

Abstract
Background
AphA is the master quorum-sensing (QS) regulator operating at low cell density in vibrios. Molecular regulation of target genes by AphA has been characterized in Vibrio harveyi and V. cholerae, but it is still poorly understood in V. parahaemolyticus.
Methodology/Principal Findings
The AphA proteins are extremely conserved in V. parahaemolyticus, Vibrio sp. Ex25, Vibrio sp. EJY3, V. harveyi, V. vulnificus, V. splendidus, V. anguillarum, V. cholerae, and V. furnissii. The above nine AphA orthologs appear to recognize conserved cis-acting DNA signals which can be represented by two consensus constructs, a 20 bp box sequence and a position frequency matrix. V. parahaemolyticus AphA represses the transcription of ahpA, qrr4, and opaR through direct AphA-target promoter DNA association, while it inhibits the qrr2-3 transcription in an indirect manner. Translation and transcription starts, core promoter elements for sigma factor recognition, Shine-Dalgarno sequences for ribosome recognition, and AphA-binding sites (containing corresponding AphA box-like sequences) were determined for the three direct AphA targets ahpA, qrr4, and opaR in V. parahaemolyticus.
Conclusions/Significance
AphA-mediated repression of ahpA, qrr2-4, and opaR was characterized in V. parahaemolyticus by using multiple biochemical and molecular experiments. The computational promoter analysis indicated the conserved mechanism of transcriptional regulation of QS regulator-encoding genes ahpA, qrr4, and opaR in vibrios.
Introduction
Quorum sensing (QS) is a process of bacterial gene regulation in response to the fluctuations in cell-population density, by synthesizing, releasing, and detecting signal molecules called autoinducers that are induced with the increasing of cell density [1]. QS is employed by bacteria to regulate a diverse array of physiological activities including symbiosis, virulence, competence, conjugation, antibiotic production, motility, and biofilm formation [1].
Vibrio harveyi, a pathogen of fishes and invertebrates, has been used as a model for QS studies. At low cell density (LCD) (Fig. 1), low concentrations of autoinducers lead to phosphorylation of LuxO (LuxO-P), and LuxO-P activates expression of the five qrr genes encoding sRNAs Qrr1-5 [2], [3]. The Qrr sRNAs promote the translation of AphA, while inhibit that of LuxR [4], [5], [6]. AphA further represses the luxR transcription, and moreover, overproduced AphA feeds back to inhibit the qrr transcription [7]. In addition, the over-production of Qrr sRNAs and LuxO-P triggers three additional feedback regulatory loops: i) LuxO-P represses the transcription of its own gene, ii) Qrr sRNAs inhibits the translation of luxO, and iii) Qrr sRNAs repress the translation of luxMN encoding the membrane-anchoring autoinducer-binding receptor protein LuxM and its cognate receptor LuxN [8], [9], [10]. The above feedbacks will contribute to control the Qrr levels within physiological states [10]. At high cell density (HCD) (Fig. 1), high concentrations of autoinducers reverse the phosphate flow in the circuit, leading to the dephosphorylation of LuxO. Dephosphorylated LuxO is inactive as a regulator, leading to the cessation of Qrr sRNA production. In the absence of Qrr sRNAs, AphA is not produced, but the translation of LuxR occurs [4], [5]. LuxR further represses the transcription of aphA, and moreover, overproduced LuxR feeds back to repress its own transcription [11]. The above regulatory circuits results in the reciprocal gradients of production of AphA and LuxR: maximal AphA and minimal LuxR production occur at LCD, while minimal AphA and maximal LuxR production at HCD [7]. Thus, AphA and LuxR represent the master QS regulators operating at LCD and HCD, respectively.
Figure 1. Model for signal transduction of QS systems in V. harveyi/V. parahaemolyticus.

The details for signal transduction during QS have been described in text. At LCD, redundant Qrr sRNAs promote AphA translation and meanwhile inhibit LuxR translation. AphA further represses the transcription of luxR/opaR, qrr2-3, and its own gene. At HCD, the cessation of Qrr sRNA production leads to no production of AphA, and LuxR/OpaR translation occurs. LuxR/OpaR in turns represses the aphA transcription, and also feeds back to inhibit its own expression. Thus, AphA acts as a master regulator of QS behaviors at LCD, and in contrast, LuxR/OpaR is the major one operating at HCD; reciprocal gradients of AphA and LuxR/OpaR are established for controlling gene expression during transition between LCD and HCD. LuxR/OpaR is also able to activate the transcription of qrr2-4 genes, leading to rapid down-regulation of luxR/opaR [47]. It should be noted that this LuxR-qrr feedback dramatically accelerates the transition HCD to LCD, but it has no effect on QS behaviors at steady-state LCD or HCD [47].The dotted lines indicated the inhibited expression of relevant regulators or the cease of relevant regulatory cascades. The blue and red lines show the regulatory cascades that were experimentally validated in V. parahaemolyticus in our previous [15] and present works, respectively.
V. parahaemolyticus is a member of the Harveyi clade that is composed of eight characterized closely related Vibrio species including V. harveyi [12]. In recent years, V. parahaemolyticus has been recognized as the leading cause of infectious gastrointestinal illness via the fecal-oral route especially in coastal countries and regions [13]. Most infections are due to the eating of raw or undercooked seafood. Less commonly, infections in the skin occur when an open wound is exposed to warm seawater containing this pathogen. All the V. harveyi QS components can be annotated to be intact and highly conserved in the genome of V. parahaemolyticus [14]. Thus, the QS signal transduction cascades should be conserved in V. harveyi and V. parahaemolyticus [15] (see also Fig. 1).
The characterized orthologs of V. harveyi LuxR often have distinct names in different vibrios, and thus, the LuxR orthologs are referred to as HMRs (HCD master regulators) herein. The four HMRs, V. parahaemolyticus OpaR, V. harveyi LuxR, V. vulnificus SmcR, and V. alginolyticus ValR, are extremely conserved (>92% identity in amino acid sequences between each other), and they recognize a cis-acting consensus that can be annotated as a 20 bp invert-repeat box sequence TATTGATAAA-TTTATCAATA [15]. The OpaR box-like sequences can be found within the promoter-proximal DNA regions of opaR, qrr2-4 and aphA in V. parahaemolyticus, and the direct transcriptional regulation of these target genes by OpaR was further experimentally validated in V. parahaemolyticus [15]. As expected, the above characterized regulatory circuits are shared by V. harveyi and V. parahaemolyticus (Fig. 1).
To the best of our knowledge, there is no report on AphA in V. parahaemolyticus. The aphA (VP2762) gene [14] of V. parahaemolyticus is composed of an open reading frame containing 540 nucleotides with a G+C content of 44.4%, and it encodes a deduced protein of 180 amino acids (a.a.) with a calculated molecular mass of 20506.36 Da and with an isolectric point of 5.501. In the present work, two consensus constructs were built to represent conserved cis-acting signals recognized by AphA orthologs from nine Vibrio species including V. parahaemolyticus, and then AphA-mediated transcriptional regulation of QS regulator-encoding genes ahpA, qrr2-4, and opaR were characterized in V. parahaemolyticus.
Materials and Methods
Bacterial Strains
The wild-type V. parahaemolyticus strain RIMD 2210633 (WT) is a pandemic O3:K6 strain isolated from a patient with traveler’s diarrhea in Japan in 1996 [14]. The base pairs (bps) 2 to 516 of coding region (540 bp in total length) of aphA was deleted from WT to generate a nonpolar mutant strain ΔaphA (see also Fig. S1), using the suicide plasmid pDS132 [16] by homologous recombination as described previously [17], [18]. Briefly, the two DNA fragments (361 and 420 bp in length, respectively) flanking the 515 bp deletion region were amplified by PCR, purified, and used as the templates to create a 791 bp deletion construct that was subsequently inserted between PstI and SphI sites of pDS132. All the primers used in the present work were listed in Table 1. Upon being verified by DNA sequencing, the recombinant vector (containing deletion construct, and sacB gene conferring sensitivity to sucrose) was introduced into Escherichia coli S17-1(pir), and then transferred into WT by conjugation. The mutant strain was selected using resistance to 10% sucrose and sensitivity to 5 µg/ml chloramphenicol, and further verified by PCR.
Table 1. Oligonucleotide primers used in this study.
| Target | Primers (forward/reverse, 5'-3') |
| Construction of mutant | |
| aphA | GTGACTGCAGCGCAGCAAATAACCAGAC/CCAATCACTTCAAGTTCTGTTGTCTTCAATCCAAATGGTC |
| aphA | GACCATTTGGATTGAAGACAACAGAACTTGAAGTGATTGG/GTGAGCATGCGTTTTCGTGACCGCTGTG |
| aphA | GTGACTGCAGCGCAGCAAATAACCAGAC/GTGAGCATGCGTTTTCGTGACCGCTGTG |
| Complementation of mutant | |
| aphA | AGCGGGATCCATGTCATTACCACACGTAATC/AGCGAAGCTTTTAACCAATCACTTCAAGTTC |
| Protein expression | |
| aphA | AGCGGGATCCATGTCATTACCACACGTAATC/AGCGAAGCTTTTAACCAATCACTTCAAGTTC |
| EMSA | |
| aphA | AACTTCCAACCACATAATTGCG/GGCTGGAGCAGGTATGATTG |
| opaR | TGTGGGTTGAGGTAGGTCG/GCCTAGTTCTAGGTCTCTTTGC |
| qrr2 | AGTGGTTGCTTATGAATC/GGTCGAGAAGTATTATGC |
| qrr3 | GGATAAGTTCAAATTGGATC/GTGGTTTCTGTGACATAC |
| qrr4 | AACCGTGAAATCCATTTAC/CGACGCATTATTAACCAG |
| DNase I footprinting | |
| aphA | AACTTCCAACCACATAATTGCG/GGCTGGAGCAGGTATGATTG |
| opaR | AGTGGGTTGAAAGTCACATCC/GCCTAGTTCTAGGTCTCTTTGC |
| qrr4 | AACCGTGAAATCCATTTAC/CGACGCATTATTAACCAG |
| Primer extension | |
| aphA | /GCTCTTACTGGCGCTTGAG |
| opaR | /ATCCATTTTCCTTGCCATTTG |
| qrr2 | /TTATTGTGAACAATCTATAT |
| qrr3 | /AATCAAGTTCACTAACAAC |
| qrr4 | /ATATACTTGTGAACAATGTG |
| LacZ fusion | |
| aphA | GCGCGTCGACCATTCGTAATACAAAAGG/GCGCGGTACCTTCCAGAAGTAACCGATGCTAG |
| opaR | GCGCGTCGACTCCATCGTGTTGCCGTAGC/GCGCGGTACCCAATATCTGCGTGACCACCAC |
| qrr2 | GCGCGTCGACAAAGTATGAAATAGTGTCGTAG/GCGCGAATTCTAGCCAACCGCAATAATC |
| qrr3 | GCGCGTCGACGTGTTGATACCCATTATTC/GCGCGAATTCTGCGATTGGCTTATATAC |
| qrr4 | GGGGTCGAC AACCGTGAAATCCATTTAC/GGGGAATTCATATACTTGTGAACAATGTG |
A PCR-generated DNA fragment containing the aphA coding region together with its promoter-proximal DNA region (499 bp upstream of the coding sequence) and transcriptional terminator (285 bp downstream) were cloned between SalI and SphI sites of the pBRMob vector which is the ligation product of a 3219 bp fragment (containing the RP4 mob DNA region for plasmid mobilization) from pDS132 digested with HindIII, and the HindIII-digested plasmid pBR328 (harboring a chloramphenicol resistance gene) [19]. Upon being verified by DNA sequencing, the recombinant plasmid was introduced into ΔaphA, yielding the complemented mutant strain C-aphA.
Bacterial Growth
For bacterial growth and maintenance, bacteria were cultivated in Luria-Bertani (LB) broth or on LB agar both with addition of 2% NaCl at 37°C. For longtime storage, bacteria were stored in Difco™ Marine (MR) broth 2216 (BD Bioscience) with addition of 30% glycerol at −85°C. Antibiotics were used at final concentrations of 100 µg/ml gentamicin, and 20 µg/ml chloramphenicol.
For the following biochemical assays, we employed a two-round design to prepare the liquid bacterial seed: firstly, the glyceric stock of bacterial cells was inoculated into 3 ml of MR broth for growing at 30°C with shaking at 200 rpm for 12 to 14 h to enter the stationary growth phase; secondly, the resulting cell culture was 50-fold diluted into 15 ml of fresh MR broth, and allowed to grow under the above conditions to reach an OD600 value of about 1.0 to 1.2. The seed culture was then 1,000-fold diluted into 30 ml of fresh MR broth for further growth under the above conditions. To determine the bacterial growth curves, the OD600 values were monitored for each culture with an 1 h interval. For the primer extension or LacZ fusion experiments, bacterial cells were harvested at an OD600 value of 0.1 to 0.15 to simulate LCD conditions.
LacZ Fusion and β-galactosidase Assay
The 300 to 800 bp promoter-proximal DNA region of each indicated gene was amplified by PCR with ExTaq™ DNA polymerase (Takara) using RIMD 2210633 genome DNA as the template. PCR fragments were then directionally cloned between SalI and EcoRI (or KpnI) sites of low-copy-number plasmid pHRP309 that harbors a gentamicin resistance gene and a promoterless lacZ reporter gene [20]. Correct cloning was verified by DNA sequencing. An empty pHRP309 plasmid was also introduced into each strain tested as the negative control. The V. parahaemolyticus strains transformed with recombinant plasmids or empty pHRP309 were grown as above to measure the β-galactosidase activity in cellular extracts using a β-Galactosidase Enzyme Assay System (Promega) [21].
RNA Isolation and Primer Extension Assay
Total bacterial RNAs were extracted using the TRIzol Reagent (Invitrogen) [15], [21]. RNA quality was monitored by agarose gel electrophoresis, and RNA quantity was determined by spectrophotometry.
For the primer extension assay [15], [21], an oligonucleotide primer complementary to a portion of RNA transcript of each indicated gene was employed to synthesize cDNAs from the RNA templates. Ten to 100 µg of total RNA from each strain was annealed with 1 pmol of [γ-32P] end-labeled reverse primer using a Primer Extension System (Promega) according to the manufacturer’s instructions. The same labeled primer was also used for sequencing with a fmol® DNA Cycle Sequencing System (Promega). The primer extension products and sequencing materials were concentrated and analyzed in a 6% polyacrylamide/8 M urea gel. The result was detected by autoradiography (Kodak film).
Preparation of Purified AphA Protein
The preparation of purified AphA protein was done as described previously [7] with the omission of thrombin cleavage procedures. The entire coding region of aphA of strain RIMD 2210633 was cloned between BamHI and HindIII sites of plasmid pET28a (Novagen). The recombinant plasmid encoding 6× His-tagged AphA protein (His-AphA) was transformed into E. coli BL21λDE3 cells, and grown in LB broth at 37°C with shaking at 200 rpm for 4 to 5 h. The resulting culture was diluted 1/100 into 200 to 300 ml of fresh LB broth, and grown under the above conditions to an OD600 of about 0.5. The culture was shifted to 18°C for 1 h, and then induced with 1 mM IPTG for 16 to 18 h with shaking at 100 rpm. Cells were collected by centrifugation and frozen at −60°C. The pellet was resuspended in 10 ml of 50 mM sodium phosphate buffer, pH 7.4, 500 mM NaCl, and 5 mM imidazole. Cells were disrupted using a cell cracker, and the insoluble material was pelleted by centrifugation at 15,000 rpm. The clarified supernatant was applied to a 3 ml Ni-NTA Agarose Column (Qiagen), and the overproduced protein was purified under native conditions. Fractions from a homogenous peak were pooled, and the final preparation was dialyzed against 10 mM Tris HCl, pH 7.5, 10 mM NaCl, 1 mM EDTA, 0.1 mM DTT, and 20% glycerol. The purified protein was stored at −60°C, and the protein purity was verified by SDS-PAGE.
Gel Mobility Shift Assay (EMSA)
The 300 to 600 bp promoter-proximal DNA region of each indicated gene was amplified by PCR. For EMSA [15], [21], the 5′ ends of DNA were labeled using [γ-32P] ATP and T4 polynucleotide kinase. DNA binding was performed in a 10 µl reaction volume containing binding buffer [1 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT, 50 mM NaCl, 10 mM Tris-HCl (pH 7.5) and 0.05 mg/ml poly-(dI-dC)], labeled DNA (1000 to 2000 c.p.m/µl), and increasing amounts of His-AphA. Three controls were included in each EMSA experiment: 1) cold probe as specific DNA competitor (the same promoter-proximal DNA region unlabeled), 2) negative probe as non-specific DNA competitor (the unlabeled coding region of the 16S rRNA gene), and 3) non-specific protein competitor [rabbit anti-F1-protein polyclonal antibodies]. The F1 protein is the protective antigen from Yersinia pestis [22]. After incubation at room temperature for 30 min, the products were loaded onto a native 4% (w/v) polyacrylamide gel, and electrophoresed in 0.5× TBE buffer for about 50 min at 220 V. Radioactive species were detected by autoradiography after exposure to Kodak film at −70°C.
DNase I Footprinting
For DNase I footprinting [15], [21], the 250 to 600 bp promoter-proximal DNA regions with a single 32P-labeled end were PCR amplified with either sense or antisense primer being end-labeled. The PCR products were purified using the QiaQuick columns (Qiagen). Increasing amounts of His-AphA were incubated with the purified, labeled DNA fragment (2 to 5 pmol) for 30 min at room temperature, in a final 10 µl reaction volume containing the binding buffer used in EMSA. Before DNA digestion, 10 µl of Ca2+/Mg2+ solution (5 mM CaCl2 and 10 mM MgCl2) was added, followed by incubation for 1 min at room temperature. The optimized RQ1 RNase-Free DNase I (Promega) was then added to the reaction mixture, and the mixture was incubated at room temperature for 40 to 90 s. The reaction was quenched by adding 9 µl of stop solution (200 mM NaCl, 30 mM EDTA, and 1% SDS), followed by incubation for 1 min at room temperature. The partially digested DNA samples were extracted with phenol/chloroform, precipitated with ethanol, and analyzed in 6% polyacrylamide/8 M urea gel. Protected regions were identified by comparison with the sequence ladders. For sequencing, we used the fmol® DNA Cycle Sequencing System (Promega). The templates for sequencing were the same as the DNA fragments for DNase I footprinting assays. Radioactive species were detected as above.
Computational Promoter Analysis
The 300 bp promoter region upstream of start codon of each indicated gene were retrieved with the ‘retrieve-seq’ program [23]. Known or predicted binding sites of AphA were collected and aligned to generate the position frequency matrix (PFM) by using the ‘matrices-consensus’ tool [23].The sequence logo representation of the above binding sites was generated by the WebLogo tool [24]. The ‘matrices-paster’ tool [23] was used to match the PFM within the promoter-proximal DNA regions tested.
Experimental Replicates and Statistical Methods
For the growth curve and LacZ fusion assays, experiments were performed with at least three independent bacterial cultures, and values were expressed as mean ± standard deviation (SD). Statistical testing of difference was made by the Student's paired t test, and a P value of <0.01 was taken as significant. For the primer extension, EMSA, and footprinting assays, representative data from at least two independent biological replicates were shown.
Results
Cis-acting Consensus Recognized by AphA
The AphA orthologs from nine Vibrio species (V. parahaemolyticus, Vibrio sp. Ex25, Vibrio sp. EJY3, V. harveyi, V. vulnificus, V. splendidus, V. anguillarum, V. cholerae, and V. furnissii) with determined whole genome sequences gave high identity (≥84%) among each other in the a.a. sequences. A phylogenetic tree (Fig. 2) was constructed from the aligned a.a. sequences of the above nine orthologous AphA proteins, with an additional AphA homologue from Photobacterium profundum SS9 [25] as the outgroup (this protein has about 66 to 70% identity to the above nine AphA orthologs). Three clades I, II, and III were arbitrarily identified for the nine Vibrio species on the basis of phylogenetic tree.
Figure 2. Phylogenetic tree of AphA orthologs.

The protein sequences were derived from V. parahaemolyticus RIMD 2210633 [14], Vibrio sp. Ex25 (Accession numbers CP001805.1, and CP001806.1), Vibrio sp. EJY3 [48], V. harveyi ATCC BAA-1116 [49], V. vulnificus YJ016 [50], V. splendidus LGP32 [51], V. anguillarum 775 [52], V. cholerae N16961 [53], V. furnissii NCTC 11218 [54], and P. profundum SS9 [25]. The a.a. sequences were aligned by the CLUSTALW [55] web server at http://align.genome.jp/. The aligned sequences were then used to construct an unrooted neighbor-joining tree using MEGA version 5.0 [56] with a bootstrap iteration number of 1000. Shown on branch points of phylogenic tree were bootstrap values (%).
Since the nine AphA orthologs are highly conserved, when they act as the DNA-binding regulatory proteins, they will recognize conserved cis-acting DNA signals in the relevant vibrios. Known or predicted binding sites of the nine AphA orthologs were collected (Table 2), and then aligned to generate an AphA consensus that manifested as a PFM (in which each row and column represents a position and a nucleotide, respectively) and as a 20 bp box sequence ATATGCAN6 TGCATAT that contained imperfect inverted repeats of ATATGCA with a 6-nt centered spacer (Fig. 3).
Table 2. Known or predicted direct targets of AphA.
| Gene name | Gene ID | AphA box-like sequence | Position& | Score | Reference | Regulation by AphA |
| V. parahaemolyticus RIMD 2210633 | ||||||
| aphA | VP2762 | GTATTCCACTTCATGCTTAT | D-238…-219 | 16.37 | This study | Negative |
| opaR | VP2516 | ATATGCACCATTACACTCAT | D-98…-79 | 18.32 | This study | Negative |
| qrr4 | VPA0199-0200 intergenic | ATATGCCACTTGTTACATTG | R-146…-127 | 10.96 | This study | Negative |
| Vibrio sp. Ex25 | ||||||
| aphA | VEA_002309 | CTATTCCACTTCATGCTTAT | D-240…-221 | 15.71 | Predicted | Negative |
| luxR | VEA_002553 | ATATGCACCATTACACTCAT | D-161…-142 | 18.32 | Predicted | Negative |
| qrr4 | VEA000800-000799 intergenic | ATATGCCACTTCATGCACTG | R-146…-127 | 14.05 | Predicted | Negative |
| Vibrio sp. EJY3 | ||||||
| aphA | VEJY3_14120 | GTATTCCACTTCATGCTTAT | D-242…-223 | 16.37 | Predicted | Negative |
| luxR | VEJY3_12990 | ATATGCACCATTACACTCAT | D-98…-79 | 18.32 | Predicted | Negative |
| qrr4 | VEJY316416-16421 intergenic | ATATGCTACCAACCGCATTG | R-145…-126 | 7.64 | Predicted | Negative |
| V. harveyi ATCC BAA-1116 | ||||||
| aphA | VIBHAR_00046 | CTATTCCACTTCATGCTTAT | D-240…-221 | 15.71 | [7] | Negative |
| luxR | VIBHAR_03459 | ATATGCACCATTACACTCAT | D-101…-82 | 18.32 | [7] | Negative |
| qrr4 | VIBHAR_06697 | ATATGCCACCATGCGCATTG | R-146…-127 | 11.01 | [7] | Negative |
| V. vulnificus YJ016 | ||||||
| aphA | VV3005 | CTATTCCACTTTATGCTTAT | D-238…-219 | 16.45 | Predicted | Negative |
| smcR | VV2770 | ATATGCACCATTACACTCAT | D-110…-91 | 18.32 | Predicted | Negative |
| qrr4 | VVA0457-0458 intergenic | ATACACACAAATTTGCATAT | R-97…-78 | 8.65 | Predicted | Negative |
| V. splendidus LGP32 | ||||||
| aphA | VS_2891 | GTATTCTACTTTATGCTTAT | D-237…-218 | 15.94 | Predicted | Negative |
| luxR | VS_2539 | ATATGCACCATTACACTCAT | D-79…-60 | 18.32 | Predicted | Negative |
| V. anguillarum 775 | ||||||
| aphA | VAA_02615 | GTATTCTACTTTATGCTTAT | D-253…-234 | 15.94 | Predicted | Negative |
| vanT | VAA_00743 | ATATGCAGCAGTACACTCAT | D-99…-80 | 13.81 | Predicted | Negative |
| V. cholerae N16961 | ||||||
| aphA | VC2647 | GTATTCCACTTTATGCTTAT | D-245…-226 | 17.11 | [30] | Negative |
| hapR | VC0583 | ATATGCACCATTACACTCAT | D-101…-82 | 18.32 | Predicted | Negative |
| pva | VCA0877 | ATATGCAACAAATTACACAT | D-178…-159 | 12.75 | [44] | Negative |
| alsR | VC1588 | ATATACACACAGATTCATAT | R-93…-74 | 8.6 | [29] | Negative |
| alsD | VC1589 | ATATACACACAGATTCATAT | D-32…-13 | 8.6 | [29] | Negative |
| vpsT | VCA0952 | ATACGCAAAAAGACTCTTAT | R-242…-223 | 10.7 | [28] | Positive |
| tcpP | VC0826 | TTATGCAATTAAGTTCTCAT | D-110…-91 | 6.71 | [27] | |
| V. furnissii NCTC 11218 | ||||||
| aphA | vfu_A00617 | GTATTCTACTTTATGCTTAT | D-246…-227 | 15.94 | Predicted | Negative |
| luxR | vfu_A00883 | ATGTACGCAATTACACTCAT | D-104…-85 | 9.66 | Predicted | Negative |
&, ‘D’ indicates the direct sequence, and the minus numbers denote nucleotide positions upstream of genes.
Figure 3. Cis-acting consensus constructs.

(a) The sequence logo representation of AphA sites (Table 2) was generated by WebLogo [24]. The 20-bp box sequence ATATGCAN6 TGCATAT contained imperfect inverted repeats of ATATGCA with a 6-nt centered spacer. (b) A position frequency matrix describes the alignment of AphA sites, and denotes the frequency of each nucleotide at each position.
The presence of AphA box-like sequences within the promoter-proximal DNA regions of aphA, qrr4, and opaR in V. parahaemolyticus (Table 2) indicated that these QS regulators-encoding genes might be the direct AphA targets in V. parahaemolyticus, which were further validated by the following gene regulation experiments.
Growth of WT and ΔahpA
The growth curves of WT and ΔaphA grown at 30°C in MR broth were determined (Fig. 4). ΔaphA grew slightly slower than WT from time of inoculation to mid-exponential growth phase; however, from mid-exponential phase to enter stationary phase, the two strains showed almost indistinguishable growth rates. These results indicated that the aphA mutation had little effect on bacterial in vitro growth when MR broth was used for cultivation.
Figure 4. Bacterial growth curves.
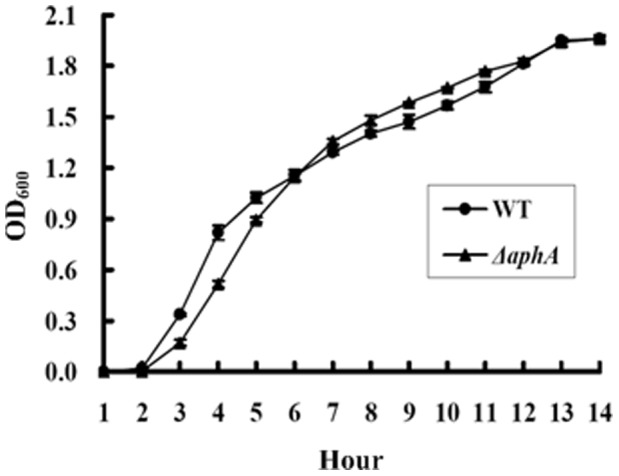
A two-round design to prepare the bacterial seed culture was employed: firstly, the glyceric stock of bacterial cells was inoculated into 3 ml of MR broth for growing at 30°C with shaking at 200 rpm for 12 to 14 h to enter the stationary growth phase; secondly, the resulting cell culture was 50-fold diluted into 15 ml of fresh MR broth, and allowed to grow under the above conditions to reach an OD600 value of about 1.0 to 1.2. The seed culture was then 1000-fold diluted into 30 ml of fresh MR broth for the further growth under the above conditions, and the OD600 values were monitored for each culture with an 1 h interval.
Cell Density-dependent Expression of aphA
The mRNA levels of aphA were measured in WT grown at various OD600 values (i.e. at different cell densities) by the primer extension assay (Fig. 5). The primer extension experiments detected a single transcription start site located at 200 bp upstream of aphA. Therefore, a single promoter was transcribed for aphA under the growth conditions tested. The aphA mRNA levels decreased dramatically with the increasing of cell density, and almost no aphA mRNA could be detected at HCD (OD600≥1.16), which supported the model (as described Fig. 1) of cell density-dependent regulation of aphA.
Figure 5. Transcriptional pattern of aphA during growth.
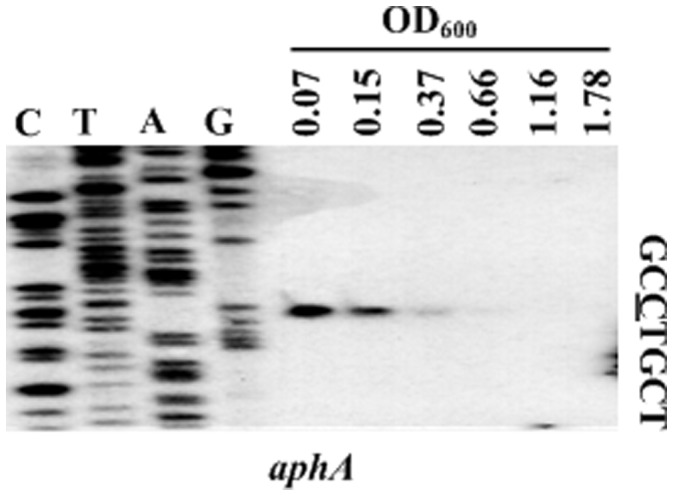
The bacterial cells were harvested at various OD600 values during growth (according to the bacterial growth curves in Fig. 4) for total RNA isolation. An oligonucleotide primer was designed to be complementary to the RNA transcript of aphA. The primer extension products were analyzed with 8 M urea-6% acrylamide sequencing gel. Lanes C, T, A, and G represented Sanger sequencing reactions. The transcription start site of aphA was underlined in the DNA sequence.
For the following gene regulation experiments, bacteria cells were harvested at an OD600 value of 0.1 to 0.15 to simulate LCD conditions at which aphA was abundantly transcribed.
Negative Auto-regulation of AphA
An aphA::lacZ fusion vector, containing a 531 bp promoter-proximal region of aphA and the promoterless lacZ, was transformed into WT or ΔaphA to compare the aphA promoter activities in these two strains (Fig. 6a). The LacZ fusion experiments disclosed that the aphA promoter activity significantly enhanced in ΔaphA relative to WT. The primer extension experiments (Fig. 6b) further disclosed that the mRNA level of aphA enhanced in ΔaphA relative to WT. A 220 bp promoter-proximal DNA region of aphA was subjected to EMSA with purified His-AphA protein (Fig. 6c). The results showed that His-AphA was able to bind to the target DNA fragment in a dose-dependent manner in vitro. As further determined by DNase I footprinting (Fig. 6d), His-AphA protected a single region from 242 to 210 bp upstream of aphA against DNase I digestion in a dose-dependent manner. This footprint contained a predicted AphA box-like sequence, and was considered as the AphA-binding site for aphA. Taken together, the AphA regulator was able to recognize the promoter of its own gene to directly repress its activity in V. parahaemolyticus.
Figure 6. Repression of its own gene by AphA. a) LacZ fusion.

The promoter-proximal DNA region of aphA was cloned into the lacZ transcriptional fusion vector pHRP309, and then transformed into WT or ΔaphA to determine the β-galactosidase activity in cellular extracts. Shown was the aphA promoter activity (Miller units) in ΔaphA or WT. b) Primer extension. An oligonucleotide primer was designed to be complementary to the RNA transcript of aphA. The primer extension products were analyzed with 8 M urea-6% acrylamide sequencing gel. Lanes C, T, A, and G represented Sanger sequencing reactions. The transcription start site of aphA was underlined in the DNA sequence. c) EMSA. The radioactively labeled DNA fragment from the 380th to 161st bp upstream of aphA was incubated with increasing amounts (lanes 1 to 4∶0, 15, 20, and 25 pmol, respectively) of purified His-AphA protein, and then subjected to 4% (w/v) polyacrylamide gel electrophoresis; the band of free DNA become weak with increasing amounts of His-AphA protein, and a retarded DNA band with decreased mobility turned up, which presumably represented the DNA-AphA complex. For lane 5, 2 pmol of cold probe, 25 pmol of His-AphA, and labeled target DNA fragment were added; the retarded DNA band disappeared due to the action of cold probe as specific DNA competitor. For lane 6, 2 pmol of negative probe, 25 pmol of His-AphA, and labeled target DNA fragment were added; the retarded DNA band occurred since the negative probe had no effect on the DNA-AphA complex. For lane 7, 20 pmol of non-specific protein competitor, and labeled target DNA fragment were added; there was no retarded DNA band observed. d) DNase I footprinting. Labeled coding or non-coding DNA probes were incubated with increasing amounts of purified His-AphA (Lanes 1, 2, 3, and 4 containing 0, 15, 20, and 25 pmol, respectively), and subjected to DNase I footprinting assay. Lanes G, A, T, and C represented Sanger sequencing reactions. The footprint regions were indicated with vertical bars. The negative numbers indicated nucleotide positions upstream of aphA.
Repression of opaR by AphA
An opaR::lacZ fusion vector, containing a 666 bp promoter-proximal region of opaR and the promoterless lacZ, was transformed into both WT and ΔaphA to compare the opaR promoter activities in these two strains (Fig. 7a). The LacZ fusion experiments disclosed that the promoter activity of opaR significantly enhanced in ΔaphA relative to WT. The primer extension experiments (Fig. 7b) detected a single transcription start site located at 74 bp upstream of opaR, and further disclosed that the mRNA level of opaR enhanced in ΔaphA relative to WT. A 334 bp promoter-proximal DNA region of opaR was subjected to EMSA with purified His-AphA protein (Fig. 7c). The results showed that His-AphA was able to bind to the DNA fragment in a dose-dependent manner in vitro. As further determined by DNase I footprinting (Fig. 7d), His-AphA protected a single region from 110 to 76 bp upstream of opaR against DNase I digestion in a dose-dependent manner. This footprint contained a predicted AphA box-like sequence, and was considered as the AphA-binding site for opaR. Taken together, AphA was able to recognize the promoter of opaR to directly repress its activity in V. parahaemolyticus.
Figure 7. Repression of qrr2-4 by OpaR.

For LacZ fusion (a1, b1, and c1), the promoter-proximal fragment of each of qrr2-4 was cloned into pHRP309, and then transformed into WT or ΔaphA to determine the β-galactosidase activity (Miller units) in cellular extracts. For primer extension (a2, b2, and c2), an oligonucleotide primer was designed to be complementary to the RNA transcript of each of qrr2-4. For EMSA (a3, b3, and c3) and DNase I footprinting (c4), the promoter-proximal fragment of each of qrr2-4 were radioactively labeled, and then incubated with increasing amounts of purified His-AphA protein. The experiments were done as described in Fig 6. The transcription start sites of qrr2-4 were underlined in the DNA sequence. Lanes G, A, T, and C represented Sanger sequencing reactions. The footprint regions were indicated with vertical bars. The negative or positive numbers indicated nucleotide positions upstream or downstream of relevant qrr gene, respectively.
Repression of qrr2-4 by AphA
The LacZ fusion vector for each of qrr2-4 was transformed into WT or ΔaphA, respectively, to compare the promoter activities of each qrr gene in the two strains (Fig. 8a1, 8b1, and 8c1). The results showed that the promoter activity of each of qrr2-4 significantly enhanced in ΔaphA relative to WT. The primer extension experiments were then conducted to compare the yields of primer extension product of each of qrr2-4 in WT and ΔaphA (Fig. 8a2, 8b2, and 8c2, respectively). The primer extension assay defined the transcription start sites for all of qrr2-4, and this assay also indicated that the mRNA level of each of qrr2-4 enhanced in ΔaphA relative to WT. Each of the promoter-proximal DNA regions of qrr2-4 was subjected to EMSA with the purified His-AphA protein (Fig. 8a3, 8b3, and 8c3, respectively). The results showed that His-AphA was able to bind to the DNA fragment of qrr4 rather than qrr2-3. As further determined by DNase I footprinting (Fig. 8c4), His-AphA protected a single region from 164 to 121 bp upstream of qrr4 against DNase I digestion in a dose-dependent manner. This footprint contained a predicted AphA box-like sequence, and was considered as the AphA-binding site for qrr4. Taken together, AphA was able to recognize the promoters of qrr4 to directly repress its activity in V. parahaemolyticus; in contrast, the qrr2-3 genes appeared to be repressed by AphA at the transcriptional level in an indirect manner.
Figure 8. Repression of opaR by AphA.

For LacZ fusion (a), the promoter-proximal DNA fragment of opaR was cloned into pHRP309, and then transformed into WT or ΔaphA to determine the β-galactosidase activity (Miller units) in cellular extracts. For primer extension (b), an oligonucleotide primer was designed to be complementary to the RNA transcript of opaR. For EMSA (c) and DNase I footprinting (d), the promoter-proximal DNA fragment of opaR was incubated with increasing amounts of purified His-AphA protein. The experiments were done as described in Fig 6. The transcription start site of opaR was underlined in the DNA sequence. Lanes G, A, T, and C represented Sanger sequencing reactions. The footprint regions were indicated with vertical bars. The negative or positive numbers indicated nucleotide positions upstream or downstream of opaR, respectively.
Discussion
Master QS Regulators at LCD and HCD
In V. harveyi and V. cholerae (the causative agent of the frequently fatal epidemic diarrheal disease cholera), AphA acts as a LCD master regulator of QS behaviors, and in contrast, LuxR/HapR is the major one operating at HCD [7]. The present work also confirmed the maximum aphA transcription at LCD and the minimum one at HCD. It seems that the reciprocal, cell density-dependent expression of AhpA and HMR is a conserve mechanism used by vibrios during the QS signal transduction.
Control of biofilm formation and virulence by HMRs and AphA has been characterized in multiple vibrios. AphA is an activator of virulence and biofilm formation in V. cholerae [26], [27], [28], [29] and V. parahaemolyticus (data unpublished). The four HMRs, HapR, OpaR, LuxR, and SmcR, are the repressors of virulence in V. cholerae [30], [31], [32], [33], V. parahaemolyticus [34], [35], V. harveyi [36], and V. vulnificus [37], respectively. HapR and OpaR also repress biofilm formation in V. cholerae [38], [39], [40] and in the pandemic O3:K6 V. parahaemolyticus (data unpublished), respectively. It should be noted that the in vivo biofilm formation has been shown to contribute to the infectivity of V. cholerae upon oral ingestion [41], [42]. Taken together, the reciprocal, cell density-dependent expression of AphA and HMR are thought to be established for controlling the virulence-related gene expression during infection: on the initial colonization (i.e., LCD) of a host by a pathogenic Vibrio species, AphA is abundantly expressed to act as a master activator of genes responsible for virulence and biofilm formation, which promotes bacterial colonization and infection; when a HCD is reached, HMR is abundantly produced to act as a master repressor of virulence and biofilm formation.
Cis-acting AphA Consensus
Two consensus constructs (a box sequence, and a PFM) were built herein to represent conserved cis-acting regulatory DNA signals recognized by the AphA orthologs from nine Vibrio species, V. parahaemolyticus, Vibrio sp. Ex25, Vibrio sp. EJY3, V. harveyi, V. vulnificus, V. splendidus, V. anguillarum, V. cholerae, and V. furnissii. The 20 bp AphA box sequence contains imperfect 7-nt inverted repeats with a 6-nt centered spacer, and this dyad symmetry structure is consistent with the fact that AphA monomers are unstable by themselves and constitute a dimer that sits on one face of the target cis-acting DNA to fit symmetrically into the DNA-binding site [43]. It should be noted that the 20 bp box herein is essentially an extended sequence of the 18 bp one previously characterized in V. harveyi [44] and V. cholerae [30].
Both box and PFM can be used to statistically predict the presence of AphA box-like elements within the target promoter-proximal DNA sequences, for instance by using the web-based Regulatory Sequence Analysis Tools [23]. For prediction with box, a concise calculation of number of mismatched nucleotides is employed to match the target DNA sequence against the box [23]. Compared to box, PFM gave a much more comprehensive description of uneven nucleotide composition in each position (i.e., some nucleotides occurred much more frequently than others); therefore, prediction with PFM will be more accurate, which generates a weight score for each target gene, and a higher score denotes the higher probability of AphA-promoter DNA association (Table 2). This assay raised several additional potential direct AphA targets especially including those responsible for virulence and biofilm formation in V. parahaemolyticus, and the validation of AphA-mediated regulation of these potential target genes are underway.
Molecular Regulation of QS Regulator-encoding Genes
The transcriptional repression of aphA by AphA/HMR has been characterized in V. harveyi [7], [11], V. cholerae [30], [33], and V. parahaemolyticus ([15] and this study). The promoter-proximal DNA regions of aphA from the nine Vibrio species were aligned in Fig. 9a, in which shown were translation and transcription starts, –35 and –10 core promoter elements for σ70 recognition, Shine-Dalgarno (SD) sequences for ribosome recognition, and AphA/OpaR box-like sequences representing conserved signals for recognition by AphA/OpaR. For all the nine Vibrio species tested, the OpaR-box like sequences were about 18 bp upstream of the AphA box-like ones that overlapped the -35 core promoter regions, indicating that the mechanism of AphA/HMR-mediated repression of aphA was conserved in vibrios.
Figure 9. Organization of promoter-proximal DNA regions.

DNA sequences were derived from V. parahaemolyticus RIMD 2210633 [14], Vibrio sp. Ex25 (Accession numbers CP001805.1, and CP001806.1), Vibrio sp. EJY3 [48], V. harveyi ATCC BAA-1116 [49], V. vulnificus YJ016 [50], V. splendidus LGP32 [51], V. anguillarum 775 [52], V. cholerae N16961 [53], and V. furnissii NCTC 11218 [54]. Shown were translation and transcription starts, SD sequences, AphA or OpaR box-like sequences, and –0/−12 and –35/−24 core promoter elements. The prediction of OpaR box-like sequences were described previously [15].
The AphA-mediated repression of HMR genes luxR and opaR has been established in V. harveyi [7] and V. parahaemolyticus (this study), respectively. In addition, the auto-repression of HMRs, OpaR, LuxR, and HapR, has been characterized in V. parahaemolyticus [15], V. harveyi [45] and V. cholerae [46], respectively. The sequence alignment (Fig. 9b) showed that the AphA box-like sequences overlapped the –10 core promoter regions while the OpaR box-like ones were downstream of the transcription start sites for HMR genes from all the above nine Vibrio species tested. The direct association between AphA/HMR and target promoters would block the entry of RNA polymerase to repress the transcription of corresponding target genes, and this mechanism appeared to be conserved in vibrios.
The AphA-mediated repression of qrr2-4 has been established in V. harveyi and V. cholerae [7], and also confirmed in V. parahaemolyticus in this study. The detected binding of AphA to the qrr4 promoter-proximal region indicated the direct regulation of qrr4 by AphA in V. harveyi [7] and V. parahaemolyticus (this study). In addition, the present study further validated that AphA repressed the qrr2-3 transcription in an indirect manner since AphA could not bound to the promoter-proximal regions of qrr2-3 (the association between qrr2-3 promoter regions and AphA were not tested in V. harveyi [7]). AphA box-like sequences were found upstream of the -24 core promoter regions of qrr4 from all five Vibrio species of Clade I (V. parahaemolyticus, Vibrio sp. Ex25, Vibrio sp. EJY3, V. harveyi, and V. vulnificus) rather than Clade II or III (Fig. 9c), indicating that AphA might directly repress the qrr4 transcription in only these five Vibrio species. AphA-mediated repression of qrr4 must occur by a highly unusual mechanism, since AphA binds a site located about one hundred nucleotides upstream of the core -24 and -12 promoter regions. In contrast, AphA box-like sequences could not be predicted for qrr2-3 from all the nine Vibrio species tested.
Supporting Information
Primer extension assay for validation of non-polar mutation. The aphA null mutant ΔaphA was generated from the wild-type (WT) strain RIMD 2210633, and then the complemented mutant strain C-aphA was constructed. As determined by several distinct methods (see text), the qrr2 transcription was under the negative control of AphA. Herein, an oligonucleotide primer, which was complementary to the RNA transcript of qrr2, was employed to detect the primer extension product that represented the relative mRNA level of qrr2 in WT, ΔaphA, and C-ΔaphA. The primer extension products were analyzed with 8 M urea–6% acrylamide sequencing gel. Lanes C, T, A, and G represented Sanger sequencing reactions. The transcription start site of qrr2 was underlined in the DNA sequence. The qrr2 mRNA level was significantly enhanced in ΔaphA relative to WT, while no obvious change in the qrr2 transcription was observed between WT and C-aphA, which confirmed that the detecting enhanced transcription of qrr2 in ΔaphA was due to the aphA mutation rather than a polar mutation.
(TIF)
Acknowledgments
We thank Professor Mitsuaki Nishibuchi from Kyoto University and Professor Hin-chung Wong from Taiwan Soochow University for kindly providing strain RIMD 2210633 and vector pBRMob, respectively. All experiments in the present work were done in State Key Laboratory of Pathogen and Biosecurity, Beijing, China.
Funding Statement
Financial support was provided by the National Natural Science Foundation of China (31170127), and by the National Basic Research Program of China (2009CB522604). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Miller MB, Bassler BL (2001) Quorum sensing in bacteria. Annu Rev Microbiol 55: 165–199. [DOI] [PubMed] [Google Scholar]
- 2. Henke JM, Bassler BL (2004) Three parallel quorum-sensing systems regulate gene expression in Vibrio harveyi . J Bacteriol 186: 6902–6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Waters CM, Bassler BL (2006) The Vibrio harveyi quorum-sensing system uses shared regulatory components to discriminate between multiple autoinducers. Genes Dev 20: 2754–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lenz DH, Mok KC, Lilley BN, Kulkarni RV, Wingreen NS, et al. (2004) The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae . Cell 118: 69–82. [DOI] [PubMed] [Google Scholar]
- 5. Tu KC, Bassler BL (2007) Multiple small RNAs act additively to integrate sensory information and control quorum sensing in Vibrio harveyi . Genes Dev 21: 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shao Y, Bassler BL (2012) Quorum-sensing non-coding small RNAs use unique pairing regions to differentially control mRNA targets. Mol Microbiol 83: 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rutherford ST, van Kessel JC, Shao Y, Bassler BL (2011) AphA and LuxR/HapR reciprocally control quorum sensing in vibrios. Genes Dev 25: 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Long T, Tu KC, Wang Y, Mehta P, Ong NP, et al. (2009) Quantifying the integration of quorum-sensing signals with single-cell resolution. PLoS Biol 7: e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tu KC, Long T, Svenningsen SL, Wingreen NS, Bassler BL (2010) Negative feedback loops involving small regulatory RNAs precisely control the Vibrio harveyi quorum-sensing response. Mol Cell 37: 567–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Teng SW, Schaffer JN, Tu KC, Mehta P, Lu W, et al. (2011) Active regulation of receptor ratios controls integration of quorum-sensing signals in Vibrio harveyi . Mol Syst Biol 7: 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pompeani AJ, Irgon JJ, Berger MF, Bulyk ML, Wingreen NS, et al. (2008) The Vibrio harveyi master quorum-sensing regulator, LuxR, a TetR-type protein is both an activator and a repressor: DNA recognition and binding specificity at target promoters. Mol Microbiol 70: 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sawabe T, Kita-Tsukamoto K, Thompson FL (2007) Inferring the evolutionary history of Vibrios by means of multilocus sequence analysis. J Bacteriol 189: 7932–7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yeung PS, Boor KJ (2004) Epidemiology, pathogenesis, and prevention of foodborne Vibrio parahaemolyticus infections. Foodborne Pathog Dis 1: 74–88. [DOI] [PubMed] [Google Scholar]
- 14. Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, et al. (2003) Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V cholerae. Lancet 361: 743–749. [DOI] [PubMed] [Google Scholar]
- 15. Zhang Y, Qiu Y, Tan Y, Guo Z, Yang R, et al. (2012) Transcriptional regulation of opaR, qrr2–4 and aphA by the master quorum-sensing regulator OpaR in Vibrio parahaemolyticus . PLoS One 7: e34622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Philippe N, Alcaraz JP, Coursange E, Geiselmann J, Schneider D (2004) Improvement of pCVD442, a suicide plasmid for gene allele exchange in bacteria. Plasmid 51: 246–255. [DOI] [PubMed] [Google Scholar]
- 17. Hiyoshi H, Kodama T, Iida T, Honda T (2010) Contribution of Vibrio parahaemolyticus virulence factors to cytotoxicity, enterotoxicity, and lethality in mice. Infect Immun 78: 1772–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Casselli T, Lynch T, Southward CM, Jones BW, DeVinney R (2008) Vibrio parahaemolyticus inhibition of Rho family GTPase activation requires a functional chromosome I type III secretion system. Infect Immun 76: 2202–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Balbas P, Soberon X, Merino E, Zurita M, Lomeli H, et al. (1986) Plasmid vector pBR322 and its special-purpose derivatives-a review. Gene 50: 3–40. [DOI] [PubMed] [Google Scholar]
- 20. Parales RE, Harwood CS (1993) Construction and use of a new broad-host-range lacZ transcriptional fusion vector, pHRP309, for gram- bacteria. Gene 133: 23–30. [DOI] [PubMed] [Google Scholar]
- 21. Zhang Y, Gao H, Wang L, Xiao X, Tan Y, et al. (2011) Molecular characterization of transcriptional regulation of rovA by PhoP and RovA in Yersinia pestis . PLoS One 6: e25484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Andrews GP, Heath DG, Anderson GW Jr, Welkos SL, Friedlander AM (1996) Fraction 1 capsular antigen (F1) purification from Yersinia pestis CO92 and from an Escherichia coli recombinant strain and efficacy against lethal plague challenge. Infect Immun 64: 2180–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Helden J (2003) Regulatory sequence analysis tools. Nucleic Acids Res 31: 3593–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14: 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vezzi A, Campanaro S, D'Angelo M, Simonato F, Vitulo N, et al. (2005) Life at depth: Photobacterium profundum genome sequence and expression analysis. Science 307: 1459–1461. [DOI] [PubMed] [Google Scholar]
- 26. Kovacikova G, Skorupski K (2001) Overlapping binding sites for the virulence gene regulators AphA, AphB and cAMP-CRP at the Vibrio cholerae tcpPH promoter. Mol Microbiol 41: 393–407. [DOI] [PubMed] [Google Scholar]
- 27. Kovacikova G, Lin W, Skorupski K (2004) Vibrio cholerae AphA uses a novel mechanism for virulence gene activation that involves interaction with the LysR-type regulator AphB at the tcpPH promoter. Mol Microbiol 53: 129–142. [DOI] [PubMed] [Google Scholar]
- 28. Yang M, Frey EM, Liu Z, Bishar R, Zhu J (2010) The virulence transcriptional activator AphA enhances biofilm formation by Vibrio cholerae by activating expression of the biofilm regulator VpsT. Infect Immun 78: 697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kovacikova G, Lin W, Skorupski K (2005) Dual regulation of genes involved in acetoin biosynthesis and motility/biofilm formation by the virulence activator AphA and the acetate-responsive LysR-type regulator AlsR in Vibrio cholerae . Mol Microbiol 57: 420–433. [DOI] [PubMed] [Google Scholar]
- 30. Lin W, Kovacikova G, Skorupski K (2007) The quorum sensing regulator HapR downregulates the expression of the virulence gene transcription factor AphA in Vibrio cholerae by antagonizing Lrp- and VpsR-mediated activation. Mol Microbiol 64: 953–967. [DOI] [PubMed] [Google Scholar]
- 31. Tsou AM, Zhu J (2010) Quorum sensing negatively regulates hemolysin transcriptionally and posttranslationally in Vibrio cholerae . Infect Immun 78: 461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhu J, Miller MB, Vance RE, Dziejman M, Bassler BL, et al. (2002) Quorum-sensing regulators control virulence gene expression in Vibrio cholerae . Proc Natl Acad Sci U S A 99: 3129–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kovacikova G, Skorupski K (2002) Regulation of virulence gene expression in Vibrio cholerae by quorum sensing: HapR functions at the aphA promoter. Mol Microbiol 46: 1135–1147. [DOI] [PubMed] [Google Scholar]
- 34. Henke JM, Bassler BL (2004) Quorum sensing regulates type III secretion in Vibrio harveyi and Vibrio parahaemolyticus . J Bacteriol 186: 3794–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gode-Potratz CJ, McCarter LL (2011) Quorum sensing and silencing in Vibrio parahaemolyticus. J Bacteriol. [DOI] [PMC free article] [PubMed]
- 36. Waters CM, Wu JT, Ramsey ME, Harris RC, Bassler BL (2010) Control of the type 3 secretion system in Vibrio harveyi by quorum sensing through repression of ExsA. Appl Environ Microbiol 76: 4996–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shao CP, Lo HR, Lin JH, Hor LI (2011) Regulation of cytotoxicity by quorum-sensing signaling in Vibrio vulnificus is mediated by SmcR, a repressor of hlyU . J Bacteriol 193: 2557–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Waters CM, Lu W, Rabinowitz JD, Bassler BL (2008) Quorum sensing controls biofilm formation in Vibrio cholerae through modulation of cyclic di-GMP levels and repression of vpsT . J Bacteriol 190: 2527–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lim B, Beyhan S, Meir J, Yildiz FH (2006) Cyclic-diGMP signal transduction systems in Vibrio cholerae: modulation of rugosity and biofilm formation. Mol Microbiol 60: 331–348. [DOI] [PubMed] [Google Scholar]
- 40. Hammer BK, Bassler BL (2003) Quorum sensing controls biofilm formation in Vibrio cholerae . Mol Microbiol 50: 101–104. [DOI] [PubMed] [Google Scholar]
- 41. Faruque SM, Biswas K, Udden SM, Ahmad QS, Sack DA, et al. (2006) Transmissibility of cholera: in vivo-formed biofilms and their relationship to infectivity and persistence in the environment. Proc Natl Acad Sci U S A 103: 6350–6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tamayo R, Patimalla B, Camilli A (2010) Growth in a biofilm induces a hyperinfectious phenotype in Vibrio cholerae . Infect Immun 78: 3560–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. De Silva RS, Kovacikova G, Lin W, Taylor RK, Skorupski K, et al. (2005) Crystal structure of the virulence gene activator AphA from Vibrio cholerae reveals it is a novel member of the winged helix transcription factor superfamily. J Biol Chem 280: 13779–13783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kovacikova G, Lin W, Skorupski K (2003) The virulence activator AphA links quorum sensing to pathogenesis and physiology in Vibrio cholerae by repressing the expression of a penicillin amidase gene on the small chromosome. J Bacteriol 185: 4825–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chatterjee J, Miyamoto CM, Meighen EA (1996) Autoregulation of luxR: the Vibrio harveyi lux-operon activator functions as a repressor. Mol Microbiol 20: 415–425. [DOI] [PubMed] [Google Scholar]
- 46. Lin W, Kovacikova G, Skorupski K (2005) Requirements for Vibrio cholerae HapR binding and transcriptional repression at the hapR promoter are distinct from those at the aphA promoter. J Bacteriol 187: 3013–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tu KC, Waters CM, Svenningsen SL, Bassler BL (2008) A small-RNA-mediated negative feedback loop controls quorum-sensing dynamics in Vibrio harveyi . Mol Microbiol 70: 896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Roh H, Yun EJ, Lee S, Ko HJ, Kim S, et al. (2012) Genome sequence of Vibrio sp. Strain EJY3, an agarolytic marine bacterium metabolizing 3,6-anhydro-L-galactose as a sole carbon source. J Bacteriol 194: 2773–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lin B, Wang Z, Malanoski AP, O'Grady EA, Wimpee CF, et al. (2010) Comparative genomic analyses identify the Vibrio harveyi genome sequenced strains BAA-1116 and HY01 as Vibrio campbellii . Environ Microbiol Rep 2: 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen CY, Wu KM, Chang YC, Chang CH, Tsai HC, et al. (2003) Comparative genome analysis of Vibrio vulnificus, a marine pathogen. Genome Res 13: 2577–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Le Roux F, Zouine M, Chakroun N, Binesse J, Saulnier D, et al. (2009) Genome sequence of Vibrio splendidus: an abundant planctonic marine species with a large genotypic diversity. Environ Microbiol 11: 1959–1970. [DOI] [PubMed] [Google Scholar]
- 52. Naka H, Dias GM, Thompson CC, Dubay C, Thompson FL, et al. (2011) Complete genome sequence of the marine fish pathogen Vibrio anguillarum harboring the pJM1 virulence plasmid and genomic comparison with other virulent strains of V. anguillarum and V. ordalii . Infect Immun 79: 2889–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, et al. (2000) DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae . Nature 406: 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lux TM, Lee R, Love J (2011) The complete genome squence of a free-living Vibrio furnissii sp. Nov. strain (NCTC 11218). J Bacteriol. [DOI] [PMC free article] [PubMed]
- 55.Thompson JD, Gibson TJ, Higgins DG (2002) Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics Chapter 2: Unit 2 3. [DOI] [PubMed]
- 56. Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer extension assay for validation of non-polar mutation. The aphA null mutant ΔaphA was generated from the wild-type (WT) strain RIMD 2210633, and then the complemented mutant strain C-aphA was constructed. As determined by several distinct methods (see text), the qrr2 transcription was under the negative control of AphA. Herein, an oligonucleotide primer, which was complementary to the RNA transcript of qrr2, was employed to detect the primer extension product that represented the relative mRNA level of qrr2 in WT, ΔaphA, and C-ΔaphA. The primer extension products were analyzed with 8 M urea–6% acrylamide sequencing gel. Lanes C, T, A, and G represented Sanger sequencing reactions. The transcription start site of qrr2 was underlined in the DNA sequence. The qrr2 mRNA level was significantly enhanced in ΔaphA relative to WT, while no obvious change in the qrr2 transcription was observed between WT and C-aphA, which confirmed that the detecting enhanced transcription of qrr2 in ΔaphA was due to the aphA mutation rather than a polar mutation.
(TIF)