

Abstract
Obese mice exhibit innate airway hyperresponsiveness (AHR), a feature of asthma. Tumor necrosis factor alpha (TNFα) is implicated in the disease progression and chronic inflammatory status of both obesity and asthma. TNF acts via two TNF receptors, TNFR1 and TNFR2. To examine the role of TNFR2 in the AHR observed in obese mice, we generated obese Cpefat mice that were either sufficient or deficient in TNFR2 (Cpefat and Cpefat/TNFR2−/− mice, respectively) and compared them with their lean controls (WT and TNFR2−/− mice). Compared to WT mice, Cpefat mice exhibited AHR to aerosolized methacholine (measured using the forced oscillation technique) which was ablated in Cpefat/TNFR2−/− mice. Bioplex or ELISA assay indicated significant increases in serum leptin, G-CSF, IL-7, IL-17A, TNFα, and KC in obese versus lean mice, as well as significant obesity-related increases in bronchoalveolar lavage fluid (BALF) G-CSF and IP-10, regardless of TNFR2 status. Importantly, BALF IL-17A was significantly increased over lean controls in Cpefat but not Cpefat/TNFR2−/− mice. Functional annotation clustering of significantly affected genes identified from microarray analysis comparing gene expression in lungs of Cpefat and WT mice, identified blood vessel morphogenesis as the gene ontology category most affected by obesity. This category included several genes associated with AHR, including endothelin and trkB. Obesity increased pulmonary mRNA expression of endothelin and trkB in TNFR2 sufficient but not deficient mice. Our results indicate that TNFR2 signaling is required for the innate AHR that develops in obese mice, and suggest that TNFR2 may act by promoting IL-17A, endothelin, and/or trkB expression.
Keywords: bronchoalveolar lavage, airway responsiveness, TNFalpha, inflammation, endothelin, trkB, IL-17A
1. INTRODUCTION
Obesity is a risk factor for asthma. Obesity increases both the prevalence and incidence of asthma, and worsens asthma control (see recent reviews [1–4]). In the obese asthmatic, weight loss improves asthma outcomes, including airway responsiveness [5, 6], although others have found no effect of obesity on responsiveness (see [7] for review). Data from animal models also support a relationship between obesity and asthma. For example, regardless of the cause of their obesity, obese mice display airway hyperresponsiveness (AHR), a characteristic feature of asthma [8–11].
In both obese humans and obese mice, even in the absence of any overt inflammatory insult, there is chronic, low-grade, systemic inflammation characterized by increased circulating leukocytes and increased serum concentrations of cytokines, chemokines, and acute phase proteins [12, 13]. Importantly, systemic inflammatory markers correlate with the presence of diseases common to obesity, including type II diabetes and atherosclerosis [14–17], suggesting that the inflammation is functionally important. We have also reported a temporal correspondence between the induction of systemic inflammation and the induction of AHR in obese Cpefat mice [18].
Of the various inflammatory moieties that are increased in the blood in obesity, TNFα is of particular interest. Several obesity-related conditions are ameliorated in obese mice lacking TNFα or TNF receptors [19–24]. Similarly, TNFα may also be relevant for obesity-related asthma. Risk estimates for asthma as a function of BMI are higher in subjects with the G/A or A/A TNFA -308 polymorphisms that lead to higher TNFα expression than in G/G subjects with lower TNFα expression, especially among those with nonatopic asthma [25]. Exogenous administration of TNFα can also induce AHR [26]. Taken together, the data suggest that augmented circulating TNFα in obesity may contribute to AHR.
TNFα can bind to either of two receptors, TNFR1 or TNFR2, which differ in their inflammatory potential, their affinity for cleaved versus membrane-associated TNFα, their cellular locus of expression, their ability to induce apoptosis, and their effects on blood vessels [27–32]. Because of the inflammatory nature of both obesity and asthma, and because TNFR1 is, in general, more pro-inflammatory than TNFR2 [31, 32], we initially examined the importance of TNFR1 for the AHR observed in obese mice. Surprisingly, our data indicated augmented airway responsiveness in Cpefat mice deficient in TNFR1 compared to Cpefat mice sufficient in TNFR1[33]. In mice lacking TNFR1, TNFα acts exclusively via TNFR2, suggesting that in the obese mice, TNFα signaling via TNFR2 may promote AHR. To address this hypothesis, airway responsiveness to aerosolized methacholine was measured in lean wildtype (WT) mice, obese Cpefat mice, lean TNFR2 deficient (TNFR2−/−) mice, as well as obese Cpefat mice that were also deficient in TNFR2 (Cpefat/TNFR2−/− mice). Our data indicated the presence of AHR in TNFR2 sufficient but not TNFR2 deficient Cpefat mice compared to their respective lean controls. To examine the mechanistic basis for this effect of TNFR2, we determined the impact of TNFR2 deficiency on the low grade systemic inflammation associated with obesity, and also examined potential TNFR2-related differences in lung inflammation. We also performed a microarray analysis comparing gene expression in lungs of WT and Cpefat mice. We then used RT-PCR to examine mRNA expression of candidates from the list of differentially expressed genes that might be contributing to obesity-related AHR.
2. METHODS
2.1 Animals
This study was approved by the Harvard Medical Area Standing Committee on Animals. Cpefat mice are genetically deficient in carboxypeptidase E (Cpe), an enzyme involved in processing prohormones and proneuropeptides that are important for appetite regulation and energy expenditure [34, Coleman, 1990 #367, 35]. Absence of Cpe leads to obesity [36, 37]. Because Cpefat mice are infertile, heterozygous Cpe+/− mice were purchased from Jackson Labs (Bar Harbor, ME). Cpe+/− mice were mated to TNFR2−/− mice, also purchased from Jackson Labs. Both types of mice were on a C57BL/6 background. Cpe+/−/TNFR2+/− offspring were bred back to TNFR2−/− mice. Cpe+/−/TNFR2−/− mice from this mating were then bred together to obtain Cpefat/TNFR2−/− mice and TNFR2−/− controls. Heterozygous Cpe+/− mice were mated to each other to generate WT and Cpefat mice. All mice were fed standard mouse chow diets. Mice were 10–12 weeks of age at the time of study. Cpefat and matched wildtype C57BL/6 mice for the microarray analysis were purchased mice from Jackson Labs.
2.2 Protocol
For studies of the role of TNFR2 in obesity-related AHR, WT, Cpefat, TNFR2−/−, and Cpefat/TNFR2−/− mice were used. Mice were anesthetized and instrumented for the measurement of pulmonary mechanics and airway responsiveness to aerosolized methacholine. Immediately after euthanasia, bronchoalveolar lavage (BAL) was performed, and the lungs were harvested. The left lung was immersed in RNAlater (Qiagen) and subsequently used to prepare RNA for measurement of gene expression by quantitative RT-PCR. In a separate cohort of WT, Cpefat, TNFR2−/−, and Cpefat/TNFR2−/− mice which did not undergo measurement of airway responsiveness, blood was obtained by cardiac puncture after euthanasia, and serum was prepared. BAL was also performed. BAL supernatants and serum from these mice were used for measurements of cytokines and chemokines by ELISA and Bioplex. In the case of BAL IL-17A, measurements were combined from these mice and the mice in which measurements of pulmonary mechanics were performed. WT and Cpefat mice for microarray analysis were euthanized, BAL was performed, and the left lung was excised and frozen in liquid nitrogen.
2.3 Measurement of pulmonary mechanics and airway responsiveness
Mice were anesthetized with xylazine (7 mg/kg) and sodium pentobarbital (50 mg/kg), ventilated and instrumented for the measurement of pulmonary mechanics as previously described [18]. The chest wall was opening bilaterally to expose the lungs to atmospheric pressure and a positive end expiratory pressure of 3 cm H2O was applied. A standardized volume history was established by twice inflating the lungs to 30 cm H2O airway opening pressure (total lung capacity (TLC)). In order to measure the static elastic properties of the lung, we then performed a slow inflation to TLC, followed by a slow deflation back to end expiratory lung volume. This quasi static pressure volume (PV) loop was repeated 3 times at one minute intervals. For each PV loop, we computed the following parameters using the Salazar-Knowles equation [38]: A, the difference between TLC and end expiratory volume; B, the difference between TLC and the predicted volume at 0 pressure; and K, a measure of the curvature of the upper portion of the deflation limb of the PV curve. The static compliance (Cstat) of the lung obtained from the lower portion of the deflationary limb of the PV loop was also obtained. Values from the 3 PV loops were averaged to obtain a mean value for each animal. Baseline pulmonary mechanics were then assessed using the forced oscillation technique [8, 18, 39, 40] as follows. Mice were given an inflation to TLC. One minute later, measurements of total lung impedance (ZL) were obtained using an 3s optimized pseudorandom signal containing frequencies ranging from 0.25 to 19.63 Hz. A parameter estimation model [41] was used to partition ZL into components representing Newtonian resistance (Rn), which largely reflects the conducting airways, and the coefficients of lung tissue damping (G) and lung tissue elastance (H), which reflect changes in the lung tissue. This sequence was repeated 3 times and the values averaged. Finally, we measured responses to inhaled aerosolized methacholine dissolved in PBS. One minute after inflation to TLC, an aerosol of PBS was delivered to the lungs for 10 seconds using an ultrasonic nebulizer (Aeroneb, SCIREQ). Rn, G and H were measured every 15 seconds for the next 3 minutes, as described above. This sequence was then repeated after delivery of increasing doses of methacholine (0.3–300 mg/ml). At each dose, the 3 highest values of Rn, G and H were averaged and used to construct dose response curves.
2.4 Bronchoalveolar lavage and serum
The lungs were lavaged twice with 1 ml PBS. The lavagates were pooled and placed on ice until centrifuged at 400 × g at 4°C for 10 min. Cell pellets were resuspended in Hank’s Balanced Salt solution and a hemocytometer was used to assess the total number of cells. Aliquots of cells were also centrifuged onto glass slides at 800 rpm for 10 min (Cytospin 3, Shandon, Sewickley, PA). The slides were air-dried, and then stained with Wright-Giemsa (LeukoStat, Fisher Scientific, Pittsburgh, PA). Cell differentials were determined by counting 300 cells under 400X magnification. Blood was collected from the heart via puncture of the right ventricle. Serum was isolated using Microtainer tubes (Becton Dickinson, NJ) and stored at −20°C until assayed for TNFα by ELISA (EBiosciences, San Diego, CA) according to the manufacturer’s instructions. Bioplex assay for 35 different cytokines, chemokines, and growth factors was performed on BAL supernatant and on serum (Eve Technologies, Calgary, Alberta, Canada). BAL IL-17A was also assessed by ELISA (BioLegend, USA).
2.5 RNA extraction and Real Time PCR
For RNA for microarray analysis, lung tissue was homogenized in 3 ml TRIzol reagent (Invitrogen; Carlsbad, CA) and total RNA subsequently extracted in accordance with the manufacturer’s instructions. Following RNA extraction, samples were run through RNeasy RNA Cleanup (QIAGEN Inc.; Valencia, CA) to increase RNA purity and then stored at −80°C.
For RT-PCR, RNA was extracted from lungs and cDNA generated as previously described [42]. Endothelin 1, TrkB, fractalkine, and adipsin mRNA expression were quantified by real time PCR (7300 Real-Time PCR Systems, Applied Biosystems, US) using SYBR-green detection. Primers are described in Table 1. The delta Ct (ΔCt) was obtained by subtracting from the gene of interest, the Ct values for a housekeeping gene, 36B4 (rplp0), a ribosomal protein. Primers for 36B4 were as described by Ohashi et al [43]. Changes in mRNA were expressed relative to values from the WT/WT air exposed mice, to obtain ΔΔCt values. Expression was calculated by RQ=2−ΔΔCt [44].
Table 1.
List of primers used for RT-PCR
| TrkB | forward reverse |
5′-CGTTGACCCGGAGAACATC-3′ 5′-AAACTTTAAGCCGGAATCCAC-3′ |
| Adipsin | forward reverse |
5′-CTGAACCCTACAAGCGATGG-3′ 5′-ACCCAACGAGGCATTCTG-3′ |
| Fractalkine | forward reverse |
5′-CCGCGTTCTTCCATTTGT-3′ 5′-GCTTCTCAAACTTGCCACCAT-3′ |
| Endothelin | forward reverse |
5′-ACT TCT GCC ACC TGG ACA TC-3′ 5′-TCT GTG GCC TTA TTG GGA AG-3′ |
| 36B4 | forward reverse |
5′-GCT CCA AGC AGA TGC AGC A-3′ 5′-CCG GAT GTG AGG CAG CAG-3′ |
2.6 Microarray
Eight RNA samples, each one pooled from 2–5 mice, were used in this analysis. Four RNA samples were from lean wildtype mice and were matched with 4 samples from Cpefat mice. Gene expression analyses on these samples were performed using the GeneChip® Mouse Genome 430A 2.0 Array platform (Affymetrix, Santa Clara, CA). Sample labeling, hybridization, and array scanning were performed by the Harvard Medical School – Partners Healthcare Center for Genetics and Genomics (HPCGG) using standard protocols (www.hpcgg.org/Microarrays/resources). For microarray analysis, expression values were extracted from .cel files using Robust Microarray Analysis (RMA) [45] and imported into the program Dchip for statistical analysis [46, 47]. We used the DAVID Bioinformatics Resources [48] (http://david.abcc.ncifcrf.gov) to classify the ontology of genes that were significantly different between WT and Cpefat mice and to perform functional annotation clustering. For the latter, we used high classification stringency and all probe sets on the 430A 2.0 chip as background.
2.7 Statistics
Differences in outcome indicators were assessed by factorial ANOVA using Cpe genotype and TNFR2 genotype as main effects, except as noted below. The Fisher LSD test was used for post hoc comparisons. For Bioplex assay, samples below the limit of detection of the assay were assigned a value of 0. In addition, serum eotaxin, G-CSF, IL-1α, IL-6, IL-9, IL-15, IP-10, KC, and VEGF, as well as BAL G-CSF were log transformed prior to statistical analysis to conform to a normal distribution. In the case of serum IL-7 and IL-17A, because neither absolute values nor log transformed values were normally distributed, a Mann Whitney U test was used instead of factorial ANOVA. Statistical analyses were carried out using SAS software (SAS Institute, Cary, NC). Results presented are mean ± SE unless otherwise indicated. p < 0.05 was considered statistically significant.
3. RESULTS
3.1 Body mass
Factorial ANOVA indicated a significant (p<0.001) effect of Cpe genotype on body mass, whereas TNFR2 genotype had no effect. Cpefat mice, whether TNFR2 deficient or not, weighed more than twice as much as wildtype controls (Fig. 1).
Figure 1.
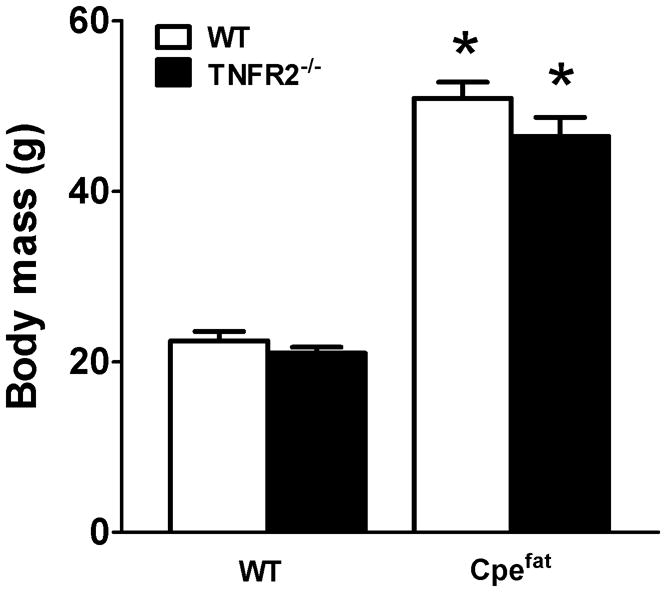
Effect of TNFR2 deficiency on body weight. Cpefat mice: mice with a genetic deficiency in the carboxypeptidase E (Cpe) gene. TNFR2−/− mice: mice genetically deficient in the tumor necrosis factor receptor 2 gene. Cpefat/TNFR2−/− mice: mice genetically deficient in both Cpe and TNFR2. Results are mean ± SE of data from 7–11 female mice in each group *p<0.001 compared to TNFR2 genotype matched lean mice
3.2 Pulmonary mechanics and airway responsiveness
PV curve parameters, A and B (measures of lung volume), and K (a measure of the curvature of the upper portion of the deflation limb) were significantly lower in obese Cpefat mice than in lean WT mice (Fig. 2A–C). Cstat was also reduced in Cpefat mice (Fig. 2D). Baseline Rn, G, and H were elevated in lungs of obese mice, consistent with the smaller lungs of these mice (Fig. 2E–G). TNFR2 genotype had no effect on PV curve parameters or on baseline oscillation pulmonary mechanics.
Figure 2.

Baseline lung volumes, oscillation mechanics, and static elastic properties of the lungs. Shown are A, the parameter from the Salazar-Knowles equation [38] indicating the difference between total lung capacity (TLC) and end expiratory volume; B, the parameter from the Salazar-Knowles equation indicating the difference between TLC and the predicted volume at 0 pressure; K, the parameter from the Salazar-Knowles equation reflecting the curvature of the upper portion of the deflation PV curve; Cstat, the static compliance of the lung, as well as the Newtonian resistance (Rn) and the coefficients of tissue damping (G) and elastance (H). Results are mean ± SE of data from 7–12 mice in each group. * p<0.001 versus TNFR2 genotype-matched lean mice.
In TNFR2 sufficient mice (Fig. 3A–C), methacholine caused a concentration dependent increase in Rn, G, and H. Baseline (PBS) values for Rn, G, and H were greater in Cpefat versus WT mice, and these changes carried through the dose response curve, although in the case of Rn and G, the obesity-related difference was amplified at higher doses of methacholine, whereas for H, the curves did not diverge further. Indeed, when we computed the change in Rn, G, and H from baseline (PBS) values, changes in G and Rn but not H were significantly greater in Cpefat than WT mice at the highest concentrations of methacholine, indicating AHR in the obese mice. In contrast, in TNFR2 deficient mice, there was no significant effect of obesity on airway responsiveness using either G or Rn as the outcome indicator (Fig. 3D–F). Indeed, in the TNFR2−/− mice, methacholine induced changes in Rn, G, and H were actually lower in the obese than the lean mice at the highest dose of methacholine.
Figure 3.

TNFR2 deficiency abolishes obesity related airway hyperresponsiveness. Methacholine induced changes in Rn, G, and H in TNFR2 sufficient (A–C) and TNFR2 deficient (D–F) wildtype (WT) and Cpefat mice. Results are mean ± SE of data from 7–12 mice per group. * p<0.05 versus wildtype for absolute values; # p<0.05 for changes in Rn, G, or H from baseline (PBS).
3.3 Serum Analyses
To confirm that serum TNFα was indeed elevated in the obese mice, serum TNFα was measured by ELISA. Factorial ANOVA indicated a significant (p<0.01) effect of Cpe genotype on serum TNFα, whereas there was no effect of TNFR2 genotype. In both TNFR2 sufficient and deficient mice, serum TNFα was approximately 2-fold greater in obese Cpefat mice than in lean controls (Fig. 4A).
Figure 4.
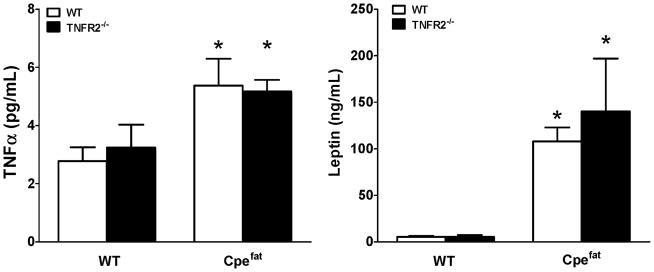
Effect of TNFR2 deficiency on serum TNFα (A) and serum leptin (B). Results are mean ± SE of data from 4–6 mice per group, *p<0.05 compared to TNFR2 genotype matched lean mice.
In cultured adipocytes, exogenous TNFα can induce the expression and/or release of many of the inflammatory moieties that are elevated in serum of obese individuals [7, 49, 50]. Consequently, it is conceivable that the lack of obesity-related AHR observed in Cpefat/TNFR2−/− mice was the result of loss of elevated TNF acting on TNFR2, not in the lung, but through changes in other aspects of the systemic inflammation of obesity. To address this issue, we performed a Bioplex assay that simultaneously measured 35 different cytokines, chemokines and growth factors. We also measured serum leptin (Fig. 4C) in WT, TNFR2−/−, Cpefat, and Cpefat/TNFR2−/− mice by ELISA. We found significant increases in serum G-CSF, IL-7, IL-17A, KC, and leptin in obese versus lean mice, regardless of TNFR2 status (Table 2, Fig. 4B, C). IL-1α and IP-10 were significantly reduced in TNFR2 deficient versus TNFR2 sufficient mice, regardless of differences in body mass (Table 2). In addition, there was a significant interaction between Cpe and TNFR2 status for eotaxin and IL-9. For both of these moieties, elevated levels in TNFR2 sufficient Cpefat versus WT mice were reversed in TNFR deficient mice (Table 2). There were no significant changes in IL-5, IL-6, IL-13, MCP-1, MIG, MIP-2, RANTES or VEGF with either obesity (Cpe status) or TNFR2 genotype, although there was a trend towards increased IL-6, IL-15, MIG, and MIP-2 in obese versus lean mice. Serum LIX was above the maximum of the standard curve in most samples. Serum GM-CSF, IFN-γ, IL-1β, IL-2, IL-3, IL-4, IL-10, IL-12 (p40 and p70), LIF, M-CSF, MIP-1α, MIP-1β, and TNFα were below the limit of detection of the Bioplex in most samples, although TNFα was detectable by ELISA, as described above.
Table 2.
Serum concentrations of cytokines and chemokines
| Cpe effect | TNFR2 effect | Cpe* TNFR2 interaction | WT | TNFR2−/− | Cpefat | Cpefat/TNFR2−/− | |
|---|---|---|---|---|---|---|---|
| Eotaxin | NS | NS | p<0.05 | 999 ± 710 | 3216 ± 1488 | 4745 ± 1867 | 869 ± 279 |
| G-CSF | p<0.02 | NS | NS | 114 ± 28 | 49 ± 16 | 296 ± 105 | 368 ± 221 |
| IL-1α | NS | p<0.02 | NS | 322 ± 102 | 89 ± 25 | 239 ± 85 | 73 ± 25 |
| IL-5 | NS | NS | NS | 1.9 ± 0.6 | 3.2 ± 1.6 | 3.1 ± 1.4 | 1.1 ± 0.3 |
| IL-6 | NS | NS | NS | 21.8 ± 15.4 | 0.7 ± 0.3 | 48.2 ± 37.9 | 33.6 ± 20.0 |
| IL-7 | p<0.02* | NS* | - | 0.9 ± 0.4 | 0.7 ± 0.6 | 3.2 ± 1.4 | 14.1 ± 7.5 |
| IL-9 | NS | NS | p<0.05 | 7.1 ± 5.9 | 25.3 ± 16.6 | 120.1 ± 52.5 | 6.4 ± 5.0 |
| IL-13 | NS | NS | NS | 37 ± 11 | 54 ± 16 | 61 ± 26 | 40 ± 19 |
| IL-15 | NS | NS | NS | 9.0 ± 3.6 | 10.6 ± 4.8 | 19.4 ± 9.5 | 21.4 ± 19.5 |
| IL-17A | p=0.050* | NS* | - | 0.3 ± 0.1 | 0.2 ± 0.1 | 7.1 ± 3.8 | 1.4 ± 0.9 |
| IP-10 | NS | P<0.05 | NS | 102 ± 28 | 63 ± 8 | 159 ± 43 | 67 ± 10 |
| KC | P<0.02 | NS | NS | 69 ± 42 | 39 ± 13 | 188 ± 94 | 101 ± 44 |
| MCP-1 | NS | NS | NS | 30 ± 23 | 24 ± 11 | 29 ± 6 | 37 ± 14 |
| MIG | NS | NS | NS | 14 ± 4 | 14 ± 5 | 26 ± 5 | 19 ± 5 |
| MIP-2 | NS | NS | NS | 93 ± 17 | 87 ± 12 | 115 ± 25 | 118 ± 29 |
| RANTES | NS | NS | NS | 7.7 ± 1.2 | 6.4 ± 1.1 | 12.6 ± 4.2 | 8.2 ± 3.9 |
| VEGF | NS | NS | NS | 1.0 ± 0.3 | 0.9 ± 0.3 | 1.3 ± 0.9 | 0.8 ± 0.4 |
Results are mean +/− SE of data from 7–8 mice in each group and are in pg/ml.
Mann Whitney U test
3.4 Bronchoalveolar lavage
Factorial ANOVA indicated a significant effect of obesity on total BAL macrophages (p<0.05). BAL neutrophils and lymphocytes were not significantly different from zero in all groups (Table 3). Bioplex analysis of BAL revealed obesity-related changes in G-CSF, IL-1α and IP-10, while TNFR2 status had no effect (Table 4). In addition, there was a significant interaction between Cpe and TNFR2 status for eotaxin and KC. For both of these moieties, reduced levels in TNFR2 sufficient Cpefat versus WT mice were reversed in TNFR deficient mice (Table 4). There were no significant obesity- or TNFR2-related changes in IL-2, IL-7, IL-9, IL-10, IL-12p40, IL-15, M-CSF, MIP1α, MIP-2, or VEGF. Other cytokines chemokines and growth factors on the Bioplex were below the limit of detection in most samples. Because obesity resulted in increased BAL G-CSF, and because of the importance of IL-17A for induction of G-CSF [51], we also examined BAL IL-17A by ELISA, which proved to be more sensitive than the Bioplex. Compared to WT mice, BAL IL-17A was increased in Cpefat mice (Fig. 5), and this increase was abolished in Cpefat/TNFR2−/− mice.
Table 3.
Bronchoalveolar cells in WT and Cpefat mice with and without TNFR2 deficiency
| Cell type | WT/WT | WT/TNFR2−/− | Cpefat/WT | Cpefat/TNFR2−/− |
|---|---|---|---|---|
| Macrophages | 3.50 ± 0.38 | 3.03 ± 0.29 | 4.18 ± 0.47 | 4.42 ± 0.38* |
| PMNs | 0.003 ± 0.002 | 0.024 ± 0.012 | 0.0 ± 0.0 | 0.011 ± 0.007 |
| Lymphocytes | 0.004 ± 0.003 | 0.0 ± 0.0 | 0.003 ± 0.002 | 0.0 ± 0.0 |
Results are mean ± SE of data from 7–11 mice per group. Results are expressed as cells ×104/ml.
p<0.05 versus WT/TNFR2−/− mice.
Table 4.
BAL concentrations of cytokines and chemokines
| Cpe effect | TNFR2 effect | Cpe*TNFR2 interaction | WT | TNFR2−/− | Cpefat | Cpefat/TNFR2−/− | |
|---|---|---|---|---|---|---|---|
| Eotaxin | NS | NS | p<0.05 | 2.8 ± 0.7 | 0.8 ± 0.3 | 1.5 ± 0.5 | 4.1 ± 1.1 |
| G-CSF | p<0.05 | NS | NS | 2.4 ± 0.6 | 0.7 ± 0.3 | 8.3 ± 6.3 | 4.3 ± 1.6 |
| IL-1α | p<0.05 | NS | NS | 6.6 ± 2.0 | 7.5 ± 1.7 | 2.6 ± 1.4 | 3.2 ± 1.6 |
| IL-2 | NS | NS | NS | 7.2 ± 1.0 | 5.5 ± 0.5 | 4.5 ± 1.2 | 6.9 ± 1.8 |
| IL-7 | NS | NS | NS | 3.1 ± 0.3 | 1.7 ± 0.6 | 1.6 ± 0.3 | 2.0 ± 1.0 |
| IL-9 | NS | NS | NS | 3.0 ± 0.6 | 1.2 ± 0.6 | 1.8 ± 0.7 | 1.8 ± 1.2 |
| IL-10 | NS | NS | NS | 1.1 ± 0.4 | 1.6 ± 0.5 | 0.7 ± 0.4 | 0.8 ± 0.4 |
| IL-12p40 | NS | NS | NS | 5.0 ± 1.9 | 2.1 ± 0.9 | 0.3 ± 0.3 | 3.6 ± 2.7 |
| IL-15 | NS | NS | NS | 7.9 ± 1.9 | 5.5 ± 2.2 | 6.6 ± 2.9 | 7.5 ± 4.9 |
| IP-10 | p<0.01 | NS | NS | 1.5 ± 0.5 | 0.8 ± 0.1 | 3.1 ± 1.5 | 6.0 ± 3.9 |
| KC | NS | NS | p<0.05 | 5.6 ± 0.8 | 1.8 ± 0.9 | 2.8 ± 1.1 | 4.4 ± 2.1 |
| M-CSF | NS | NS | NS | 2.0 ± 0.5 | 1.4 ± 0.4 | 1.6 ± 0.4 | 1.7 ± 1.7 |
| MIP-1β | NS | NS | NS | 21.6 ± 1.7 | 18.4 ± 3.7 | 16.8 ± 2.0 | 16.6 ± 6.7 |
| MIP-2 | NS | NS | NS | 22.4 ± 2.6 | 11.3 ± 6.3 | 15.4 ± 6.6 | 18.8 ± 9.5 |
| VEGF | NS | NS | NS | 4.6 ± 2.1 | 5.5 ± 0.3 | 4.7 ± 2.4 | 3.0 ± 0.6 |
Results are mean ± SE of data from 3–5 mice in each group and are in pg/ml.
Figure 5.
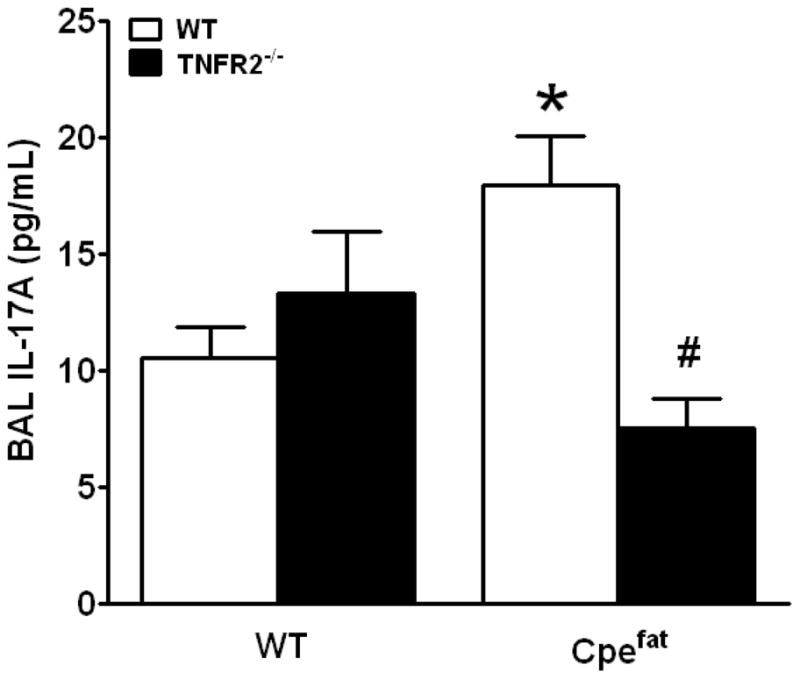
Effect of obesity and TNFR2 deficiency on BAL IL-17A. Results are mean ± SE of data from 11–14 mice per group, #p<0.05 compared to Cpefat mice.
3.5 Microarray
To obtain a comprehensive picture of differences in gene expression in the lungs of Cpefat and WT mice, we performed a microarray analysis. Comparison of expression values of arrays from Cpefat and WT mice indicated 52 Affy IDs that were significantly altered by Cpe genotype (p<0.05) with at least a 1.5-fold change. Of these, 25 were reduced, and 27 were increased in Cpefat versus WT mice (Table 5). We also performed functional annotation clustering on the genes listed in Table 5, to determine whether there were gene ontology (GO) categories that were enriched in this gene set. Four annotation clusters had significant (p<0.05) enrichment scores (Table 6). Of these, the cluster with the greatest enrichment score included the GO term “blood vessel morphogenesis”, and comprised 8 unique genes. Remarkably, several genes in this category have been linked to AHR in animal models, including endothelin-1, neurotrophic tyrosine kinase, receptor, type 2 (trkB), and chemokine (C-X3-C motif) ligand 1 (Cx3cl1, fractalkine). Other genes in this category were SRY-box containing gene 17 (Sox17), delta-like 4 (Drosophila)(Dll4), endoglin, endomucin, and sema domain, immunoglobulin domain (Ig), short basic domain, secreted (semaphorin) 3C (Sema3C). There were additional genes affected by Cpe genotype (Table 5) that, though not classified as related to blood vessel morphogenesis, are either strongly expressed in endothelial cells, including Vcam1, Esm1, and Plvap, or can impact growth of endothelial cells (Fgfbp1). Other annotation clusters significantly affected by Cpe genotype included two clusters that contained genes with transmembrane regions, and a cluster related to tube morphogenesis. Genes included within these clusters are indicated in the right most column of Table 5.
Table 5.
Genes with expression values that were significantly different in lungs of wildtype and Cpefat mice
| Affymetrix ID | Gene name | Symbol | Cpefat/WT | Cluster |
|---|---|---|---|---|
| 1420465_s_at | major urinary protein 1/2 | Mup1/2 | 0.12 | |
| 1417867_at | complement factor D (adipsin) | Cfd | 0.33 | |
| 1453128_at | lymphatic vessel endothelial hyaluronan receptor 1 | Lyve1 | 0.37 | 2,3 |
| 1418090_at | plasmalemma vesicle associated protein | Plvap | 0.37 | 2,3 |
| 1449280_at | endothelial cell-specific molecule 1 | Esm1 | 0.42 | |
| 1415994_at | cytochrome P450, family 2, subfamily E, polypeptide 1 | Cyp2e1 | 0.51 | |
| 1434528_at | alanine and arginine rich domain containing protein | Aard | 0.55 | |
| 1415803_at | chemokine (C-X3-C motif) ligand 1 | Cx3cl1 | 0.59 | 1,2,3 |
| 1421826_at | delta-like 4 (Drosophila) | Dll4 | 0.60 | 1,2,3 |
| 1422340_a_at | actin, gamma 2, smooth muscle, enteric | Actg2 | 0.60 | |
| 1417917_at | calponin 1 | Cnn1 | 0.61 | |
| 1423121_at | insulin degrading enzyme | Ide | 0.61 | |
| 1418554_at | G protein-coupled receptor 182 | Gpr182 | 0.61 | 2,3 |
| 1423555_a_at | interferon-induced protein 44 | Ifi44 | 0.61 | |
| 1420798_s_at | protocadherin alpha cluster | Pcdha@ | 0.62 | 2,3 |
| 1426172_a_at | CD209a antigen | Cd209a | 0.62 | 2,3 |
| 1419086_at | fibroblast growth factor binding protein 1 | Fgfbp1 | 0.63 | |
| 1422837_at | sciellin | Scel | 0.63 | |
| 1427809_at | latrophilin 3 | Lphn3 | 0.63 | 2,3 |
| 1448690_at | potassium channel, subfamily K, member 1 | Kcnk2 | 0.65 | 2,3 |
| 1452359_at | RELT-like 1 | Rell1 | 0.65 | 2,3 |
| 1448162_at | vascular cell adhesion molecule 1 | Vcam1 | 0.65 | 2,3 |
| 1432176_a_at | endoglin | Eng | 0.66 | 1,2,3,4 |
| 1424525_at | gastrin releasing peptide | Grp | 0.66 | |
| 1417429_at | flavin containing monooxygenase 1 | Fmo1 | 0.66 | 3 |
| 1423506_a_at | neuronatin | Nnat | 1.50 | |
| 1422562_at | Ras-related associated with diabetes | Rrad | 1.52 | |
| 1421928_at | Eph receptor A4 | Epha4 | 1.53 | 2,3 |
| 1448662_at | frizzled homolog 6 (Drosophila) | Fzd6 | 1.55 | 2,3,4 |
| 1417251_at | palmdelphin; similar to palmdelphin | Palmd | 1.62 | |
| 1423294_at | mesoderm specific transcript | Mest | 1.63 | 2,3 |
| 1449305_at | coagulation factor X | F10 | 1.64 | |
| 1426328_a_at | sodium channel, voltage gated, type III, beta | Scn3b | 1.72 | 2,3 |
| 1425582_a_at | endomucin | Emcn | 1.76 | 1,2,3 |
| 1422474_at | phosphodiesterase 4B, cAMP specific | Pde4b | 1.80 | |
| 1417396_at | podocalyxin-like | Podxl2 | 1.85 | 2,3 |
| 1451245_at | leucine rich repeat containing 3B | Lrrc3b | 1.85 | 2,3 |
| 1451415_at | RIKEN cDNA 1810011010 gene | 1810011O10Rik | 1.86 | |
| 1427495_at | sodium channel, voltage gated, type VII, alpha | Scn7a | 1.93 | 3 |
| 1424010_at | microfibrillar-associated protein 4 sema domain, immunoglobulin (Ig) domain, short basic |
Mfap4 | 1.99 | |
| 1420696_at | domain, secreted, (semaphorin) 3C solute carrier family 6 (neurotransmitter transporter, |
Sema3C | 2.00 | 1 |
| 1421346_a_at | taurine), member 6 v-myc myelocytomatosis viral related oncogene, |
Slc6a6 | 2.13 | 2,3 |
| 1417155_at | neuroblastoma derived (avian) | Mycn | 2.14 | 4 |
| 1431213_a_at | hypothetical gene LOC433762 | LOC433762 | 2.15 | |
| 1429177_x_at | SRY-box containing gene 17 | Sox17 | 2.16 | 1 |
| 1451924_a_at | endothelin 1 | Edn1 | 2.53 | 1,4 |
| 1455869_at | calcium/calmodulin-dependent protein kinase II, beta | Camk2b | 2.55 | |
| 1448484_at | S-adenosylmethionine decarboxylase 1/2 | Amd1/2 | 2.69 | |
| 1421657_a_at | SRY-box containing gene 17 | Sox17 | 2.78 | 1 |
| 1451447_at | CUE domain containing 1 | Cuedc1 | 2.79 | |
| 1416835_s_at | S-adenosylmethionine decarboxylase 1,2 | Amd1/2 | 3.53 | |
| 1420838_at | neurotrophic tyrosine kinase, receptor, type 2 | Ntrk2 | 3.81 | 1,2,3 |
Table 6.
Clusters (by enrichment score)* significantly affected by Cpefat genotype
| Cluster Category | Enrichment Score | Gene Count | P value |
|---|---|---|---|
|
| |||
| Cluster 1 | 4.44 | ||
| Blood vessel morphogenesis | 8 | 1.4E-5 | |
| Blood vessel development | 8 | 5.4E-5 | |
| Vasculature development | 8 | 6.4E-5 | |
|
| |||
| Cluster 2 | 3.45 | ||
| Topological domain: extracellular | 18 | 3.4E-5 | |
| Topological domain: cytoplasmic | 19 | 3.4E-4 | |
| Transmembrane region | 21 | 3.9E-3 | |
|
| |||
| Cluster 3 | 2.39 | ||
| Transmembrane | 23 | 1.1E-3 | |
| Transmembrane region | 21 | 3.9E-3 | |
| Integral to membrane | 23 | 5.8E-3 | |
| Intrinsic to membrane | 23 | 1.1E-2 | |
|
| |||
| Cluster 4 | 1.26 | ||
| Tube morphogenesis | 4 | 3.1E-2 | |
| Branching morphogenesis of a tube | 3 | 5.9E-2 | |
| Tube developement | 4 | 8.8E-2 | |
Functional annotation clustering was performed using the program DAVID on high stringency.
3.6 Gene expression
To determine whether TNFR2 deficiency affected obesity-related changes in expression of genes identified on the microarray (Table 5), we used RT-PCR to measure pulmonary mRNA expression of endothelin, trkB, adipsin, and fractalkine. We chose endothelin, trkb, and fractalkine because each has been shown to impact airway responsiveness in mice [52–55]. Adipsin was chosen because it is a member of the complement family of proteins and because complement has been shown to play a key role in airway responsiveness [56]. Consistent with the results of the microarray, we observed greater expression of endothelin and trkB and reduced expression of adipsin and fractalkine in obese Cpefat versus lean WT mice (Fig. 5). TNFR2 deficiency ablated the obesity-related increases in endothelin and trkB expression, whereas it had no effect on adipsin expression and further exacerbated obesity-related declines in fractalkine.
4. DISCUSSION
Our results indicated that serum TNFα was increased in obese Cpefat mice (Fig. 4), and that the AHR observed in Cpefat mice was ablated if the mice were TNFR2 deficient (Fig. 3). Both obesity and TNFR2 deficiency had effects on serum and BAL cytokines and chemokines (Tables 2, 4 and Figures 4, 5) that could be contributing to the attenuated AHR observed in obese TNFR2 deficient mice. For example, BAL IL-17A was elevated in Cpefat but not Cpefat/TNFR2−/− mice (Figure 5). Microarray analysis demonstrated obesity-related changes in gene expression in the lung (Table 4), including increases in endothelin and trkB, genes that have been linked to AHR in other models. Importantly, obesity-related increases in endothelin and trkB were attenuated in TNFR2 deficient mice (Fig. 5). The results are consistent with the hypothesis that in obese mice, TNFα signaling via TNFR2 contributes to AHR and suggest TNFR2-dependent candidates that may be involved in these events.
Lung volume parameters A and B were reduced in lungs of obese versus lean mice (Fig. 2A, B), indicating smaller lungs in the obese mice, but TNFR2 genotype had no effect on these outcomes. Obese ob/ob and db/db mice also have smaller lungs than their wildtype controls [9, 57]. Reductions in Cstat (Fig. 2D) and elevations in Rn, G, and H in the obese mice (Fig. 2E–G) were likely the result of the smaller lungs of these animals, although Rochester et al suggested that reductions in Cstat in human obesity might be the result of increased pulmonary blood volume [58]. We frequently observed large amounts of adipose tissue in the thoracic cavity of Cpefat mice and in other types of obese mice, and it is conceivable that this mass impinges upon lung expansion. TLC is also reduced in morbidly obese humans, but not in subjects with milder obesity [59].
Compared to WT mice, obese Cpefat mice exhibited AHR (Fig. 3), consistent with previous reports using both these mice [8, 18] and other types of obese mice [9–11, 60]. Increased responsiveness was observed when either Rn or G but not H was used as the outcome indicator. A similar pattern of responsiveness, with greater aerosolized methacholine induced changes in Rn and G, but not H, is observed in the innately hyperresponsive A/J mouse. Using computational modeling, others have concluded that such a pattern is indicative of increased airway smooth muscle contractility [61]. Interesting, when A/J mice are challenged with i.v. instead of aerosolized methacholine, augmented responsiveness is detected only with Rn, but not G or H [61]. We have previously reported similar results in Cpefat mice challenged with i.v. methacholine [8].
TNFα was elevated in the serum of obese Cpefat mice (Fig. 4A), consistent with data from obese humans and other types of obese mice [62–64]. Elevations in serum TNFα appeared to be functionally important, since signaling via TNFR2 was required for obesity-related AHR (Fig. 3): obesity-related augmented Rn and G responses to methacholine were not observed in TNFR2 deficient mice. Indeed, in the TNFR2 deficient mice, methacholine induced changes in Rn, G, and H were actually reduced in obese versus lean mice at the highest concentration of methcholine (Fig. 3). It is conceivable that such changes are the result of increased TNFα signaling via TNFR1 in these mice, since we have reported that signaling via TNFR1 is actually protective against AHR in obese mice [33]. The decrease in obesity-related AHR observed in TNFR2−/− mice was not the result of milder obesity in the Cpefat/TNFR2−/− mice, since body weight was virtually identical in the two strains (Fig. 1). Similarly, Uysal et al [19, 20] reported no difference in body weight of obese leptin deficient (ob/ob) mice, and ob/ob mice that were genetically deficient in TNFR2, TNFR1, or TNFα itself. It is also unlikely that the TNFR2 mediated effects that contribute to obesity-related AHR are mediated by TNFα binding to airway smooth muscle. TNFα has been shown to act directly on airway smooth muscle, increasing its contractility [65], but these effects are mediated via TNFR1 not TNFR2 [65, 66].
It is conceivable that the effects of TNFR2 that contribute to AHR are related to interactions between TNFR2 and other aspects of the inflammation of obesity. Although most studies indicate that inflammation induced by TNFα is largely mediated through TNFR1 activation, TNFR2 can also contribute[31]. In cultured adipocytes, exogenous TNFα can induce many of the inflammatory moieties that are elevated in serum of obese individuals [7]. Consequently, we examined the impact of TNFR2 deficiency on systemic and pulmonary inflammation in obese Cpefat mice using Bioplex and ELISA assay. Our results confirm previous observations indicating that serum leptin is increased in serum of obese Cpefat mice, and extend them to include TNFα, G-CSF, IL-7, and IL-17A (Fig. 4, Table 2). Our results also indicate increased G-CSF, IP-10, and IL-17A in BAL of obese Cpefat mice (Table 4, Fig. 5), as well as increases in BAL macrophages (Table 3).
Of the various factors measured in serum of these mice, only IP-10 and IL-1α were significantly affected by TNFR2 deficiency (Fig. 4, Table 2), and these were not genes that were impacted by obesity. While these results suggest that signaling through TNFR2 does not have a major impact on the systemic inflammation of obesity, it is possible that IP-10 and/or IL-1α synergize with other aspects of obesity-related systemic inflammation to promote AHR. Under such circumstances, loss of TNFR2 and consequent reductions in IP-10 and IL-1α (Table 2) would be expected to attenuate obesity-related AHR. There was also an interaction between obesity and TNFR deficiency on serum eotaxin and IL-9 (Table 2). Both moieties have been associated with AHR in mouse models of allergic inflammation [67, 68]. Hence, obesity/TNFR2 elated changes in eotaxin or IL-9 could have the capacity to influence AHR in these obese mice.
We observed significant increases in IL-17A in both serum and BAL of obese mice (Tables 2 and 4; Fig. 5). G-CSF is strongly induced by IL-17A [51], so the elevations in BAL and serum G-CSF in obese mice (Tables 2 and 4) are consistent with the elevations in IL-17A. Serum IL-17A is also elevated in obese versus lean human subjects [69]. Compared to lean mice, obese mice also have an increased capacity to generate IL-17A from a variety of cellular sources [70, 71]. Although we do not know the cellular source of this IL-17, it is interesting to note that in adipose tissue, γ/δ T cells are the T cell type with the greatest capacity to produce IL-17A [72]. Our data indicated increased serum IL-7 and a trend towards increased serum IL-15 in obese mice (Table 2). Both IL-7 and IL-15 promote proliferation of γ/δ T cells [73, 74].
It is conceivable that the increased BAL IL-17A observed in obese Cpefat mice contributes to their AHR: obesity-related increases in BAL IL-17A were attenuated in TNFR2−/− mice (Fig. 5), as was AHR (Fig. 3). In mice, IL-17A acts directly on airway smooth muscle to increased its contractility [75]. Changes in airway smooth muscle contractility would be expected to increase changes in Rn and G but not H induced by aerosolized methacholine [61] which is consistent with our observations in Cpefat versus WT mice (Fig. 3).
We also performed gene expression profiling of the lungs of obese and lean mice in order to uncover potential mechanisms for the AHR observed in obese mice. Among the genes significantly impacted by obesity, functional annotation clustering indicated overrepresentation of genes involved in blood vessel morphogenesis (Table 5). It is perhaps not surprising that effects of obesity on the pulmonary vasculature were observed, since obesity also has a major impact on the systemic vasculature [76], and since obesity is more common in patients with idiopathic pulmonary arterial hypertension [77]. Notably, TNFα has also been shown to play a role in the systemic vascular changes associated with obesity [78], and the ability of TNFα to promote angiogenesis is largely TNFR2 dependent [27–29].
Given the observed changes in some BAL cytokines and chemokines (Table 4, Fig. 5), we were surprised to find that pulmonary expression of genes related to inflammation and/or innate immunity was not significantly impacted by obesity. We considered the possibility that changes occurred, but that the magnitude of the changes did not meet our criterion of at least a 1.5 fold change. However, when we relaxed the criteria to include genes in which at least a 1.25 fold change occurred, genes related to inflammation, immunity and host defense were still not on the list of significantly affected GO categories (data not shown). Inflammatory cells and/or cells expressing inflammatory genes may make up a very small percentage of the total lung cells. Microarray analysis is not sensitive to genes with low overall expression.
We do not know the mechanistic basis for the apparent effects on blood vessels, or whether an anatomical correlate of these changes exists. Cpefat mice are not only obese, but also hyperinsulinemic and hyperlipidemic [35, 79], and it is possible that these conditions contributed to the observed effects on pulmonary gene expression. In addition, others have reported airway closure in spontaneously breathing obese versus lean human subjects [80, 81]. We do not know whether airway closure normally exists in obese mice, but such closure would be expected to result in reductions in PO2 in the alveoli subtended by those airways, and it is possible that alterations of genes involved in blood vessel morphogenesis results from this hypoxia.
RT-PCR on lungs of mice from a separate cohort of mice confirmed the results of the microarray: both RT-PCR and microarray indicated that endothelin and trkB mRNA expression were increased in lungs of obese mice, whereas expression of fractalkine and adipsin were decreased (Table 5, Figure 6). Although changes in mRNA expression do not necessarily translate into changes in protein expression, the observations that myocardial and vascular endothelial expression of endothelin [82, 83], as well as plasma endothelin [84, 85] are increased in obese mice support the obesity-related increases in pulmonary endothelin mRNA expression observed in these mice (Fig. 5A). In contrast, no obesity-related increase in endothelin was observed in the lungs of TNFR2 deficient mice (Fig. 5A). These results are consistent with the ability of TNFα to induce endothelin [86, 87], and with the observation that TNFα-induced reductions in cerebral blood volume require endothelin and are TNFR2 dependent [88]. Data support a role for endothelin in other obesity-related conditions including hypertension and insulin resistance [89–92], and it is conceivable that increased pulmonary endothelin expression (Fig. 5A) contributes to obesity-related AHR: endothelin causes AHR when administered intranasally in mice [52], and endothelin receptor antagonists attenuate AHR induced by allergen sensitization and challenge [52, 53]. Furthermore, both obesity-related AHR and increases in endothelin mRNA were attenuated in TNFR2−/− mice (Figs. 3 and 6A).
Figure 6.

Pulmonary mRNA expression of endothelin (panel A), trkB (panel B), adipsin (panel C) and fractalkine (panel D) in lean wildtype (WT) and obese Cpefat mice either sufficient or deficient in TNFR2. Results are mean ± SE of data from 8–12 mice per group. * p<0.01 versus TNFR2 genotype-matched WT mice.
We also observed a marked increase in trkB mRNA expression in lungs of obese TNFR2 sufficient but not deficient Cpefat mice (Fig. 6B). In guinea pigs, exogenous administration of BDNF, one of the ligands for trkB, causes AHR [54] and BDNF also contributes to neuronal hyperreactivity in the airways of allergen challenged mice [93]. Futhermore, TNFα, via changes in TrkB expression, augments the ability of BDNF to augment intracellular Ca++ fluxes induced by bronchoconstricting agonists in human airway smooth muscle cells [94]. Thus, it is conceivable that obesity-related elevations in trkB could augment AHR, and that loss of trkB expression in Cpefat/TNFR2−/− mice might explain the corresponding reduction in AHR in these mice.
Varying effects on asthma outcomes of anti-TNFα strategies including etanercept, infliximab, and golimumab have been reported [95]. Since none of these studies stratified their subjects on the basis of body mass index (BMI), and since the asthma phenotype appears to vary with BMI [96], the therapeutic benefit of anti-TNF strategies in obesity-related asthma is unknown. However, it is important to note that the TNF based therapeutics currently available are not specific for TNFR2 versus TNFR1 mediated events. Studies with mice may not necessarily be translatable to human subjects, especially given the heterogeneity of asthmatic responses. Nevertheless, our data showing reduced AHR in obese TNFR2 deficient mice (Fig. 3), in conjunction with previous observations showing augmented AHR in obese TNFR1 deficient mice[33] suggest that strategies that specifically interfere with TNFR2, rather than pan TNF-blocking strategies may have therapeutic potential in obesity-related asthma. Alternatively, strategies aimed at possible downstream targets of TNFR2, such as endothelin, may also have therapeutic potential.
In summary, our results indicate that TNFR2 signaling is required for the innate airway hyperresponsiveness that develops in obese mice and suggest TNFR2 dependent candidates, including IL-17A, endothelin, and TrkB, that may be involved in these events.
Acknowledgments
This work was supported by the U.S. National Institute of Health grants ES-013307, HL-084044, and ES-00002.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beuther DA, Sutherland ER. Overweight, obesity, and incident asthma: a meta-analysis of prospective epidemiologic studies. Am J Respir Crit Care Med. 2007;175:661–6. doi: 10.1164/rccm.200611-1717OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ford ES. The epidemiology of obesity and asthma. J Allergy Clin Immunol. 2005;115:897–909. doi: 10.1016/j.jaci.2004.11.050. quiz 10. [DOI] [PubMed] [Google Scholar]
- 3.Litonjua AA, Gold DR. Asthma and obesity: common early-life influences in the inception of disease. J Allergy Clin Immunol. 2008;121:1075–84. doi: 10.1016/j.jaci.2008.03.005. quiz 85–6. [DOI] [PubMed] [Google Scholar]
- 4.Shore SA, Johnston RA. Obesity and asthma. Pharmacol Ther. 2006;110:83–102. doi: 10.1016/j.pharmthera.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 5.Maniscalco M, Zedda A, Faraone S, Cerbone MR, Cristiano S, Giardiello C, et al. Weight loss and asthma control in severely obese asthmatic females. Respir Med. 2008;102:102–8. doi: 10.1016/j.rmed.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 6.Dixon AE, Pratley RE, Forgione PM, Kaminsky DA, Whittaker-Leclair LA, Griffes LA, et al. Effects of obesity and bariatric surgery on airway hyperresponsiveness, asthma control, and inflammation. J Allergy Clin Immunol. 2011;128:508–15. e2. doi: 10.1016/j.jaci.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shore SA. Obesity, airway hyperresponsiveness, and inflammation. J Appl Physiol. 2009:00749.2009. doi: 10.1152/japplphysiol.00749.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnston RA, Theman TA, Shore SA. Augmented Responses to Ozone in Obese Carboxypeptidase E-Deficient Mice. Am J Physiol Regul Integr Comp Physiol. 2006;290:R126–33. doi: 10.1152/ajpregu.00306.2005. [DOI] [PubMed] [Google Scholar]
- 9.Lu FL, Johnston RA, Flynt L, Theman TA, Terry RD, Schwartzman IN, et al. Increased pulmonary responses to acute ozone exposure in obese db/db mice. Am J Physiol Lung Cell Mol Physiol. 2006;290:L856–65. doi: 10.1152/ajplung.00386.2005. [DOI] [PubMed] [Google Scholar]
- 10.Shore SA, Rivera-Sanchez YM, Schwartzman IN, Johnston RA. Responses to ozone are increased in obese mice. J Appl Physiol. 2003;95:938–45. doi: 10.1152/japplphysiol.00336.2003. [DOI] [PubMed] [Google Scholar]
- 11.Johnston RA, Theman TA, Lu FL, Terry RD, Williams ES, Shore SA. Diet-induced obesity causes innate airway hyperresponsiveness to methacholine and enhances ozone-induced pulmonary inflammation. J Appl Physiol. 2008;104:1727–35. doi: 10.1152/japplphysiol.00075.2008. [DOI] [PubMed] [Google Scholar]
- 12.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6:772–83. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 13.Scherer PE. Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes. 2006;55:1537–45. doi: 10.2337/db06-0263. [DOI] [PubMed] [Google Scholar]
- 14.Pickup JC, Mattock MB, Chusney GD, Burt D. NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia. 1997;40:1286–92. doi: 10.1007/s001250050822. [DOI] [PubMed] [Google Scholar]
- 15.Vozarova B, Weyer C, Hanson K, Tataranni PA, Bogardus C, Pratley RE. Circulating interleukin-6 in relation to adiposity, insulin action, and insulin secretion. Obes Res. 2001;9:414–7. doi: 10.1038/oby.2001.54. [DOI] [PubMed] [Google Scholar]
- 16.Bastard JP, Maachi M, Van Nhieu JT, Jardel C, Bruckert E, Grimaldi A, et al. Adipose tissue IL-6 content correlates with resistance to insulin activation of glucose uptake both in vivo and in vitro. J Clin Endocrinol Metab. 2002;87:2084–9. doi: 10.1210/jcem.87.5.8450. [DOI] [PubMed] [Google Scholar]
- 17.Teramoto S, Yamamoto H, Ouchi Y. Increased C-reactive protein and increased plasma interleukin-6 may synergistically affect the progression of coronary atherosclerosis in obstructive sleep apnea syndrome. Circulation. 2003;107:E40–0. doi: 10.1161/01.cir.0000053956.46188.5f. [DOI] [PubMed] [Google Scholar]
- 18.Johnston RA, Zhu M, Hernandez CB, Williams ES, Shore SA. Onset of obesity in carboxypeptidase E-deficient mice and effect on airway responsiveness and pulmonary responses to ozone. J Appl Physiol. 2010;108:1812–9. doi: 10.1152/japplphysiol.00784.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uysal KT, Wiesbrock SM, Hotamisligil GS. Functional analysis of tumor necrosis factor (TNF) receptors in TNF-alpha-mediated insulin resistance in genetic obesity. Endocrinology. 1998;139:4832–8. doi: 10.1210/endo.139.12.6337. [DOI] [PubMed] [Google Scholar]
- 20.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389:610–4. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- 21.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bouter B, Geary N, Langhans W, Asarian L. Diet-genotype interactions in the early development of obesity and insulin resistance in mice with a genetic deficiency in tumor necrosis factor-alpha. Metabolism. 2010;59:1065–73. doi: 10.1016/j.metabol.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 23.Liang H, Yin B, Zhang H, Zhang S, Zeng Q, Wang J, et al. Blockade of tumor necrosis factor (TNF) receptor type 1-mediated TNF-alpha signaling protected Wistar rats from diet-induced obesity and insulin resistance. Endocrinology. 2008;149:2943–51. doi: 10.1210/en.2007-0978. [DOI] [PubMed] [Google Scholar]
- 24.De Taeye BM, Novitskaya T, McGuinness OP, Gleaves L, Medda M, Covington JW, et al. Macrophage TNF-alpha contributes to insulin resistance and hepatic steatosis in diet-induced obesity. Am J Physiol Endocrinol Metab. 2007;293:E713–25. doi: 10.1152/ajpendo.00194.2007. [DOI] [PubMed] [Google Scholar]
- 25.Castro-Giner F, Kogevinas M, Imboden M, de Cid R, Jarvis D, Machler M, et al. Joint effect of obesity and TNFA variability on asthma: two international cohort studies. Eur Respir J. 2009;33:1003–9. doi: 10.1183/09031936.00140608. [DOI] [PubMed] [Google Scholar]
- 26.Thomas PS, Yates DH, Barnes PJ. Tumor necrosis factor-alpha increases airway responsiveness and sputum neutrophilia in normal human subjects. American Journal of Respiratory and Critical Care Medicine. 1995;152:76–80. doi: 10.1164/ajrccm.152.1.7599866. [DOI] [PubMed] [Google Scholar]
- 27.Goukassian DA, Qin G, Dolan C, Murayama T, Silver M, Curry C, et al. Tumor necrosis factor-alpha receptor p75 is required in ischemia-induced neovascularization. Circulation. 2007;115:752–62. doi: 10.1161/CIRCULATIONAHA.106.647255. [DOI] [PubMed] [Google Scholar]
- 28.Kishore R, Tkebuchava T, Sasi SP, Silver M, Gilbert HY, Yoon YS, et al. Tumor necrosis factor-alpha signaling via TNFR1/p55 is deleterious whereas TNFR2/p75 signaling is protective in adult infarct myocardium. Adv Exp Med Biol. 2011;691:433–48. doi: 10.1007/978-1-4419-6612-4_45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sasi SP, Yan X, Enderling H, Park D, Gilbert HY, Curry C, et al. Breaking the ‘harmony’ of TNF-alpha signaling for cancer treatment. Oncogene. 2011 doi: 10.1038/onc.2011.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vandenabeele P, Declercq W, Beyaert R, Fiers W. Two tumour necrosis factor receptors: structure and function. Trends Cell Biol. 1995;5:392–9. doi: 10.1016/s0962-8924(00)89088-1. [DOI] [PubMed] [Google Scholar]
- 31.MacEwan DJ. TNF receptor subtype signalling: differences and cellular consequences. Cell Signal. 2002;14:477–92. doi: 10.1016/s0898-6568(01)00262-5. [DOI] [PubMed] [Google Scholar]
- 32.Naude PJ, den Boer JA, Luiten PG, Eisel UL. Tumor necrosis factor receptor cross-talk. FEBS J. 2011;278:888–98. doi: 10.1111/j.1742-4658.2011.08017.x. [DOI] [PubMed] [Google Scholar]
- 33.Zhu M, Williams ES, Shore SA. Role of TNFR1 in the Pulmonary Phenotype of Obese Mice. Am J Respir Crit Care Med. 2008;176:A835. [Google Scholar]
- 34.Leibel RL, Chung WK, Chua SC., Jr The molecular genetics of rodent single gene obesities. J Biol Chem. 1997;272:31937–40. doi: 10.1074/jbc.272.51.31937. [DOI] [PubMed] [Google Scholar]
- 35.Coleman DL, Eicher EM. Fat (fat) and tubby (tub): two autosomal recessive mutations causing obesity syndromes in the mouse. J Hered. 1990;81:424–7. doi: 10.1093/oxfordjournals.jhered.a111019. [DOI] [PubMed] [Google Scholar]
- 36.Johnston RA, Zhu M, Hernandez CB, Williams ES, Shore SA. Onset of obesity in carboxypeptidase E-deficient mice and effect on airway responsiveness and pulmonary responses to ozone. J Appl Physiol. 2010:00784.2009. doi: 10.1152/japplphysiol.00784.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnston RA, Theman TA, Shore SA. Augmented responses to ozone in obese carboxypeptidase E-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2006;290:R126–33. doi: 10.1152/ajpregu.00306.2005. [DOI] [PubMed] [Google Scholar]
- 38.Salazar E, Knowles JH. An Analysis of Pressure-Volume Characteristics of the Lungs. J Appl Physiol. 1964;19:97–104. doi: 10.1152/jappl.1964.19.1.97. [DOI] [PubMed] [Google Scholar]
- 39.Shore SA, Williams ES, Zhu M. No effect of metformin on the innate airway hyperresponsiveness and increased responses to ozone observed in obese mice. J Appl Physiol. 2008;105:1127–33. doi: 10.1152/japplphysiol.00117.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rivera-Sanchez YM, Johnston RA, Schwartzman IN, Valone J, Silverman ES, Fredberg JJ, et al. Differential effects of ozone on airway and tissue mechanics in obese mice. J Appl Physiol. 2004;96:2200–6. doi: 10.1152/japplphysiol.00960.2003. [DOI] [PubMed] [Google Scholar]
- 41.Hantos Z, Daroczy B, Suki B, Nagy S, Fredberg JJ. Input impedance and peripheral inhomogeneity of dog lungs. J Appl Physiol. 1992;72:168–78. doi: 10.1152/jappl.1992.72.1.168. [DOI] [PubMed] [Google Scholar]
- 42.Shore SA, Williams ES, Chen L, Benedito LA, Kasahara DI, Zhu M. Impact of aging on pulmonary responses to acute ozone exposure in mice: role of TNFR1. Inhal Toxicol. 2011;23:878–88. doi: 10.3109/08958378.2011.622316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N, et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J Biol Chem. 2010;285:6153–60. doi: 10.1074/jbc.M109.088708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 45.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li C, Hung Wong W. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2:RESEARCH0032. doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A. 2001;98:31–6. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 49.Katsuki A, Sumida Y, Murashima S, Murata K, Takarada Y, Ito K, et al. Serum Levels of Tumor Necrosis Factor-α Are Increased in Obese Patients with Noninsulin-Dependent Diabetes Mellitus. Journal of Clinical Endocrinology & Metabolism. 1998;83:859–62. doi: 10.1210/jcem.83.3.4618. [DOI] [PubMed] [Google Scholar]
- 50.Yamakawa T, Tanaka S-I, Yamakawa Y, Kiuchi Y, Isoda F, Kawamoto S, et al. Augmented Production of Tumor Necrosis Factor-α in Obese Mice. Clinical Immunology and Immunopathology. 1995;75:51–6. doi: 10.1006/clin.1995.1052. [DOI] [PubMed] [Google Scholar]
- 51.Mei J, Liu Y, Dai N, Hoffmann C, Hudock KM, Zhang P, et al. Cxcr2 and Cxcl5 regulate the IL-17/G-CSF axis and neutrophil homeostasis in mice. The Journal of Clinical Investigation. 2012;122:974–86. doi: 10.1172/JCI60588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richter M, Cloutier S, Sirois P. Endothelin, PAF and thromboxane A2 in allergic pulmonary hyperreactivity in mice. Prostaglandins Leukot Essent Fatty Acids. 2007;76:299–308. doi: 10.1016/j.plefa.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 53.Landgraf RG, Jancar S. Endothelin A receptor antagonist modulates lymphocyte and eosinophil infiltration, hyperreactivity and mucus in murine asthma. Int Immunopharmacol. 2008;8:1748–53. doi: 10.1016/j.intimp.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 54.Bennedich Kahn L, Gustafsson LE, Olgart Hoglund C. Brain-derived neurotrophic factor enhances histamine-induced airway responses and changes levels of exhaled nitric oxide in guinea pigs in vivo. Eur J Pharmacol. 2008;595:78–83. doi: 10.1016/j.ejphar.2008.07.041. [DOI] [PubMed] [Google Scholar]
- 55.Tighe RM, Li Z, Potts EN, Frush S, Liu N, Gunn MD, et al. Ozone inhalation promotes CX3CR1-dependent maturation of resident lung macrophages that limit oxidative stress and inflammation. J Immunol. 2011;187:4800–8. doi: 10.4049/jimmunol.1101312. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 56.Lajoie S, Lewkowich IP, Suzuki Y, Clark JR, Sproles AA, Dienger K, et al. Complement-mediated regulation of the IL-17A axis is a central genetic determinant of the severity of experimental allergic asthma. Nat Immunol. 2010;11:928–35. doi: 10.1038/ni.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang K, Rabold R, Abston E, Schofield B, Misra V, Galdzicka E, et al. Effects of leptin deficiency on postnatal lung development in mice. J Appl Physiol. 2008;105:249–59. doi: 10.1152/japplphysiol.00052.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rochester DF, Enson Y. Current concepts in the pathogenesis of the obesity-hypoventilation syndrome. Mechanical and circulatory factors. Am J Med. 1974;57:402–20. doi: 10.1016/0002-9343(74)90135-1. [DOI] [PubMed] [Google Scholar]
- 59.Ray CS, Sue DY, Bray G, Hansen JE, Wasserman K. Effects of obesity on respiratory function. Am Rev Respir Dis. 1983;128:501–6. doi: 10.1164/arrd.1983.128.3.501. [DOI] [PubMed] [Google Scholar]
- 60.Johnston RA, Zhu M, Rivera-Sanchez YM, Lu FL, Theman TA, Flynt L, et al. Allergic airway responses in obese mice. Am J Respir Crit Care Med. 2007;176:650–8. doi: 10.1164/rccm.200702-323OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wagers SS, Haverkamp HC, Bates JH, Norton RJ, Thompson-Figueroa JA, Sullivan MJ, et al. Intrinsic and antigen-induced airway hyperresponsiveness are the result of diverse physiological mechanisms. J Appl Physiol. 2007;102:221–30. doi: 10.1152/japplphysiol.01385.2005. [DOI] [PubMed] [Google Scholar]
- 62.Yamakawa T, Tanaka S, Yamakawa Y, Kiuchi Y, Isoda F, Kawamoto S, et al. Augmented production of tumor necrosis factor-alpha in obese mice. Clin Immunol Immunopathol. 1995;75:51–6. doi: 10.1006/clin.1995.1052. [DOI] [PubMed] [Google Scholar]
- 63.Katsuki A, Sumida Y, Murashima S, Murata K, Takarada Y, Ito K, et al. Serum levels of tumor necrosis factor-alpha are increased in obese patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1998;83:859–62. doi: 10.1210/jcem.83.3.4618. [DOI] [PubMed] [Google Scholar]
- 64.Moon YS, Kim DH, Song DK. Serum tumor necrosis factor-alpha levels and components of the metabolic syndrome in obese adolescents. Metabolism. 2004;53:863–7. doi: 10.1016/j.metabol.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 65.Chen H, Tliba O, Van Besien CR, Panettieri RA, Jr, Amrani Y. TNF-[alpha] modulates murine tracheal rings responsiveness to G-protein-coupled receptor agonists and KCl. J Appl Physiol. 2003;95:864–72. doi: 10.1152/japplphysiol.00140.2003. discussion 3. [DOI] [PubMed] [Google Scholar]
- 66.Amrani Y, Panettieri RA, Jr, Frossard N, Bronner C. Activation of the TNF alpha-p55 receptor induces myocyte proliferation and modulates agonist-evoked calcium transients in cultured human tracheal smooth muscle cells. Am J Respir Cell Mol Biol. 1996;15:55–63. doi: 10.1165/ajrcmb.15.1.8679222. [DOI] [PubMed] [Google Scholar]
- 67.Kung TT, Luo B, Crawley Y, Garlisi CG, Devito K, Minnicozzi M, et al. Effect of Anti-mIL-9 Antibody on the Development of Pulmonary Inflammation and Airway Hyperresponsiveness in Allergic Mice. American Journal of Respiratory Cell and Molecular Biology. 2001;25:600–5. doi: 10.1165/ajrcmb.25.5.4533. [DOI] [PubMed] [Google Scholar]
- 68.Mould AW, Ramsay AJ, Matthaei KI, Young IG, Rothenberg ME, Foster PS. The Effect of IL-5 and Eotaxin Expression in the Lung on Eosinophil Trafficking and Degranulation and the Induction of Bronchial Hyperreactivity. The Journal of Immunology. 2000;164:2142–50. doi: 10.4049/jimmunol.164.4.2142. [DOI] [PubMed] [Google Scholar]
- 69.Sumarac-Dumanovic M, Stevanovic D, Ljubic A, Jorga J, Simic M, Stamenkovic-Pejkovic D, et al. Increased activity of interleukin-23/interleukin-17 proinflammatory axis in obese women. Int J Obes. 2008;33:151–6. doi: 10.1038/ijo.2008.216. [DOI] [PubMed] [Google Scholar]
- 70.Winer S, Paltser G, Chan Y, Tsui H, Engleman E, Winer D, et al. Obesity predisposes to Th17 bias. European Journal of Immunology. 2009;39:2629–35. doi: 10.1002/eji.200838893. [DOI] [PubMed] [Google Scholar]
- 71.Pini M, Fantuzzi G. Enhanced production of IL-17A during zymosan-induced peritonitis in obese mice. J Leukoc Biol. 2009;87:51–8. doi: 10.1189/jlb.0309188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zuniga LA, Shen W-J, Joyce-Shaikh B, Pyatnova EA, Richards AG, Thom C, et al. IL-17 Regulates Adipogenesis, Glucose Homeostasis, and Obesity. The Journal of Immunology. 2010;185:6947–59. doi: 10.4049/jimmunol.1001269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.He YW, Malek TR. Interleukin-7 receptor alpha is essential for the development of gamma delta + T cells, but not natural killer cells. J Exp Med. 1996;184:289–93. doi: 10.1084/jem.184.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Inagaki-Ohara K, Nishimura H, Mitani A, Yoshikai Y. Interleukin-15 preferentially promotes the growth of intestinal intraepithelial lymphocytes bearing gamma delta T cell receptor in mice. Eur J Immunol. 1997;27:2885–91. doi: 10.1002/eji.1830271121. [DOI] [PubMed] [Google Scholar]
- 75.Kudo M, Melton AC, Chen C, Engler MB, Huang KE, Ren X, et al. IL-17A produced by [alpha][beta] T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat Med. 2012 doi: 10.1038/nm.2684. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Willett WC, Dietz WH, Colditz GA. Guidelines for healthy weight. N Engl J Med. 1999;341:427–34. doi: 10.1056/NEJM199908053410607. [DOI] [PubMed] [Google Scholar]
- 77.Burger CD, Foreman AJ, Miller DP, Safford RE, McGoon MD, Badesch DB. Comparison of body habitus in patients with pulmonary arterial hypertension enrolled in the Registry to Evaluate Early and Long-term PAH Disease Management with normative values from the National Health and Nutrition Examination Survey. Mayo Clin Proc. 2011;86:105–12. doi: 10.4065/mcp.2010.0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kleemann R, Zadelaar S, Kooistra T. Cytokines and atherosclerosis: a comprehensive review of studies in mice. Cardiovasc Res. 2008;79:360–76. doi: 10.1093/cvr/cvn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nishina PM, Lowe S, Wang J, Paigen B. Characterization of plasma lipids in genetically obese mice: the mutants obese, diabetes, fat, tubby, and lethal yellow. Metabolism. 1994;43:549–53. doi: 10.1016/0026-0495(94)90194-5. [DOI] [PubMed] [Google Scholar]
- 80.Hakala K, Mustajoki P, Aittomaki J, Sovijarvi AR. Effect of weight loss and body position on pulmonary function and gas exchange abnormalities in morbid obesity. Int J Obes Relat Metab Disord. 1995;19:343–6. [PubMed] [Google Scholar]
- 81.Rorvik S, Bo G. Lung volumes and arterial blood gases in obesity. Scand J Respir Dis Suppl. 1976;95:60–4. [PubMed] [Google Scholar]
- 82.Adiarto S, Emoto N, Iwasa N, Yokoyama M. Obesity-induced upregulation of myocardial endothelin-1 expression is mediated by leptin. Biochem Biophys Res Commun. 2007;353:623–7. doi: 10.1016/j.bbrc.2006.12.066. [DOI] [PubMed] [Google Scholar]
- 83.Silver AE, Beske SD, Christou DD, Donato AJ, Moreau KL, Eskurza I, et al. Overweight and obese humans demonstrate increased vascular endothelial NAD(P)H oxidase-p47(phox) expression and evidence of endothelial oxidative stress. Circulation. 2007;115:627–37. doi: 10.1161/CIRCULATIONAHA.106.657486. [DOI] [PubMed] [Google Scholar]
- 84.Pontiroli AE, Pizzocri P, Koprivec D, Vedani P, Marchi M, Arcelloni C, et al. Body weight and glucose metabolism have a different effect on circulating levels of ICAM-1, E-selectin, and endothelin-1 in humans. Eur J Endocrinol. 2004;150:195–200. doi: 10.1530/eje.0.1500195. [DOI] [PubMed] [Google Scholar]
- 85.Ferri C, Bellini C, Desideri G, Baldoncini R, Properzi G, Santucci A, et al. Circulating endothelin-1 levels in obese patients with the metabolic syndrome. Exp Clin Endocrinol Diabetes. 1997;105 (Suppl 2):38–40. doi: 10.1055/s-0029-1211794. [DOI] [PubMed] [Google Scholar]
- 86.Kahaleh MB, Fan PS. Effect of cytokines on the production of endothelin by endothelial cells. Clin Exp Rheumatol. 1997;15:163–7. [PubMed] [Google Scholar]
- 87.Lees DM, Pallikaros Z, Corder R. The p55 tumor necrosis factor receptor (CD120a) induces endothelin-1 synthesis in endothelial and epithelial cells. Eur J Pharmacol. 2000;390:89–94. doi: 10.1016/s0014-2999(00)00022-4. [DOI] [PubMed] [Google Scholar]
- 88.Sibson NR, Blamire AM, Perry VH, Gauldie J, Styles P, Anthony DC. TNF-alpha reduces cerebral blood volume and disrupts tissue homeostasis via an endothelin- and TNFR2-dependent pathway. Brain. 2002;125:2446–59. doi: 10.1093/brain/awf256. [DOI] [PubMed] [Google Scholar]
- 89.Cardillo C, Campia U, Iantorno M, Panza JA. Enhanced vascular activity of endogenous endothelin-1 in obese hypertensive patients. Hypertension. 2004;43:36–40. doi: 10.1161/01.HYP.0000103868.45064.81. [DOI] [PubMed] [Google Scholar]
- 90.Mather KJ, Mirzamohammadi B, Lteif A, Steinberg HO, Baron AD. Endothelin contributes to basal vascular tone and endothelial dysfunction in human obesity and type 2 diabetes. Diabetes. 2002;51:3517–23. doi: 10.2337/diabetes.51.12.3517. [DOI] [PubMed] [Google Scholar]
- 91.Ahlborg G, Shemyakin A, Bohm F, Gonon A, Pernow J. Dual endothelin receptor blockade acutely improves insulin sensitivity in obese patients with insulin resistance and coronary artery disease. Diabetes Care. 2007;30:591–6. doi: 10.2337/dc06-1978. [DOI] [PubMed] [Google Scholar]
- 92.Weil BR, Westby CM, Van Guilder GP, Greiner JJ, Stauffer BL, DeSouza CA. Enhanced endothelin-1 system activity with overweight and obesity. Am J Physiol Heart Circ Physiol. 2011;301:H689–95. doi: 10.1152/ajpheart.00206.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Braun A, Lommatzsch M, Neuhaus-Steinmetz U, Quarcoo D, Glaab T, McGregor GP, et al. Brain-derived neurotrophic factor (BDNF) contributes to neuronal dysfunction in a model of allergic airway inflammation. British Journal of Pharmacology. 2004;141:431–40. doi: 10.1038/sj.bjp.0705638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Prakash YS, Thompson MA, Pabelick CM. Brain-derived neurotrophic factor in TNF-alpha modulation of Ca2+ in human airway smooth muscle. Am J Respir Cell Mol Biol. 2009;41:603–11. doi: 10.1165/rcmb.2008-0151OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Matera MG, Calzetta L, Cazzola M. TNF-alpha inhibitors in asthma and COPD: We must not throw the baby out with the bath water. Pulmonary Pharmacology & Therapeutics. 2010;23:121–8. doi: 10.1016/j.pupt.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 96.Van Veen IH, Ten Brinke A, Sterk PJ, Rabe KF, Bel EH. Airway inflammation in obese and nonobese patients with difficult-to-treat asthma. Allergy. 2008;63:570–4. doi: 10.1111/j.1398-9995.2007.01597.x. [DOI] [PubMed] [Google Scholar]