

Abstract
Background
We recently characterized a specific inorganic triphosphatase (PPPase) from Nitrosomonas europaea. This enzyme belongs to the CYTH superfamily of proteins. Many bacterial members of this family are annotated as predicted adenylate cyclases, because one of the founding members is CyaB adenylate cyclase from A. hydrophila. The aim of the present study is to determine whether other members of the CYTH protein family also have a PPPase activity, if there are PPPase activities in animal tissues and what enzymes are responsible for these activities.
Methodology/Principal Findings
Recombinant enzymes were expressed and purified as GST- or His-tagged fusion proteins and the enzyme activities were determined by measuring the release of inorganic phosphate. We show that the hitherto uncharacterized E. coli CYTH protein ygiF is a specific PPPase, but it contributes only marginally to the total PPPase activity in this organism, where the main enzyme responsible for hydrolysis of inorganic triphosphate (PPPi) is inorganic pyrophosphatase. We further show that CyaB hydrolyzes PPPi but this activity is low compared to its adenylate cyclase activity. Finally we demonstrate a high PPPase activity in mammalian and quail tissue, particularly in the brain. We show that this activity is mainly due to Prune, an exopolyphosphatase overexpressed in metastatic tumors where it promotes cell motility.
Conclusions and General Significance
We show for the first time that PPPase activities are widespread in bacteria and animals. We identified the enzymes responsible for these activities but we were unable to detect significant amounts of PPPi in E. coli or brain extracts using ion chromatography and capillary electrophoresis. The role of these enzymes may be to hydrolyze PPPi, which could be cytotoxic because of its high affinity for Ca2+, thereby interfering with Ca2+ signaling.
Introduction
Recently, we have demonstrated for the first time that a bacterial enzyme hydrolyzed inorganic triphosphate (tripolyphosphate, PPPi) with high specificity and catalytic efficiency [1]. This enzyme from Nitrosomonas europaea (referred to as NeuTTM) belongs to the CYTH protein superfamily identified in 2002 by Iyer and Aravind [2]. According to these authors the catalytic domains of human 25-kDa thiamine triphosphatase (hThTPase, [3]) and CyaB-like adenylyl cyclase from Aeromonas hydrophila (AC2, [4]) define a novel superfamily of domains supposed to bind “organic phosphates”. This superfamily was therefore called “CYTH” (CyaB-THiamine triphosphatase), and the presence of orthologs was demonstrated in all three superkingdoms of life. Using multiple alignments and secondary structure predictions, Iyer and Aravind showed that the catalytic core of CYTH enzymes contained a novel α+β scaffold with 6 conserved acidic and 4 basic residues. At least 4 of the acidic residues (generally glutamates) are likely to chelate 1 or 2 divalent cations that are required for catalysis, as is the case for nucleotide cyclases, polymerases and some phosphohydrolases [2], [5], [6]. Independently of those bioinformatic studies, Shuman and coworkers investigated the first step of RNA capping in fungi and protozoa catalyzed by RNA triphosphatases. They showed that the yeast RNA triphosphatase Cet1 belongs to a family of metal-dependent phosphohydrolases, whose active site is located within a topologically closed hydrophilic β-barrel (composed of 8 antiparallel β strands) that they called the “triphosphate tunnel” [7]. Strikingly similar structures were found for several bacterial and archaeal proteins of unknown function [7]. Since all those proteins carry the CYTH signature, Gong et al., [7] coined the name “Triphosphate Tunnel Metalloenzyme” (TTM) for a superfamily including Cet-1-like RNA triphosphatases and all other CYTH proteins. However, the NeuTTM PPPase does not exhibit the closed tunnel structure [1]. On the other hand, bacterial CYTH enzymes have generally been annotated as adenylyl cyclases, but only AC2 from A. hydrophila [4] and YpAC4 form Y. pestis [8] have been found to exhibit adenylyl cyclase activity, and its physiological significance in these organisms is unclear.
Only two other bacterial CYTH enzymes have been functionally characterized: NeuTTM [1] and CthTTM from Clostridium thermocellum [9], [10]. Both have a high PPPase activity (though CthTM was less specific) but neither had any significant adenylyl cyclase activity [1], [9]. We thus suspect that PPPase activity might be the ancestral role of CYTH proteins, but the significance of such an activity is by no means clear. Indeed, in contrast to high molecular weight polyphosphates, no data are available concerning the presence or the possible role of PPPi (and other short chain polyphosphates) in either prokaryotic or eukaryotic cells. However, this lack of data is essentially due to the current absence of sensitive and specific methods to detect short chain polyphosphates. Actually, it is not excluded that these compounds might play very important roles in cell physiology.
The aim of the present investigation was to check whether significant PPPase activities exist in E. coli but also in mammalian tissues and what enzymes are responsible for these activities.
Results
Characterization of recombinant E. coli ygiF protein
As we have previously shown that the N. europaea CYTH protein is a specific tripolyphosphatase [1], we wanted to check whether the E. coli ortholog ygiF [2], labeled as a predicted adenylate cyclase, had a similar activity and substrate specificity. YgiF was overexpressed in E. coli as a GST-fusion protein and purified (Figure S1). The purified fusion protein had indeed a high PPPase activity ( Figure 1 ) with a V max of 27±2 µmoles min−1 mg−1 and the K m was 270±25 µM. The optimum pH was 8.5. The enzyme was activated by Mg2+ (EC50 = 1.3±0.2 mM) and inhibited by Ca2+ (IC50 = 0.6±0.3 mM). Among other substrates tested, only ThTP was slowly hydrolyzed. No hydrolysis was observed with ATP, ITP, GTP, PPi or polyphosphate (65±5 residues) as substrates ( Figure 2 ).
Figure 1. Enzymatic characterization of GST-ygiF PPPase activity.

(A) Enzyme activity as a function of PPPi concentration. The curve was obtained by non-linear fitting to the Michaelis-Menten equation. The Mg2+ concentration was 5 mM. (B) PPPase activity as a function of pH at 0.5 mM Mg2+. (C) PPPase activity as a function of Mg2+ concentration. (D) Inhibition of PPPase activity by Ca2+ in the presence of Mg2+ (5 mM). If not otherwise stated, the substrate concentration was 0.5 mM and the incubation was carried out at 50°C and at pH 8.5. (Means ± SD, n = 3).
Figure 2. Specificity of GST-ygiF.

The various substrates were tested at a concentration of 0.5 mM in the presence of 5 mM Mg2+. The incubation temperature was 50°C and the pH 8.5. (Means ± SD, n = 3).
These results suggest that ygiF, like NeuTTM, is a specific tripolyphosphatase, with the characteristic properties of the CYTH protein family, i. e. alkaline optimum pH, activation by Mg2+ and inhibition by Ca2+.
In E. coli supernatants, hydrolysis of PPPi is mainly catalyzed by inorganic pyrophosphatase
We first estimated the PPPase activity of a crude extract from wild-type MG1655 E. coli (supernatant obtained after sonication and centrifugation at 40,000×g). We found a relatively high Mg2+-dependent PPPase activity with alkaline pH optimum: the specific activity was about 0.2 µmol min−1 mg−1 at 37°C (with [PPPi] = 1 mM, [Mg2+] = 5 mM, pH 9.1). The activity was remarkably heat-stable (up to 80°C).
Those properties are reminiscent of E. coli inorganic pyrophosphatase (EcPPase) [11], [12]. We therefore suspected that PPPi hydrolysis by E. coli extracts might be mainly catalyzed by the PPase, which is abundant in the bacterial cytoplasm [13].
We partially purified the enzyme responsible for the high PPPase activity in E. coli supernatants using various chromatographic techniques (Table S1), followed by native electrophoresis on agarose gels and in-gel activity determination. The band containing the activity was then excised. Analysis by LC-ESI/MS/MS identified inorganic pyrophosphatase (PPase) as a major protein present in the fraction.
We therefore used a commercially available preparation of purified EcPPase to characterize its PPPase activity ( Figure 3 ). This preparation indeed hydrolyzed PPPi under the conditions observed above. As expected, it was less efficient for the hydrolysis of PPPi than for PPi, considered its natural substrate. With PPPi, we found a V max = 50 µmol min−1 mg−1 at 25°C about 40% of Vmax measured with PPi under similar conditions (V max = 125 µmol min−1 mg−1). The Km for PPPi was 0.91±0.09 mM, three orders of magnitude higher than for PPi (≤10−3 M compared to ≤10−6 M) [14].
Figure 3. Enzyme activity of commercial E. coli recombinant pyrophosphatase as a function of PPPi concentration.

The enzyme was incubated in the presence of 10 mM Mg2+ in 50 mM CHES buffer at pH 9.1, for 30 min at 25°C. (Means ± SD, n = 3).
Considering a molecular mass of 20 kDa per subunit, we estimated that kcat = 16.7 s−1 at 25°C for PPPi, compared to 42 s−1 for PPi. Using a different preparation of recombinant E. coli pyrophosphatase, Avaeva et al. [14] reported a substantially higher value for k cat (389 s−1 with PPi). In any event, due to the very large difference in K m values for PPi and PPPi, the catalytic efficiency (k cat/K m) is much lower for PPPi (18.103 s M−1) than for PPi (≥42.106 s M−1).Considering that the intracellular PPi concentration is relatively high, except under severe energy stress [13], it is likely that most of the active sites of pyrophosphatase remain saturated with PPi and unavailable for PPPi hydrolysis. In addition, the catalytic efficiency for PPPi hydrolysis is low. Therefore, it may be argued that, in vivo, the pyrophosphatase is uneffective as a PPPase and ygiF, which is much more specific, might be the main enzyme responsible for PPPi hydrolysis in vivo. We thus estimated the PPPase activity in the ygiF knockout strain JW 3026-2. No decrease of specific PPPase activity in crude extracts was observed (Figure S2), indicating that the contribution of ygiF to PPPase activity is, at best, marginal. This is not surprising as PPase is highly expressed in E. coli with nearly 400 copies per cell [15]. On the other hand, ygiF is expressed at only 20 copies per cell. It is therefore not surprising that ygif does not contribute significantly to PPPi hydrolysis in a bacterial supernatant fraction. For ygiF to play a significant role in PPPi hydrolysis, it would require at least a 20-fold upregulation of its expression. This is however not very plausible as it is constitutively cotranscribed with glutamine synthetase adenylyl transferase [16]. Also in contrast to inorganic pyrophosphatase, ygiF is not an essential protein.
As E. coli also contains an exopolyphosphatase which might be responsible for some PPPi hydrolysis, we used a mutant strain devoid of both polyphosphate kinase and exopolyphosphatase (Δppk-ppx::km). However, PPPase activity was the same in this strain as in the wild-type MG1655 strain (Figure S3), suggesting that exopolyphosphatase does not contribute significantly to PPPi hydrolysis in an E. coli supernatant, in agreement with previous results that showed that PPPi is not a good substrate for this enzyme [17].
We also tested whether PPase and PPPase activity changed during growth of E. coli, but both PPase and PPPase activities remained relatively constant (Figure S4).
CyaB adenylate cyclase has a significant triphosphatase activity but lacks specificity
The A. hydrophila CyaB protein, one of the founding members of the CYTH protein superfamily has an adenylate cyclase activity, but is different from class 1 adenylyl cyclases present in most organisms [4]. Furthermore, CyaB was not expressed under normal laboratory growth conditions. As for ygiF, we wanted to test whether this enzyme is able to hydrolyze triphosphates. Our results show that it indeed hydrolyzes substrates such as ATP, ThTP and PPPi but at a low rate ( Figure 4A ). In the presence of Mg2+ it has no preference for any of these substrates, while it has a slight preference for ThTP over ATP and PPPi in the presence of Mn2+. The rate of hydrolysis of these substrates is at least an order of magnitude lower than its adenylate cyclase activity previously reported [4] and respectively 2 and 3 orders of magnitude lower than the PPPase activity of ygiF (this study) and NeuTTM [1]. Therefore, this activity is probably not of physiological importance. With ThTP as substrate, the optimum pH is alkaline ( Figure 4B ) as is usually observed with enzymes of the CYTH superfamily and the activity is highest at a temperature of 50°C ( Figure 4C ).
Figure 4. Enzymatic hydrolysis of ATP, PPPi and ThTP by recombinant CyaB protein.

(A) Specificity of CyaB in the presence of 5 mM Mg2+ or Mn2+ at pH 9.1. (B) pH dependence of ThTPase activity. (C) Temperature dependence of ThTPase activity at pH 7.5 and 9.1. If not otherwise stated, the substrate concentration was 0.5 mM, the Mg2+ or Mn2+ concentration was 5 mM and a temperature of 50°C was used. (Means ± SD, n = 3).
PPPase activities in mammalian tissues
As the above results suggest that PPPase activities are rather common in bacteria, we wanted to test whether such activities are also present in mammalian tissues. We measured PPPi hydrolysis in the supernatant fraction of several rat and quail tissues as well as pig brain ( Figure 5A ). The specific PPPase activity was highest in brain, only 2–3 times lower than in E. coli supernatants under identical conditions. The activity was not strongly pH-dependent but was highest around pH 7 ( Figure 5B ). PPPase activity was activated by Mg2+, to a lesser extent by Mn2+, but was insensitive to Ca2+ ( Figure 5C ).
Figure 5. PPPase activities in animal tissue supernatant fractions.

(A) PPPi hydrolysis in various organs of the rat, and the quail, and in pig brain at pH 7.0 in the presence of 5 mM Mg2+ (n = 3–9). (B) pH dependence of PPPase hydrolysis in rat brain in the presence of 5 mM Mg2+ (n = 3). (C) Activation of PPPase activity by divalent cations. The PPPi concentration was 0.5 mM and the incubation was carried out at 37°C. (Means ± SD, n = 3–9).
In order to identify the enzyme responsible for PPPase hydrolysis in mammalian brain, we used a combination of chromatographic techniques and native polyacrylamide gel electrophoresis, and we carried out the identification by LC-ESI/MS/MS. A major protein identified was the pig homologue of the Drosophila protein prune. It was recently shown that human prune is an exopolyphosphatase with higher activity towards short chain polyphosphates such as PPPi and P4 [18]. It also hydrolyzes very efficiently adenosine and guanosine tetraphosphates. For PPPi, the k cat was 13 s−1 and the K m was as low as 2.2 µM. This suggest that most of the PPPase activity in mammalian tissues is due to h-Prune. The latter belongs to the DHH protein superfamily named after the characteristic N-terminal Asp-His-His motif. It has no sequence similarities with proteins of the CYTH superfamily (Figure S5).
h-Prune is often overexpressed in metastatic cancer and interacts with the metastasis tumor suppressor nm23-H1, coding for nucleoside diphosphate kinase A (NDPK-A). It was shown that phosphorylation of nm23-H1 by casein kinase 1 leads to complex formation with h-Prune, hence promoting cell motility in breast cancer cells. The role of h-Prune in normal adult brain cells is unknown. H-Prune and nm23-H1 are co-expressed during embryonic development and may play a role in mammalian brain development, while in the adult brain their role may be predominantly linked to their respective enzyme activities [19].
Is PPPi present in living cells?
Until now PPPi has not been shown to exist in living cells except for very specialized organelles such as protozoan acidocalcisomes [20]. One of the problems when determining PPPi is the absence of a specific and sensitive detection method. We first tested a chromatographic separation technique using a Dionex device equipped with a conductivity detector. Such a method was already used to detect exogenously added tripolyphosphate in frozen cod and scallop adductor [21]. We were able to detect in E. coli a small peak at the retention time of PPPi corresponding to a concentration of 1 µM, while no peak was detected in rat brain extracts ( Figure 6 ).
Figure 6. Separation of a E. coli extract on a Dionex column.

The sample was spiked with exogenous PPPi (10 mg/L). It can be estimated that peak 11 accounts for less than 0.5 mg/L.
A method using capillary electrophoresis (CE) coupled to a diode array detector was also optimized to detect PPPi with a limit of detection estimated to 0.5 µM. After fractionation of E. coli supernatants on G-25 column, no PPPi peak could be observed ( Figure 7 ), suggesting that PPPi, if it exists in living cells, is not a very abundant compound and its concentrations are probably in the low micromolar or even sub-micromolar range.
Figure 7. Separation of PPPi by capillary electrophoresis.

Electropherograms of (a) E. coli supernatant after fractionation on G-25 column and (b) E. coli supernatant after fractionation on G-25 column spiked with 3 µM PPPi.
Discussion
Next to carbon, oxygen, nitrogen and hydrogen, phosphorus is the most abundant element in living organisms. Phosphorus is found mainly under the form of phosphate in many organic compounds such as nucleotides, metabolic intermediates (intermediates of glycolysis such as phosphoenolpyruvate for instance), phospholipids or in bones as calcium phosphate salts. In addition to be present in organic molecules, phosphates may exist in free forms. Hydrolysis of ATP yields Pi whose intracellular concentration may exceed 1 mM. Pi is recycled mainly by regeneration of ATP via FoF1-ATP synthase according to the reaction ADP+Pi
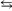 ATP.
ATP.
Inorganic pyrophosphate (PPi) is released in different anabolic processes such as the synthesis of ribo- and deoxyribonucleotides ((NMP)n+NTP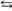 (NMP)n+1+PPi) or the synthesis of aminoacyl-tRNA (amino acid+tRNA+ATP
(NMP)n+1+PPi) or the synthesis of aminoacyl-tRNA (amino acid+tRNA+ATP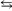 aminoacyl-tRNA+AMP+PPi). In each case, PPi is rapidly hydrolyzed to 2 Pi by an inorganic PPase (EC 3.6.1.1), rendering these reactions globally irreversible. PPases are widely distributed in all organisms and here, we show that at least E. coli PPase also has an important PPPase activity. Hence, PPi is constantly formed in all organisms, but it is not synthesized de novo, at least in animal cells [22], though a pyrophosphate synthase, related to V-type ATPases, has been characterized from Rhodospirillum rubrum
[23].
aminoacyl-tRNA+AMP+PPi). In each case, PPi is rapidly hydrolyzed to 2 Pi by an inorganic PPase (EC 3.6.1.1), rendering these reactions globally irreversible. PPases are widely distributed in all organisms and here, we show that at least E. coli PPase also has an important PPPase activity. Hence, PPi is constantly formed in all organisms, but it is not synthesized de novo, at least in animal cells [22], though a pyrophosphate synthase, related to V-type ATPases, has been characterized from Rhodospirillum rubrum
[23].
Long chain polyphosphates have been discovered in many organisms [24], [25]. These polyphosphates are linear chains of up to 1000 Pi residues linked by phosphoanhydride bonds. They may play multiple roles as energy sources, phosphate reservoirs, phosphate donors and chelators of divalent cations. In E. coli, they are synthesized according to the reaction (polyP)n+ATP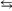 (polyP)n+1+ADP. A recent study suggested that polyP could be synthesized in mammalian cells by a mechanism requiring FoF1-ATP synthase [26]. Polyphosphates may be hydrolyzed by dedicated exopolyphosphatases, but less specific enzymes such as Clostridium thermocellum CYTH protein [10] or h-Prune for instance [18] may also act as exopolyphosphatases.
(polyP)n+1+ADP. A recent study suggested that polyP could be synthesized in mammalian cells by a mechanism requiring FoF1-ATP synthase [26]. Polyphosphates may be hydrolyzed by dedicated exopolyphosphatases, but less specific enzymes such as Clostridium thermocellum CYTH protein [10] or h-Prune for instance [18] may also act as exopolyphosphatases.
PPPi has never been demonstrated to exist in living organisms with the exception of the very specialized protozoan acidocalcisomes [20]. PPPi can be generated as the final product of polyP-glucose phosphotransferase (EC 2.7.1.63), present in various bacteria (Mycobacterium tuberculosis, Propionobacterium shermanii…). This enzyme uses polyP to form glucose 6-phosphate from glucose ((polyP)n+D-glucose???(polyP)n−1+D-glucose 6-phosphate). In animals, an endopolyphosphatase (EC 3.6.1.10) may produce PPPi [27]. PPPi may also serve as a substrate for the phosphorylation of membrane proteins in rat liver microsomes in vitro [28]. This activity is probably not of physiological significance as it is inhibited by micromolar concentrations of ADP and ATP.
The observation that NeuTTM is a highly specific PPPase raises the question of the existence of this compound in living cells [1]. Moreover, our present results show that ygiF, which is commonly annotated a predicted adenylate cyclase, is also a relatively specific PPPase. Its activity is however insignificant compared to PPPase activity resulting from inorganic PPase in E. coli.
Figure 8 shows the comparison of the sequences of NeuTTM and ygiF. It is obvious that ygiF is about twice as long as NeuTTM. We previously suggested that the catalytic dyad in NeuTTM is formed by Tyr-28 and Lys-52. Though the equivalent of Tyr-28 is present in ygiF, the equivalent of Lys-52 seems to be absent. Furthermore, the specific activity of ygiF is one or two orders of magnitude lower than that of NeuTTM. These results suggest that the physiological role of ygiF might be different from PPPi hydrolysis, but it is probably not an adenylate cyclase, as we were unable to detect such activity in purified ygiF.
Figure 8. Sequence alignment of NeuTTM and ygiF.

The sequence alignment was made using CLC Sequence Viewer 6 (CLC bio A/S, 8200 Aarhus, Denmark).
We also studied another protein from the CYTH protein family: the A. hydrophyla CyaB protein, which was previously shown to have a significant adenylate cyclase activity. Our data show that CyaB also has PPPase activity, but this activity is lower than its adenylate cyclase activity [4]. Finally, we show for the first time a high PPPase activity in animal tissues, and in particular in brain. This PPPase activity is catalyzed by the homologs of the Drosophila exopolyphosphatase prune.
Our results show a high PPPase activity in many cells from bacteria to birds and mammals. However, few of these activities come from specific PPPases: the only known specific PPPases are NeuTTM and ygiF, though it is questionable that for the latter, this activity is of physiological importance.
In view of the many and important PPPase activities present in cells, the issue is to know whether PPPi exists as such in living cells. This compound is difficult to detect in small amounts: there are no specific enzymatic assays and it does not contain any chromophore or fluorophore. We first tested an anion exchange separation method coupled to electrochemical detection. This method is routinely used to test the composition of industrial preparations of small-chain polyphosphates. A small peak with a retention time corresponding to PPPi was detected in E. coli but not in brain extracts. We then developed a method based on a separation of phosphates by capillary electrophoresis. Again, no definite conclusion could be drawn as to the existence of PPPi in living cells. However, our data allow us to draw the conclusion that if it exists, intracellular concentrations hardly exceed 1 µM. This would be in agreement with the high PPPase activities observed. Furthermore, many PPPi hydrolyzing enzymes are proteins that are constitutively expressed with a relatively constant expression (inorganic pyrophosphatase, mammalian prune). It is therefore not likely that intracellular PPPi concentrations undergo large variations, as it would be the case for a signaling molecule for instance.
PPPi has a very strong chelating power for divalent cations and in particular Ca2+, which is the reason why it was extensively used in detergents to control water hardness. The dissociation constant K d for Ca2+ is approximately 10−7 M [29]. This would mean that if it accumulated in cells, it would be expected to interfere with Ca2+ signaling. It is therefore possible that the existence of PPPase activities of widely expressed less specific phosphohydrolases is a protective means to prevent accumulation of intracellular PPPi that might accumulate from degradation of long chain polyphosphates for instance. From this point of view, specific PPPases such as NeuTTM could thus be considered as metabolite proofreading enzymes, an expression recently coined by Van Schaftingen and colleagues, by analogy with the proofreading enzymes involved in DNA repair for instance [30], [31], and which should protect cells from unwanted toxic metabolic side-products.
On the other hand, it cannot be excluded that PPPi, which is an energy-rich compound and the simplest triphosphate that can be imagined, may have played a role in energy metabolism in the earliest organisms. Therefore, we recently suggested that PPPi hydrolysis could be the primitive activity of the CYTH protein family [1]. But with the appearance of PPPase side-activity in other abundant phosphohydrolases, CYTH proteins could have evolved towards more complex activities, such as adenylate cyclase in A. hydrophila [4], mRNA–triphosphatase in S. cerevisiae and some protozoans [7], [32], [33] or thiamine triphosphatase in mammals [3], [34].
Materials and Methods
Materials
Sodium triphosphate (PPPi), sodium trimetaphosphate (Cyclic PPPi), polyphosphate (sodium phosphate glass, 65±5 residues), ATP, ITP, GTP, guanosine 5′- tetraphosphate (Gp4, tris salt) and E. coli pyrophosphatase were from Sigma-Aldrich NV/SA (Bornem, Belgium). Thiamine triphosphate (ThTP) was synthesized and purified as previously described [35]. The CyaB expression plasmid pTRC99ACyaB was a gift of Drs Antoine Danchin and Agnieszka Sekowska (AMAbiotics SAS, Genopole Campus 1 - Genavenir 8, 5, rue Henri Desbruères, 91030 EVRY Cedex, France). All animal experiments were made in accordance with the directives of the committee for animal care and use of the University of Liège and in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC). The protocols were approved by the Committee on the Ethics of Animal Experiments of the University of Liège (# 823 for rats and # 727 for quails). The animals were killed by decapitation and all efforts were made to minimize suffering. Pig brains were obtained from the local slaughterhouse (Abattoirs Publics de Liège & Waremme SC, rue de Droixhe 15, 4020 Liège, Belgium) with the permission to be used for experimental purposes at the University of Liège.
Bacterial strains
JW3026-2 (F-, Δ(araD-araB)567, ΔlacZ4787(::rrnB-3), λ−, ΔygiF747::kan, rph-1, Δ(rhaD-rhaB)568, hsdR514, CGSC#10312) was from the E. coli Genetic Stock Center (Yale University, New Haven CT, USA). The CF5802 strain lacking polyP kinase (PPK) and exopolyphosphatase (Δppk-ppx::km) [36] was a gift from Dr. M. Cashel (Laboratory of Molecular Genetics, NICHD, National Institutes of Health, Bethesda, MA).
Determination of phosphohydrolase activities
The standard incubation medium contained 50 mM buffer, 5 mM MgCl2, 0.5 mM PPPi and 20 µL of the enzyme at the adequate concentration in a total volume of 100 µL. The mixture was incubated at 37 or 50°C, the reaction was stopped by addition of 1 mL phosphate reagent [37] and the absorbance (UV-1800 Shimadzu UV spectrophotometer) was read at 635 nm after 30 min and compared with a standard curve (0–0.5 mM K2HPO4) to estimate the release of inorganic phosphate. The buffers used for incubation at different pH values were: Na-MOPS (pH 7.0–7.5), Na-HEPES (pH 7.5–8.0), Na-TAPS (pH 8.0–9.0), Na-CHES (pH 9.0–10.0) and Na-CAPS (pH 10.0–10.5).
Purification of PPPase activity from E. coli supernatants
E. coli wild type strain MG1655 was grown overnight in LB medium at 37°C under agitation (250 rpm). The bacteria were harvested by centrifugation for 10 minutes, at 8000×g at 4°C. The pellet was suspended in Hepes-buffer (30 mM Hepes, 50 mM NaCl, pH 7.2–7.4) and cells were lysed by high pressure using a French press. The lysate was centrifuged for 30 minutes at 40,000×g at 18°C. The supernatant (called S1) was brought to 50% saturation with ammonium sulfate and kept under constant stirring for one hour at room temperature. After centrifugation for 30 minutes at 20,000×g at 18°C, the supernatant (called S50) was brought to 80% saturation with ammonium sulfate and kept under constant stirring for one hour at room temperature. After centrifugation 30 minutes at 20,000×g at 18°C, the pellet was suspended in Hepes-buffer. The preparation obtained (called P80) was loaded on a monoQ column coupled to a Fast Protein Liquid Chromatography (FPLC) apparatus and elution was carried out with a linear NaCl gradient from 50 to 500 mM. The fractions with the highest PPPase activity were pooled (called F1) and placed on a Sephadex G200 column (1×25 cm) equilibrated with Hepes-buffer. Elution was performed in the same buffer. The fractions with the highest PPPase activity were pooled (called F2) and used to perform a non-denaturing polyacrylamide gel separation and the PPPase activity was determined as described below. The bands containing PPPase activity were cut and analyzed by liquid chromatography coupled on-line to positive mode electrospray ionization tandem mass spectrometry (UltiMate 3000 Nano LC Systems, Dionex, AmaZone, Bruker) for protein identification.
In-gel enzyme activities
Non-denaturating polyacrylamide gels (10%) were run in Tris-Glycine buffer (25 mM Tris, 190 mM glycine). After incubation for 30 minutes in a bath containing PPPi (0.5 mM), buffer (20 mM), and MgCl2 (1 mM), the gel was colored with phosphate precipitation reagent (1% v/v ammonium heptamolybdate, 1% v/v triethylamine, 1 N HNO3) [38]. For E. coli PPPase, the incubation temperature was 50°C and buffer was Na-CHES (pH 9.5) while for pig brain PPPase incubation temperature was 37°C and buffer was Na-MOPS (pH 7.1).
Overexpression and purification of recombinant ygiF
YgiF was produced in E. coli as a GST fusion protein, as previously described [39]. Genomic DNA was isolated from E. coli DH5α bacteria using a standard protocol (Invitrogen). YgiF coding sequence was amplified from 1 µg of genomic DNA using Pfu polymerase and 35 PCR cycles of denaturation (94°C, 20 s), annealing (64°C, 30 s) and elongation (72°C, 90 s) using forward (5′-TTTGGATCCATGGCTCAGGAAATCGAATTAAAG-3′) and reverse (5′-TTTGCGGCCGCTTAACGTTTTCCGCTGTGCAACC-3′) primers. The fragment was then incubated for 15 minutes at 72°C in the presence of Taq polymerase and dATP to add a polyA tail. The 1.3 kb PCR fragment was cloned by TA cloning in the pGEM-T vector (Promega). The plasmid containing the insert was sequenced as described above and then digested by BamHI and NotI. The released fragment was purified on agarose gel and subcloned into pGEX-4T2 (GE Healthcare) to produce the GST-ygiF fusion protein in E. coli DH5α. The GST-fusion protein was purified using a 1 mL GSTrap HP column (GE Healthcare).
Cloning of the A. hydrophilia CyaB gene in the PVP16 vector
The plasmid pTRC99ACyaB (gift from A. Danchin and A. Sekowska AMAbiotics SAS, Genopole Campus 1 - Genavenir 8, 91030 EVRY Cedex, France), containing untagged CyaB, was used as starting material. The A. hydrophila CyaB gene (Sismeiro et al. 1998) was amplified by polymerization chain reaction and inserted in the PVP16 vector (Song et al. 2008). To confirm the insertion of the gene, the plasmid was sequenced. The two primers that were used to amplify the gene are: CyaB Forward: 5′-TTGGCGCGCCCAGAAAACCTGTACTTCCAGTCCATGTCATCACAACACTTTCAGG-3′ and CyaB Reverse: 5′-TTACTAGTTCATGAGGGCACTTTCTCTGAG-3′. The ATG underlined in the sequence of CyaB Forward corresponds to the codon that encoded the first methionine in the protein sequence.
Overexpression and purification of recombinant A. hydrophila CyaB
BL21DE3 were transformed with PVP16CyaB Vector and the expression of the protein was induced by using IPTG. This vector allows the expression of 8-histidine-tagged maltose binding protein (8HIS-MBP) fusion protein. After a first purification on a nickel column (1 mL HisTrap™ HP column (GE Healthcare) [1]), the tag was removed by Tobacco Etch Virus digestion and the purity was checked using Coomassie blue staining after SDS-PAGE.
Purification of PPPase activity from pig brain
PPPase was purified from about 500 g of pig brain. All the purification steps were carried out at 4°C. The tissues were homogenized in Tris-HCl buffer (30 mM Tris-HCl, 50 mM, NaCl, 0.1 mM EDTA, 1.5 mM DTT, pH 7.4). The homogenate was centrifuged at 20,000×g for 20 minutes. The supernatant was brought to 80% saturation in ammonium sulfate and kept under constant stirring for 1 h. After centrifugation at 27,000×g for 30 minutes, the pellet was suspended in buffer A (20 mM Tris-HCl, 1 mM DTT, pH 7.4) and 5 ml aliquots were placed on a Sephadex G 75 column (3×50 cm, GE Healthcare Life Sciences) equilibrated with buffer A. The proteins were eluted with the same buffer at a flow rate of 2 mL/min and all the fractions with high PPPase activity were pooled. The pool was loaded on a DEAE-Sephacel anion exchange column (1.4×24 cm, GE Healthcare Life Sciences) equilibrated with buffer A. The proteins were eluted with an exponential concave NaCl gradient from 0 to 0.4 M in buffer A. Fractions with highest PPPase activity were pooled and centrifuged in Amicon ultra-15 centrifugal filter (Ultracel-10 membrane) units (Merck Millipore SA/NV, Overijse, Belgium). The retained proteins were dissolved in buffer B containing 20 mM Tris-HCl and 50 mM NaCl. 25 mg of protein were loaded on a Mono Q column (GE Healthcare Life Sciences) coupled to a Fast Protein Liquid Chromatography apparatus (ÄKTApurifier, Amersham) and elution was made with buffer B using a linear NaCl gradient from 50 to 500 mM. The fraction with the highest PPPase activity was pooled and DTT was added to reach a concentration of 1 mM. The proteins were then separated by non-denaturing polyacrylamide gel electrophoresis. The band with PPPase activity was cut and analyzed by liquid chromatography- mass spectrometry as described above for E. coli PPPase activity.
Detection of PPPi using anion exchange column and electrochemical detection
Ion chromatography coupled with a conductivity detector was used to detect the tripolyphosphate anion. The chromatograph (Dionex ICS-2000) was equipped with an Ion Pac AS 16 2×250 mm column, an Ion Pac AG16 2×50 mm guard column and an ARS 300 suppressor. The KOH gradient necessary for ion separation was generated by an EluGen EGC-KOH cartridge. The gradient profile was the following: start at 20 mM KOH, linear ramp to 45 mM for 4 minutes, step at this level during one minute to fully resolve the tri-meta phosphate from pyrophosphate followed by a second linear ramp up to 70 mM KOH during 3 minutes to elute highly charged polyphosphate ions. The ions were detected by an integrated Dionex DS6 conductivity detector after eluent suppression. The injection volume was 25 µL and the flow rate was 0.36 mL/min.
Development of a method for PPPi determination by capillary electrophoresis (CE)
CE experiments were carried out on a HP3DCE system (Agilent Technologies, Waldbronn, Germany) equipped with an incorporated diode array detector and a temperature control system (15–60°C±0.1°C). A CE Chemstation (Hewlett-Packard, Palo Alto, CA, USA) was used for instrument control, data acquisition and data handling. Fused-silica capillaries were provided by ThermoSeparation Products (San Jose, CA, USA).
Electrophoretic separations were carried out with uncoated fused-silica capillaries having 50 µm internal diameter and 70 cm length (61.5 cm to the detector). At the beginning of each working day, the capillary was washed with 1 N NaOH, water and the background electrolyte (BGE) for 10 min. Before each injection, the capillary was washed successively with water, for 1 min, 1 N NaOH for 3 min, water for 1 min and then equilibrated with the BGE for 5 min. The applied voltage was 12 kV in the negative polarity mode and UV detection was set at 260 nm. Injections were made by applying a voltage of −10 kV for a period of 12 s and the capillary was thermostated at 30°C. The composition of the BGE was optimized according to sensitivity and selectivity: the PPPi peak (N: 233.000) was well separated from PPi and Pi. The BGE used for electrophoretic experiments was made up of 3 mM sodium molybdate in 200 mM malonate buffer adjusted to pH 3: acetonitrile (ACN) (80∶20; v/v). The determination of PPPi is based on the in-capillary formation of anionic polyoxomolybdate complex in the presence of ACN, as already described for phosphonate, Pi and PPi [40]. E. coli supernatants were injected into the CE system after fractionation on a G-25 column.
Preparation of samples for PPPi determination
E. coli (MG1655 strain) were grown overnight in 50 ml Luria Bertani medium and centrifuged (10 min at 8000×g). The pellet was suspended in 500 µL 20 % TCA and centrifuged for 5 minutes at 13,000×g. The TCA suspension was then extracted 10 times with 3 volumes of diethyl ether. The sample was injected onto a 3.9 mL G-25 column and fractions of 500 µl were collected in 10 mM Hepes pH 7. In the control samples, 83 µL of 72% TCA were added to 417 µL 10 mM PPPi and treated as above.
Supporting Information
Purification of tripolyphosphatase activity from E. coli soluble fraction.
(DOCX)
Purification of the GST-ygiF fusion protein. (Lane 1, molecular mass weight markers; Lane 2, bacterial supernatant; lane 3, purified protein after the GST column [2]).
(TIFF)
PPPase activity as a function of pH in the supernatants of MG1655 (WT) and JW3026-2 (YgiF KO) strains at 37°C (0.5 mM PPPi, 5 mM Mg2+).
(TIFF)
PPPase activity in the supernatants of MG1655 (WT) and DPPK-PPX (CF5802) and DygiF (JW3026-2) strains. The supernatants were incubated for 20 min at 50°C (5 mM PPPi, 5 mM Mg2+ in TAPS buffer at pH 9.5). (Means ± SD, n = 3).
(TIFF)
PPase (blue) and PPPase (red) activities as a function of growth in E. coli . Growth was measured by following the absorbance at 600 nm (black curve).
(TIFF)
Sequence alignment of Drosophila , pig and human prune. The sequence alignment was made using CLC Sequence Viewer 6 (CLC bio A/S, 8200 Aarhus, Denmark).
(TIFF)
Acknowledgments
Nucleic acid sequencing was made by the Genotranscriptomics Platform and protein identification by the LC-ESI/MS/MS by the Proteomics Platform, GIGA, University of Liège (http://www.giga.ulg.ac.be/).
Funding Statement
LB is a Research Director and BL is a Research Associate at the Funds for Scientific Research – FNRS (Belgium). DD is a Research Fellows of the Fonds pour la Formation à la Recherche dans l'Industrie et dans l'Agriculture (F.R.I.A.). This work was supported by grants of the “Fonds de la Recherche Fondamendale collective” (Grants and 2.4558.04 and 2.4508.10). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Delvaux D, Murty MR, Gabelica V, Lakaye B, Lunin VV, et al. (2011) A specific inorganic triphosphatase from Nitrosomonas europaea: structure and catalytic mechanism. J Biol Chem 286: 34023–34035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Iyer LM, Aravind L (2002) The catalytic domains of thiamine triphosphatase and CyaB-like adenylyl cyclase define a novel superfamily of domains that bind organic phosphates. BMC Genomics 3: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lakaye B, Makarchikov AF, Antunes AF, Zorzi W, Coumans B, et al. (2002) Molecular characterization of a specific thiamine triphosphatase widely expressed in mammalian tissues. J Biol Chem 277: 13771–13777. [DOI] [PubMed] [Google Scholar]
- 4. Sismeiro O, Trotot P, Biville F, Vivares C, Danchin A (1998) Aeromonas hydrophila adenylyl cyclase 2: a new class of adenylyl cyclases with thermophilic properties and sequence similarities to proteins from hyperthermophilic archaebacteria. J Bacteriol 180: 3339–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aravind L, Koonin EV (1998) A novel family of predicted phosphoesterases includes Drosophila prune protein and bacterial RecJ exonuclease. Trends Biochem Sci 23: 17–19. [DOI] [PubMed] [Google Scholar]
- 6. Tesmer JJ, Sunahara RK, Johnson RA, Gosselin G, Gilman AG, et al. (1999) Two-metal-ion catalysis in adenylyl cyclase. Science 285: 756–760. [DOI] [PubMed] [Google Scholar]
- 7. Gong C, Smith P, Shuman S (2006) Structure-function analysis of Plasmodium RNA triphosphatase and description of a triphosphate tunnel metalloenzyme superfamily that includes Cet1-like RNA triphosphatases and CYTH proteins. RNA 12: 1468–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gallagher DT, Kim SK, Robinson H, Reddy PT (2011) Active-site structure of class IV adenylyl cyclase and transphyletic mechanism. J Mol Biol 405: 787–803. [DOI] [PubMed] [Google Scholar]
- 9. Keppetipola N, Jain R, Shuman S (2007) Novel triphosphate phosphohydrolase activity of Clostridium thermocellum TTM, a member of the triphosphate tunnel metalloenzyme superfamily. J Biol Chem 282: 11941–11949. [DOI] [PubMed] [Google Scholar]
- 10. Jain R, Shuman S (2008) Polyphosphatase activity of CthTTM, a bacterial triphosphate tunnel metalloenzyme. J Biol Chem 283: 31047–31057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Josse J (1966) Constitutive inorganic pyrophosphatase of Escherichia coli. 1. Purification and catalytic properties. J Biol Chem 241: 1938–1947. [PubMed] [Google Scholar]
- 12. Zyryanov AB, Shestakov AS, Lahti R, Baykov AA (2002) Mechanism by which metal cofactors control substrate specificity in pyrophosphatase. Biochem J 367: 901–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kukko-Kalske E, Lintunen M, Inen MK, Lahti R, Heinonen J (1989) Intracellular PPi concentration is not directly dependent on amount of inorganic pyrophosphatase in Escherichia coli K-12 cells. J Bacteriol 171: 4498–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Avaeva S, Ignatov P, Kurilova S, Nazarova T, Rodina E, et al. (1996) Escherichia coli inorganic pyrophosphatase: site-directed mutagenesis of the metal binding sites. FEBS Lett 399: 99–102. [DOI] [PubMed] [Google Scholar]
- 15. Taniguchi Y, Choi PJ, Li GW, Chen H, Babu M, et al. (2010) Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 329: 533–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van Heeswijk WC, Rabenberg M, Westerhoff HV, Kahn D (1993) The genes of the glutamine synthetase adenylylation cascade are not regulated by nitrogen in Escherichia coli. Mol Microbiol 9: 443–457. [DOI] [PubMed] [Google Scholar]
- 17. Akiyama M, Crooke E, Kornberg A (1993) An exopolyphosphatase of Escherichia coli. The enzyme and its ppx gene in a polyphosphate operon. J Biol Chem 268: 633–639. [PubMed] [Google Scholar]
- 18. Tammenkoski M, Koivula K, Cusanelli E, Zollo M, Steegborn C, et al. (2008) Human metastasis regulator protein H-prune is a short-chain exopolyphosphatase. Biochemistry 47: 9707–9713. [DOI] [PubMed] [Google Scholar]
- 19. Carotenuto P, Marino N, Bello AM, D'Angelo A, Di Porzio U, et al. (2006) PRUNE and NM23-M1 expression in embryonic and adult mouse brain. J Bioenerg Biomembr 38: 233–246. [DOI] [PubMed] [Google Scholar]
- 20. Moreno B, Urbina JA, Oldfield E, Bailey BN, Rodrigues CO, et al. (2000) 31P NMR spectroscopy of Trypanosoma brucei, Trypanosoma cruzi, and Leishmania major. Evidence for high levels of condensed inorganic phosphates. J Biol Chem 275: 28356–28362. [DOI] [PubMed] [Google Scholar]
- 21. Cui H, Cai F, Xu Q (2000) Determination of tripolyphosphate in frozen cod and scallop adductor by ion chromatography. J Chromatogr A 884: 89–92. [DOI] [PubMed] [Google Scholar]
- 22. Terkeltaub RA (2001) Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol Cell Physiol 281: C1–C11. [DOI] [PubMed] [Google Scholar]
- 23. Baltscheffsky M, Nadanaciva S, Schultz A (1998) A pyrophosphate synthase gene: molecular cloning and sequencing of the cDNA encoding the inorganic pyrophosphate synthase from Rhodospirillum rubrum. Biochim Biophys Acta 1364: 301–306. [DOI] [PubMed] [Google Scholar]
- 24. Kumble KD, Kornberg A (1995) Inorganic polyphosphate in mammalian cells and tissues. J Biol Chem 270: 5818–5822. [DOI] [PubMed] [Google Scholar]
- 25. Ault-Riché D, Fraley CD, Tzeng CM, Kornberg A (1998) Novel assay reveals multiple pathways regulating stress-induced accumulations of inorganic polyphosphate in Escherichia coli. J Bacteriol 180: 1841–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pavlov E, Aschar-Sobbi R, Campanella M, Turner RJ, Gomez-Garcia MR, et al. (2010) Inorganic polyphosphate and energy metabolism in mammalian cells. J Biol Chem 285: 9420–9428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kornberg SR, Lehman IR, Bessman MJ, Simms ES, Kornberg A (1958) Enzymatic cleavage of deoxyguanosine triphosphate to deoxyguanosine and tripolyphosphate. J Biol Chem 233: 159–162. [PubMed] [Google Scholar]
- 28. Tsutsui K (1986) Tripolyphosphate is an alternative phosphodonor of the selective protein phosphorylation of liver microsomal membrane. J Biol Chem 261: 2645–2653. [PubMed] [Google Scholar]
- 29. Chang DM (1983) The binding of free calcium ions in aquous solution using chelating agents, phosphates and poly(acrylic acid). JAOCS 60: 618–622. [Google Scholar]
- 30. Linster CL, Noel G, Stroobant V, Vertommen D, Vincent MF, et al. (2011) Ethylmalonyl-CoA decarboxylase, a new enzyme involved in metabolite proofreading. J Biol Chem 286: 42992–43003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marbaix AY, Noel G, Detroux AM, Vertommen D, Van Schaftingen E, et al. (2011) Extremely Conserved ATP- or ADP-dependent Enzymatic System for Nicotinamide Nucleotide Repair. J Biol Chem 286: 41246–41252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lima CD, Wang LK, Shuman S (1999) Structure and mechanism of yeast RNA triphosphatase: an essential component of the mRNA capping apparatus. Cell 99: 533–543. [DOI] [PubMed] [Google Scholar]
- 33. Gong C, Martins A, Shuman S (2003) Structure-function analysis of Trypanosoma brucei RNA triphosphatase and evidence for a two-metal mechanism. J Biol Chem 278: 50843–50852. [DOI] [PubMed] [Google Scholar]
- 34. Song J, Bettendorff L, Tonelli M, Markley JL (2008) Structural basis for the catalytic mechanism of mammalian 25-kDa thiamine triphosphatase. J Biol Chem 283: 10939–10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bettendorff L, Nghiêm HO, Wins P, Lakaye B (2003) A general method for the chemical synthesis of gamma-32P-labeled or unlabeled nucleoside 5(′)-triphosphates and thiamine triphosphate. Anal Biochem 322: 190–197. [DOI] [PubMed] [Google Scholar]
- 36. Kuroda A, Murphy H, Cashel M, Kornberg A (1997) Guanosine tetra- and pentaphosphate promote accumulation of inorganic polyphosphate in Escherichia coli. J Biol Chem 272: 21240–21243. [DOI] [PubMed] [Google Scholar]
- 37. Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA (1979) An improved assay for nanomole amounts of inorganic phosphate. Anal Biochem 100: 95–97. [DOI] [PubMed] [Google Scholar]
- 38. Simonovic AD, Gaddameedhi S, Anderson MD (2004) In-gel precipitation of enzymatically released phosphate. Anal Biochem 334: 312–317. [DOI] [PubMed] [Google Scholar]
- 39. Makarchikov AF, Lakaye B, Gulyai IE, Czerniecki J, Coumans B, et al. (2003) Thiamine triphosphate and thiamine triphosphatase activities: from bacteria to mammals. Cell Mol Life Sci 60: 1477–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Himeno S, Inazuma N, Kitano E (2007) Simultaneous determination of phosphonate, phosphate, and diphosphate by capillary electrophoresis using in-capillary complexation with Mo(VI). J Sep Sci 30: 1077–1081. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Purification of tripolyphosphatase activity from E. coli soluble fraction.
(DOCX)
Purification of the GST-ygiF fusion protein. (Lane 1, molecular mass weight markers; Lane 2, bacterial supernatant; lane 3, purified protein after the GST column [2]).
(TIFF)
PPPase activity as a function of pH in the supernatants of MG1655 (WT) and JW3026-2 (YgiF KO) strains at 37°C (0.5 mM PPPi, 5 mM Mg2+).
(TIFF)
PPPase activity in the supernatants of MG1655 (WT) and DPPK-PPX (CF5802) and DygiF (JW3026-2) strains. The supernatants were incubated for 20 min at 50°C (5 mM PPPi, 5 mM Mg2+ in TAPS buffer at pH 9.5). (Means ± SD, n = 3).
(TIFF)
PPase (blue) and PPPase (red) activities as a function of growth in E. coli . Growth was measured by following the absorbance at 600 nm (black curve).
(TIFF)
Sequence alignment of Drosophila , pig and human prune. The sequence alignment was made using CLC Sequence Viewer 6 (CLC bio A/S, 8200 Aarhus, Denmark).
(TIFF)