the phage restriction assay. In effect, these Ocr mutations are creating an r+/−m+/− phenotype for the EcoKI RM system depending upon the experimental conditions where concentrations of unmodified DNA targets sites, EcoKI RM enzyme, M.EcoKI and Ocr are varying.
How did Ocr evolve?
Evolutionary theory predicts that improved fitness of one species will be at the expense of another species in the same environment (the Red Queen hypothesis; 43). Bacteriophages are a primary predator of bacteria and the pairwise interaction of phage and host drives evolution of new traits that protect the bacterium from predation, while the predator evolves to overcome new defences (44). Bacteria have evolved numerous strategies to prevent infection, ranging from phase-variable surface receptors and polysaccharides to RM, CRISPR-Cas and toxin-antitoxin systems (45). These mechanisms have been collectively referred to as the ‘resistome’ (46).
Following phage adsorption, the injected DNA will be recognized as ‘foreign’ by the resident RM systems of the infected bacterium. Phages have evolved counter-strategies, such as inclusion of unusual bases in their DNA and anti-RM proteins, e.g., Ocr. The amino acid composition of Ocr is far from that expected for a soluble globular protein with a chain length of 116 amino acids. Negative residues are over-represented (34 instead of 12 expected), and positive residues under-represented (6 instead of 12 expected). If one presumes that the progenitor of Ocr had a more average amino acid composition and no anti-RM activity then mutations would have to accumulate to obtain activity. The reduction in the proportion of basic residues during the course of evolution is probably not too significant or unlikely. The acquisition of many extra acidic residues, however, if all were required for anti-RM activity, would seem less likely. Our mutagenesis and chemical modification data (16) indicate that far fewer than 34 acidic residues are required for anti-RM activity. Sequence alignment of Ocr homologues supports this notion, with only ∼15 conserved or nearly conserved acidic residues present over the homologous sequences (Supplementary Figure S2). It is therefore possible that Ocr evolved from a protein scaffold that already contained a significant number of acidic residues and which interacted with another DNA binding protein. We note early work that showed showing a strong interaction of Ocr with the RNA polymerase of E. coli (47). Furthermore, a number of the acidic residues in first and fourth helices of Ocr (4) appear to contain motifs that are present in other DNA-mimicking proteins, e.g. DinI and UGI (3). DinI mimics single-stranded DNA and interacts with RecA during SOS induction in E. coli (48). It is possible that T7 acquired a gene encoding a DinI-like homologue which subsequently evolved a minimal anti-RM activity and continued to rapidly mutate, under coevolutionary pressure, to acquire greater anti-RM activity. We note that genes encoding DinI homologues are present in some phage genomes, e.g. the cytotoxin converting phage CDT-1ϕ (49).
The switch from original function to anti-RM would appear, from our data, to require only a few amino acid changes. Thus, a minimal DNA mimic anti-RM protein should arise easily. Although it also creates a problem for cell viability this should not be a significant problem for a lytic phage such as T7. Selection for reduced toxicity, better DNA mimicry and improved anti-RM function can occur subsequently to improve the binding affinity of the anti-RM protein for the Type I RM enzyme to such a degree that the RM enzyme:anti-RM complex becomes more stable than the RM enzyme:DNA complex: the situation now observed for the interaction of Ocr with Type I RM enzymes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1 and 2.
FUNDING
Wellcome Trust [GR080463MA, 090288/Z/09/ZA]; RASOR grant from the BBSRC [BB/C511599/1]. Funding for open access charge: Wellcome Trust and the Biotechnology and Biological Sciences Research Council.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
N.K. thanks the EPSRC/BBSRC RASOR Doctoral Training Centre in Cell & Proteomic Technologies for a studentship. We thank Prof Wilson Poon and Mr Ryan Morris (School of Physics, University of Edinburgh) for use of the plate reader and instruction in its use.
REFERENCES
- 1.Putnam DC, Tainer AJ. Protein mimicry of DNA and pathway regulation. DNA Repair. 2005;4:1410–1420. doi: 10.1016/j.dnarep.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 2.Tock MR, Dryden DTF. The biology of restriction and anti-restriction. Curr. Opin. Microbiol. 2005;8:466–472. doi: 10.1016/j.mib.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Dryden DTF. DNA mimicry by proteins and the control of enzymatic activity on DNA. Trends Biotechnol. 2006;24:378–382. doi: 10.1016/j.tibtech.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Walkinshaw MD, Taylor P, Sturrock SS, Atanasiu C, Berge T, Henderson RM, Edwardson JM, Dryden DTF. Structure of Ocr from bacteriophage T7, a protein that mimics B-form DNA. Mol. Cell. 2002;9:187–194. doi: 10.1016/s1097-2765(02)00435-5. [DOI] [PubMed] [Google Scholar]
- 5.McMahon SA, Roberts GA, Johnson KA, Cooper LP, Liu H, White JH, Carter LG, Sanghvi B, Oke M, Walkinshaw MD, et al. Extensive DNA mimicry by the ArdA anti-restriction protein and its role in the spread of antibiotic resistance. Nucleic Acids Res. 2009;37:4887–4897. doi: 10.1093/nar/gkp478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas CM, Nielsen KM. Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 2005;3:711–721. doi: 10.1038/nrmicro1234. [DOI] [PubMed] [Google Scholar]
- 7.Wilkins BM. Plasmid promiscuity: meeting the challenge of DNA immigration control. Environ. Microbiol. 2002;4:495–500. doi: 10.1046/j.1462-2920.2002.00332.x. [DOI] [PubMed] [Google Scholar]
- 8.Studier FW. Gene 0.3 of bacteriophage T7 acts to overcome the DNA restriction system of the host. J. Mol. Biol. 1975;94:283–295. doi: 10.1016/0022-2836(75)90083-2. [DOI] [PubMed] [Google Scholar]
- 9.Kruger DH, Schroeder C, Hansen S, Rosenthal HA. Active protection by bacteriophages T3 and T7 against E. coli B- and K-specific restriction of their DNA. Mol. Gen. Genet. 1977;153:99–106. doi: 10.1007/BF01036001. [DOI] [PubMed] [Google Scholar]
- 10.Dunn JJ, Elzinga M, Mark KK, Studier FW. Amino acid sequence of the gene 0.3 protein of bacteriophage T7 and nucleotide sequence of its mRNA. J. Biol. Chem. 1981;256:2579–2585. [PubMed] [Google Scholar]
- 11.Mark KK, Studier FW. Purification of the gene 0.3 protein of bacteriophage T7, an inhibitor of the DNA restriction system of Escherichia coli. J. Biol. Chem. 1981;256:2573–2578. [PubMed] [Google Scholar]
- 12.Bandyopadhyay PK, Studier FW, Hamilton DL, Yuan R. Inhibition of the type I restriction-modification enzymes EcoB and EcoK by the gene 0.3 protein of bacteriophage T7. J. Mol. Biol. 1985;182:567–578. doi: 10.1016/0022-2836(85)90242-6. [DOI] [PubMed] [Google Scholar]
- 13.Atanasiu C, Byron O, McMiken H, Sturrock SS, Dryden DTF. Characterisation of the structure of Ocr, the gene 0.3 protein of bacteriophage T7. Nucleic Acids Res. 2001;29:3059–3068. doi: 10.1093/nar/29.14.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atanasiu C, Su T-J, Sturrock SS, Dryden DTF. Interaction of the Ocr gene 0.3 protein of bacteriophage T7 with EcoKI restriction/modification enzyme. Nucleic Acids Res. 2002;30:3936–3944. doi: 10.1093/nar/gkf518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blackstock JJ, Egelhaaf SU, Atanasiu C, Dryden DTF, Poon WCK. Shape of Ocr, the gene 0.3 protein of bacteriophage T7, modeling based on light scattering experiments. Biochemistry. 2001;40:9944–9949. doi: 10.1021/bi010587+. [DOI] [PubMed] [Google Scholar]
- 16.Stephanou AS, Roberts GA, Cooper LP, Clarke DJ, Thomson AR, MacKay CL, Nutley M, Cooper A, Dryden DTF. Dissection of the DNA mimicry of the bacteriophage T7 Ocr protein using chemical modification. J. Mol. Biol. 2009;391:565–576. doi: 10.1016/j.jmb.2009.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stephanou AS, Roberts GA, Tock MR, Pritchard EH, Turkington R, Nutley M, Cooper A, Dryden DTF. A mutational analysis of DNA mimicry by Ocr, the gene 0.3 antirestriction protein of bacteriophage T7. Biochem. Biophys. Res. Commun. 2009;378:129–132. doi: 10.1016/j.bbrc.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 18.Zavilgelsky GB, Kotova VY, Rastorguev SM. Comparative analysis of anti-restriction activities of ArdA (ColIb-P9) and Ocr (T7) proteins. Biochemistry. 2008;73:906–911. doi: 10.1134/s0006297908080087. [DOI] [PubMed] [Google Scholar]
- 19.Zavilgelsky GB, Kotova VY, Rastorguev SM. Antirestriction and antimodification activities of T7 Ocr: effects of amino acid substitutions in the interface. Mol. Cell. Biol. 2009;43:93–100. [PubMed] [Google Scholar]
- 20.Zavilgelsky GB, Rastorguev SM. Antirestriction proteins ArdA and Ocr as efficient inhibitors of Type I restriction–modification enzymes. Mol. Biol. 2009;43:241–248. [PubMed] [Google Scholar]
- 21.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2010;38:D234–D236. doi: 10.1093/nar/gkp874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dryden DTF. Bacterial DNA methyltransferases. In: Cheng X, Blumenthal RM, editors. S-Adenosylmethionine-Dependent Methyltransferases: Structures and Functions. Singapore: World Scientific Publishing; 1999. pp. 283–340. [Google Scholar]
- 23.Murray NE. Type I restriction systems: sophisticated molecular machines (a legacy of Bertani and Weigle) Microbiol. Mol. Biol. Rev. 2000;64:412–434. doi: 10.1128/mmbr.64.2.412-434.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murray NE. Immigration control of DNA in bacteria: self versus non-self. Microbiology. 2002;148:3–20. doi: 10.1099/00221287-148-1-3. [DOI] [PubMed] [Google Scholar]
- 25.Loenen WAM. Tracking EcoKI and DNA fifty years on: a golden story full of surprises. Nucleic Acids Res. 2003;31:7059–7069. doi: 10.1093/nar/gkg944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kennaway CK, Taylor JE, Song CF, Potrzebowski W, Nicholson W, White JH, Swiderska A, Obarska-Kosinska A, Callow P, Cooper LP, et al. Structure and operation of the DNA-translocating type I DNA restriction enzymes. Genes Dev. 2012;26:92–104. doi: 10.1101/gad.179085.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kennaway CK, Obarska-Kosinska A, White JH, Tuszynska I, Cooper LP, Bujnicki JM, Trinick J, Dryden DTF. The structure of M.EcoKI Type I DNA methyltransferase with a DNA mimic antirestriction protein. Nucleic Acids Res. 2009;37:762–770. doi: 10.1093/nar/gkn988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su T-J, Tock MR, Egelhaaf SU, Poon WCK, Dryden DTF. DNA bending by M.EcoKI methyltransferase is coupled to nucleotide flipping. Nucleic Acids Res. 2005;33:3235–3244. doi: 10.1093/nar/gki618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blakely GW, Murray NE. Control of the endonuclease activity of type I restriction-modification systems is required to maintain chromosome integrity following homologous recombination. Mol. Microbiol. 2006;60:883–893. doi: 10.1111/j.1365-2958.2006.05144.x. [DOI] [PubMed] [Google Scholar]
- 30.Davies GP, Kemp P, Molineux IJ, Murray NE. The DNA translocation and ATPase activities of restriction-deficient mutants of EcoKI. J. Mol. Biol. 1999;292:787–796. doi: 10.1006/jmbi.1999.3081. [DOI] [PubMed] [Google Scholar]
- 31.Amann E, Ochs B, Abel KJ. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene. 1988;69:301–315. doi: 10.1016/0378-1119(88)90440-4. [DOI] [PubMed] [Google Scholar]
- 32.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd edn. New York: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 33.Serfiotis-Mitsa D, Roberts GA, Cooper LP, White JH, Nutley M, Cooper A, Blakely GW, Dryden DTF. The Orf18 gene product from conjugative transposon Tn916 is an ArdA antirestriction protein that inhibits Type I DNA restriction-modification systems. J. Mol. Biol. 2008;383:970–981. doi: 10.1016/j.jmb.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 34.Makovets S, Titheradge AJ, Murray NE. ClpX and ClpP are essential for the efficient acquisition of genes specifying type IA and IB restriction systems. Mol. Microbiol. 1998;28:25–35. doi: 10.1046/j.1365-2958.1998.00767.x. [DOI] [PubMed] [Google Scholar]
- 35.Dryden DTF, Cooper LP, Murray NE. Purification and characterization of the methyltransferase from the Type-I restriction and modification system of Escherichia-coli K12. J. Biol. Chem. 1993;268:13228–13236. [PubMed] [Google Scholar]
- 36.Dryden DTF, Cooper LP, Thorpe PH, Byron O. The in vitro assembly of the EcoKI type I DNA restriction/modification enzyme and its in vivo implications. Biochemistry. 1997;36:1065–1076. doi: 10.1021/bi9619435. [DOI] [PubMed] [Google Scholar]
- 37.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 38.Whitmore L, Wallace BA. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32:W668–W673. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Powell LM, Connolly BA, Dryden DTF. The DNA binding characteristics of the trimeric EcoKI methyltransferase and its partially assembled dimeric form determined by fluorescence polarisation and DNA footprinting. J. Mol. Biol. 1998;283:947–961. doi: 10.1006/jmbi.1998.2142. [DOI] [PubMed] [Google Scholar]
- 40.Chen K, Roberts GA, Stephanou AS, Cooper LP, White JH, Dryden DTF. Fusion of GFP to the M.EcoKI DNA methyltransferase produces a new probe of Type I DNA restriction and modification enzymes. Biochem. Biophys. Res. Commun. 2010;398:254–259. doi: 10.1016/j.bbrc.2010.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Makovets S, Powell LM, Titheradge AJB, Blakely GW, Murray NE. Is modification sufficient to protect a bacterial chromosome from a resident restriction endonuclease? Mol. Microbiol. 2004;51:135–147. doi: 10.1046/j.1365-2958.2003.03801.x. [DOI] [PubMed] [Google Scholar]
- 42.Webb JL, King G, Ternent D, Titheradge AJ, Murray NE. Restriction by EcoKI is enhanced by co-operative interactions between target sequences and is dependent on DEAD box motifs. EMBO J. 1996;15:2003–2009. [PMC free article] [PubMed] [Google Scholar]
- 43.Van Valen L. Molecular evolution as predicted by natural selection. J. Mol. Evol. 1974;3:89–101. doi: 10.1007/BF01796554. [DOI] [PubMed] [Google Scholar]
- 44.Paterson S, Vogwill T, Buckling A, Benmayor R, Spiers AJ, Thomson NR, Quail M, Smith F, Walker D, Libberton B, et al. Antagonistic coevolution accelerates molecular evolution. Nature. 2010;464:275–278. doi: 10.1038/nature08798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Makarova KS, Wolf YI, Snir S, Koonin EV. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J. Bacteriol. 2011;193:6039–6056. doi: 10.1128/JB.05535-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoskisson PA, Smith MC. Hypervariation and phase variation in the bacteriophage ‘resistome’. Curr. Opin. Microbiol. 2007;10:396–400. doi: 10.1016/j.mib.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 47.Ratner D. The interaction bacterial and phage proteins with immobilized Escherichia coli RNA polymerase. J. Mol Biol. 1974;88:373–383. doi: 10.1016/0022-2836(74)90488-4. [DOI] [PubMed] [Google Scholar]
- 48.Ramirez BE, Voloshin ON, Camerini-Otero RD, Bax A. Solution structure of DinI provides insight into its mode of RecA inactivation. Protein Sci. 2000;9:2161–2169. doi: 10.1110/ps.9.11.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Asakura M, Hinenoya A, Alam MS, Shima K, Zahid SH, Shi L, Sugimoto N, Ghosh AN, Ramamurthy T, Faruque SM, et al. An inducible lambdoid prophage encoding cytolethal distending toxin (Cdt-I) and a type III effector protein in enteropathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA. 2007;104:14483–14488. doi: 10.1073/pnas.0706695104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.