

Abstract
TAS-102 is an oral anticancer drug composed of trifluorothymidine (TFT) and TPI (an inhibitor of thymidine phosphorylase that strongly inhibits the biodegradation of TFT). Similar to 5-fluorouracil (5FU) and 5-fluoro-2′-deoxyuridine (FdUrd), TFT also inhibits thymidylate synthase (TS), a rate-limiting enzyme of DNA biosynthesis, and is incorporated into DNA. TFT exhibits an anticancer effect on colorectal cancer cells that have acquired 5FU and/or FdUrd resistance as a result of the overexpression of TS. Therefore, we examined the mode of action of TFT-induced DNA damage after its incorporation into DNA. When HeLa cells were treated with TFT, the number of ring-open aldehyde forms at apurinic/apyrimidinic sites increased in a dose-dependent manner, although we previously reported that no detectable excisions of TFT paired to adenine were observed using uracil DNA glycosylases, thymine DNA glycosylase or methyl-CpG binding domain 4 and HeLa whole cell extracts. To investigate the functional mechanism of TFT-induced DNA damage, we measured the phosphorylation of ATR, ATM, BRCA2, chk1 and chk2 in nuclear extracts of HeLa cells after 0, 24, 48 or 72 h of exposure to an IC50 concentration of TFT, FdUrd or 5FU using Western blot analysis or an enzyme-linked immunosorbent assay (ELISA). Unlike FdUrd and 5FU, TFT resulted in an earlier phosphorylation of ATR and chk1 proteins after only 24 h of exposure, while phosphorylated ATM, BRCA2 and chk2 proteins were detected after more than 48 h of exposure to TFT. These results suggest that TFT causes single-strand breaks followed by double-strand breaks in the DNA of TFT-treated cells. TFT (as TAS-102) showed a more potent antitumor activity than oral 5FU on CO-3 colon cancer xenografts in mice, and such antitumor potency was supported by the increased number of double-strand breaks occurring after single-strand breaks in the DNA of the TFT-treated tumors. These results suggest that TFT causes single-strand breaks after its incorporation into DNA followed by double-strand breaks, resulting in DNA damage. This effect of TFT on DNA may explain its potent anticancer activity in cancer therapy.
Keywords: trifluorothymidine, 5-fluorouracil, 5-fluoro-2′-deoxyuridine, DNA double-strand break
Introduction
Trifluorothymidine (TFT), which was originally synthesized by Heidelberger et al (1), has been shown to exert a potent suppressive effect on many transplanted tumors in mice. TFT is reportedly phosphorylated by thymidine kinase to yield TFT-monophosphate (2), which directly inhibits thymidylate synthase (TS) in the absence of reduced folate (3,4). The antimetabolite 5-fluorouracil (5FU), which is currently used to treat patients with gastric, colorectal, head/neck, breast and other types of cancer, is also thought to exert its antitumor activity mainly by inhibiting TS via the formation of a ternary-complex with methylenetetrahydrofolate and 5-fluorodeoxyuridine monophosphate (FdUMP) derived from 5FU (5). However, we found that when tumor cells were treated with higher concentrations of TFT for short periods, TFT was rapidly incorporated into the DNA and exerted a cytocidal activity by causing DNA fragmentation rather than by inhibiting TS in the tumor cells (6–8). Based on these observations, TAS-102 was developed as a novel anticancer drug composed of a combination of TFT and TPI, a specific inhibitor of thymidine phosphorylase that strongly inhibits the biodegradation of TFT. TPI is expected to reinforce the uptake of TFT into tumor DNA, thereby promoting the function of TFT against cancer cells with high expression levels of TS that have a low sensitivity and/or are resistant to 5FU.
We previously reported (9) that no detectable excisions of TFT paired to adenine were observed using uracil DNA glycosylases (UDG), thymine DNA glycosylase (TDG), methyl-CpG binding domain 4 (MBD4) and HeLa whole cell extracts. However, TDG and MBD4 were able to excise the TFT paired to guanine in DNA. Additional data also indicated that the small-interfering RNA-mediated knockdown of TDG or MBD4 significantly increased resistance to the cytotoxic effects of 5-fluoro-2′-deoxyuridine (FdUrd), but not to those of TFT. These data suggest that the inhibitory effects of TFT on DNA replication and repair enzymes are apparently distinct from those of 5FU and FdUrd.
In the present study, we showed that the double-strand breaks induced by TFT in the DNA of tumor cells were important for the exertion of the potent anticancer activity of TFT.
Materials and methods
Materials
FdUrd and EMEM were obtained from Sigma-Aldrich Japan (Tokyo, Japan). 5FU was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). TFT and TPI [5-chloro-6-(2-iminopyrrolidin-1-yl) methyl-2,4 (1H,3H)-pyrimidinedione hydrochloride] were synthesized at Taiho Pharmaceutical Co., Ltd. (Tokyo, Japan). Capecitabine, an oral 5FU prodrug, was obtained from BePharm Ltd. (Shanghai, China). TAS-102 was prepared by mixing TFT and TPI at a molar ratio of 1:0.5 in 0.5% hydroxypropyl methylcellulose. Fetal calf serum (FCS) was obtained from JRH Bioscience (Lenexa, KS, USA).
Cell lines and cultures
The HeLa human cervical cancer cell line was purchased from Dai-Nippon Pharmaceutical Co., Ltd. (Osaka, Japan) and cultured in EMEM medium with 10% FCS.
Apurinic/apyrimidinic site measurements
The number of apurinic/apyrimidinic (AP) sites was determined using a commercially available DNA damage quantification kit (Dojindo Molecular Technology, Kumamoto, Japan). Briefly, a Get pure DNA kit (Dojindo Molecular Technology) was used to extract and purify the genomic DNA from the indicated HeLa cell line according to the manufacturer's specifications. Purified DNA (1 μg) was then incubated for 1 h at 37°C with an aldehyde reactive probe (ARP) reagent (N'-aminooxymethyl-carbonyl-hydrazino-D-biotin), which reacts specifically with the ring-open aldehyde form of an AP site (10). After an overnight fixation step, biotin-tagged AP sites were quantified using colorimetric detection with peroxidase-conjugated streptavidin at 450 nm with a SpectraMax340 microplate spectrophotometer (Molecular Devices).
Western blot analysis
Nuclear cell extracts were obtained using an NE-PER Nuclear and Cytoplasmic Extraction kit (Thermo Fisher Scientific, MA, USA). Western blot analysis was performed using anti-phospho-ATM, anti-phospho-ATR, anti-phospho-chk1 and anti-phospho-chk2 (Cell Signaling Technology, MA, USA), as well as anti-phospho-BRCA2 (Millipore, CA, USA). A total protein control for phosphorylated protein was performed using anti-ATM, anti-ATR, anti-BRCA2, anti-chk1 and anti-chk2 (Cell Signaling Technology).
Evaluation of antitumor activity
Five-week-old male nude mice (BALB/c nu/nu) were purchased from Clea Japan, Inc. (Tokyo, Japan) and raised in the specific pathogen-free animal quarters of our laboratory. The CO-3 human colorectal cancer cell line was obtained from the Central Institute for Experimental Animals (Kawasaki, Japan). Tumor specimens (∼3-mm cubic fragments) were transplanted subcutaneously into the dorsal region of each animal. Approximately 1 week later, the animals were grouped according to tumor size, so that the mean and standard deviation of the tumor volume would be as uniform as possible in all of the groups. The tumor volume was calculated using the equation: (V) = 1/2 × S (shorter diameter)2 × L (longer diameter). TAS-102 and capecitabine were administered orally once daily for 14 consecutive days. The rate of tumor growth (IR) inhibition was regarded as a measure of the antitumor efficacy and was calculated using the relative tumor volume (RTV) in the drug-treated groups (T) compared to that in the control group (C) using the following equation: IR (%) = (1 – T/C) × 100. All of the protocols and procedures were approved by the Institutional Animal Care and Use Committee of Taiho Pharmaceuticals.
Pulse field gel electrophoresis
To detect high molecular weight DNA fragmentation, genomic DNA in the CO-3 tumor cells was extracted and purified using a Get pure DNA kit, loaded with exactly 1.2% agarose and 10 μg of DNA in each well; the DNA was then separated using pulse field gel electrophoresis (PFGE) using a CHEF-DR II system (Bio-Rad Laboratories, Tokyo, Japan) at 1.5 V/cm for 20 h at 25°C in 0.5X TBE buffer as previously described (11). A comparison of the DNA concentrations among the different samples was performed using ethidium bromide staining. The intensity of each band was quantified fluorometrically, and the percentage of double-strand break fragments vs. the number in an untreated control was expressed for each lane.
Statistical analysis
Data concerning the relative tumor volume for all of the mice in all groups were analyzed using a MultiStaff program (Dunnett's t-test for comparison among multiple groups) to test for the significance of inter-group differences in antitumor efficacy. Data for groups in which premature deaths occurred were excluded from this analysis.
Results
DNA damage by TFT or FdUrd on AP sites in HeLa cellular DNA
Since AP sites are an intermediate of many DNA N-glycosylases, the number of AP sites estimated using the APR assay can be a useful indicator of the level of DNA damage in the cells. Similar to FdUrd, TFT induced DNA damage in a concentration-dependent manner (Fig. 1).
Figure 1.
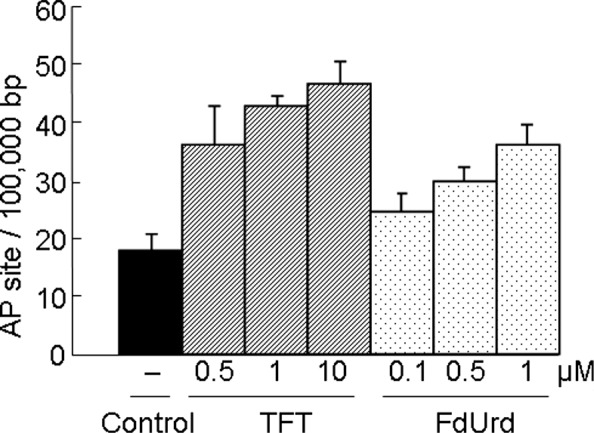
HeLa cells were treated with an IC50 concentration of TFT (0.5, 1 or 10 μM) or FdUrd (0.1, 0.5 or 1 μM). After 72 h, the cells were harvested in PBS, and their genomic DNA was extracted as described in Materials and methods. The number of AP sites was determined using an ARP reagent (N'-aminooxymethyl-carbonyl-hydrazino-D-biotin) and an ELISA. The IC50 value using the MTT assay for 72 h in HeLa cells was 1 μM for TFT and 0.5 μM for FdUrd. The values are the mean ± SD (n=6).
Phosphorylation of ATR, ATM and BRCA2
To investigate the mechanism of TFT-induced DNA damage, we measured the phosphorylation of ATR, ATM and BRCA2 using Western blot analysis after 0, 24, 48 or 72 h of exposure to IC50 concentrations of TFT, FdUrd or 5FU in HeLa cell lysates (Fig. 2). The phosphorylation of ATR was detected after 24 h of exposure to TFT, but ATM and BRCA2 were detected after >48 h of exposure to TFT. Exposure to FdUrd resulted in the weak phosphorylation of ATR at 24 h, while phosphorylated ATM was not observed between 24 and 72 h. The phosphorylations of ATR, ATM and BRCA2 by 5FU was modestly detected between 24 and 48 h.
Figure 2.
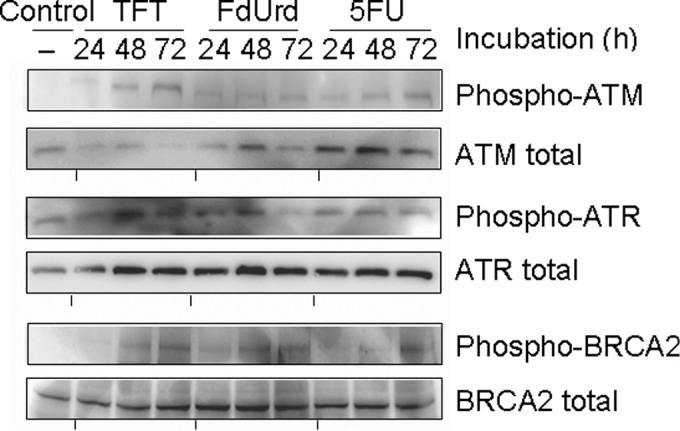
Western blot analysis of the phosphorylation levels for ATM, ATR and BRCA3 after 24, 48 or 72 h of exposure to the IC50 concentrations of TFT, FdUrd or 5FU in HeLa cell nuclear extracts. The blots are representative of three independent experiments.
Phosphorylation of chk1 and chk2 as observed using Western blot analysis
To investigate the checkpoint mechanism of TFT-induced DNA damage, we measured the phosphorylation of chk1 and chk2 using Western blot analysis after 0, 24, 48 or 72 h of exposure to IC50 concentrations of TFT, FdUrd or 5FU in HeLa cell lysates (Fig. 3). The phosphorylation of chk1 protein was detected after 24 h of exposure to TFT, while the phosphorylation of chk2 was only detected after 48 h of exposure to TFT. By contrast, exposure to FdUrd or 5FU did not result in the phosphorylation of chk1 at 48 h.
Figure 3.
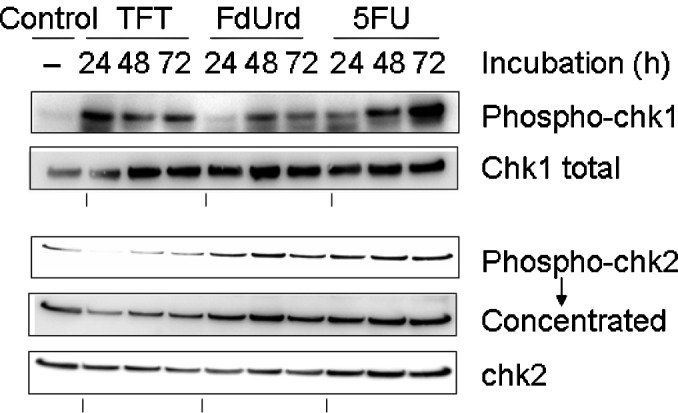
Western blot analysis of the phosphorylation levels for the cell cycle-regulating proteins chk1 and chk2 after 24, 48 or 72 h of exposure to the IC50 concentrations of TFT, FdUrd or 5FU in HeLa cell nuclear extracts. The blots are representative of three independent experiments.
Antitumor activity of TAS-102 and capecitabine against colorectal cancer CO-3 xenografts in a nude mouse model
The antitumor activities of TFT (as TAS-102) and capecitabine against CO-3 human colorectal carcinoma are shown in Table I. TFT was administered in combination with TPI as TAS-102 to increase the TFT levels in the serum. TAS-102 exhibited a dose-dependent and significant antitumor effect when administered orally at a dose of 75 or 150 mg/kg (Dunnett's t-test). Capecitabine also produced a significant antitumor effect compared to that in the untreated control group (Student's t-test). The weight loss at the end of the dosing period relative to the body weight at the beginning of the experiment was −7.86% in the untreated control group, and ranged from −10.35 to −13.17% in the oral administration group. The weight loss in all of the treatment groups did not differ significantly from that in the untreated control group.
Table I.
Antitumor effect of TAS-102 and capecitabine in human colorectal cancer CO-3 cells.
| Drug | Dose (mg/kg/day) | RTVa | IRb (%) | BWCc (%) |
|---|---|---|---|---|
| Control | - | 22.09±1.80 | - | −7.86 |
| TAS-102 | 75 | 12.27±0.86d | 44.5 | −10.35 |
| TAS-102 | 150 | 10.26±0.73d | 53.5 | −13.17 |
| Capecitabine | 360 | 13.62±0.97e | 38.4 | −11.06 |
Relative tumor volume on day 15.
Tumor growth inhibition rate {IR, % = [1− (average RTV in the treatment group/average RTV in the control group)] × 100}.
Percent weight change from pre-treatment on day 15.
P<0.01 vs. the control group (Dunnett's t-test).
P<0.01 vs. the control group (Student's t-test).
Effects of TAS-102 and capecitabine on AP sites and damage to bases in cellular DNA
To investigate whether the incorporation of TFT into DNA and the subsequent DNA damage may explain the potent antitumor activity of TFT, we first measured the number of AP sites and the amount of damage to the bases in cellular DNA from colorectal cancer CO-3 xenografts in a nude mouse model (Fig. 4). Similar to the in vitro activity (Fig. 1), the administration of TAS-102 and capecitabine significantly increased the number of AP sites and/or the amount of base damage formed in the cellular DNA of the CO-3 xenografts (Dunnett's t-test). These results for TFT-induced DNA damage are consistent with those of a previous report (8). The numbers of AP sites induced by TAS-102 (75 or 150 mg/kg) and capecitabine (360 mg/kg) were 40.9±16.3, 49.1±13.8 and 33.1±11.3 AP sites/100,000 bp, respectively; these values were higher than that for the untreated control group (15.2±10.2 AP sites/100,000 bp).
Figure 4.
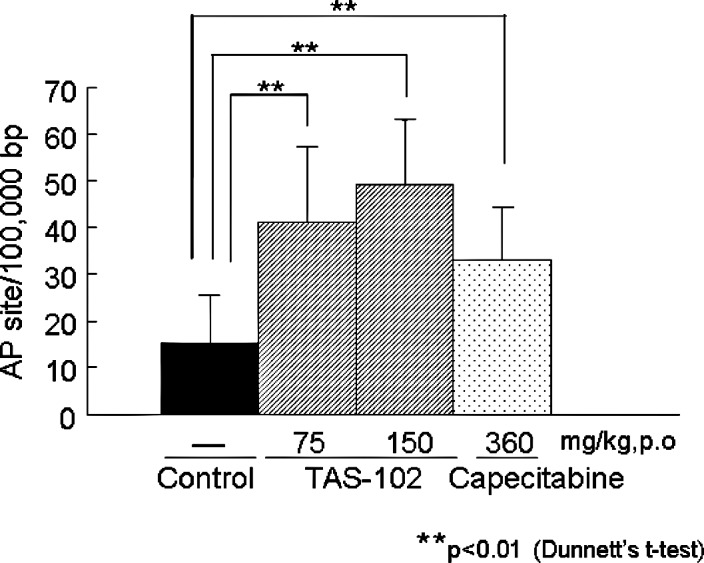
TAS-102 or capecitabine was administered orally for 14 consecutive days to nude mice transplanted with CO-3 xenografts. The tumors were collected and genomic DNA. The number of AP sites and/or the amount of base damage in the cellular DNA of the CO-3 tumor cells was measured, as described in Materials and methods. The values are the mean ± SD (n=8).
Effects of TAS-102 and capecitabine on double-strand DNA breaks
We previously reported that the DNA fragmentation rate for small molecular weight DNA in a TAS-102-treated group was significantly higher than that in an untreated control group in vivo (8). Therefore, in the present experiment, we measured the number of double-strand breaks in large molecular weight DNA using PFGE to investigate TFT-induced DNA damage in detail (Fig. 5). PFGE revealed more marked DNA smearing (>2 kbp) in the TAS-102-treated group than in any other group. The relative DNA fragmentation rates (DFR), compared to the control group for each fragmented area, were 126.1% in the TAS-102-treated group (150 mg/kg) and 93.3% in the capecitabine-treated group (360 mg/kg). The DFR% in the TAS-102-treated group was significantly higher than that in the control group. By contrast, the DFR% in the capecitabine-treated group did not differ significantly from that in the untreated control group.
Figure 5.

Double-strand breaks in the DNA were measured using pulse field gel electrophoresis (PFGE). DNA extracted from CO-3 cells was purified and separated using PFGE as described in Materials and methods. The intensity of each band was quantified, and the percentage of double-strand break fragments vs. that in the untreated control in each lane (DNA fragmentation rate, DFR) was calculated.
Discussion
5FU is widely used as an antitumor pyrimidine for the treatment of patients with gastric, colorectal, head and neck and breast cancers. The cytotoxic mechanism of 5FU involves the inhibition of TS activity by the formation of a ternary complex with 5-fluoro-2′-deoxyuridine-5′-monophosphate and methylenetetrahydrofolate as well as its incorporation into RNA and the subsequent inhibition of RNA maturation (12–15). Similar to 5FU, TFT reportedly exerts its cytocidal action by inhibiting TS (5) and/or through its incorporation into DNA (6,7). We previously demonstrated that when tumor cells are treated with high concentrations of TFT for short periods, TFT mainly manifests its antitumor activity though the induction of DNA fragmentation after its incorporation into the DNA of cancer cells (8). Therefore, we further examined the mode of action of TFT-induced DNA damage in cancer cells.
When HeLa cells were treated with TFT, β-elimination in the genomic DNA was detected in a dose-dependent manner (Fig. 1).
We showed in a previous report (9) that no detectable excisions of TFT paired to adenine were observed using UDG, TDG and MBD4, and HeLa whole cell extracts. However, TDG and MBD4 were able to excise the TFT paired to guanine in DNA. Although the precise mechanism for TFT-induced β-elimination in genomic DNA remains to be elucidated, the present results suggest the following points.
First, the majority of TFT is incorporated at T-sites (T/A, not T/G base pairs) in the cellular DNA. However, as TFT is massively incorporated into DNA, miss-incorporated TFT that has bound to guanine may be excised by TDG or MBD4.
Second, DNA-incorporated TFT may be converted into new structures, such as 5-carboxy-2′-deoxyuridine residues (16,17). The reactivity of the trifluoromethyl group of 5-trifluoro-methyluracil towards nucleophiles has been well documented (18). The metabolism of TFT incorporated into DNA requires further investigation. The repair of single- and double-strand breaks is critical for genetic integrity and cell survival. We showed that the phosphorylation of ATR and chk1 proteins in HeLa cells was detected after exposure to IC50 concentrations of TFT for more than 24 h, while the phosphorylation of ATM, BRCA2 and chk2 proteins in HeLa cells was only detected after more than 48 h of exposure to the same concentration of TFT (Fig. 3). The phosphorylation rates for chk1 and chk2 were consistent with those in a previous report (19). Thus, TFT apparently induces cell cycle arrest during the G2/M-phase of the cell cycle, while 5FU reportedly arrests cells during the G1 or S-phase (20). The G2/M-phase arrest induced by TFT may be accompanied by the activation of chk1, probably in an ATR-dependent manner during the initial step, as ATR initiates checkpoint responses to various agents that stall replication forks and damage DNA in multiple ways (21). The ATM kinase is activated by double-strand breaks, such as ionizing radiation. Furthermore, the ATM signals induce the association of homologous recombination repair components, such as Rad51, Xrcc3, BRCA2 and H2AX (22,23). Our results suggest that single-strand breaks induced by the incorporation of TFT into DNA may lead to to double-strand breaks when the cells progress to a subsequent DNA replication phase (24). In an in vivo experiment, TFT (as TAS-102) showed a more potent antitumor activity than the 5FU-derivative capecitabine when evaluated using equitoxic doses. Unlike capecitabine, this antitumor potency of TFT may originate from the double-strand breaks, rather than the single-strand breaks, in DNA. The presently reported functional mechanisms of TFT-induced DNA damage may be responsible for the potent anticancer activity of TFT against 5FU-resistant tumors.
References
- 1.Heidelberger C, Parsons DG, Remy DC. Syntheses of 5-trifluoromethyluracil and 5-trifluoromethyl-2′-deoxyuridine. J Med Chem. 1964;7:1–5. doi: 10.1021/jm00331a001. [DOI] [PubMed] [Google Scholar]
- 2.Bresnick E, Wiliams SS. Effects of 5-trifluoromethyldeoxy-uridine upon deoxythymidine kinase. Biochem Pharmacol. 1967;16:503–507. doi: 10.1016/0006-2952(67)90097-4. [DOI] [PubMed] [Google Scholar]
- 3.Reyers P, Heidelberger C. Fluorinated pyrimidines: XXVI. Mammalian thymidylate synthetase: its mechanism of action and inhibition by fluorinated nucleotides. Mol Pharmacol. 1965;1:14–30. [PubMed] [Google Scholar]
- 4.Eckstein JW, Foster PG, Finer-Moore J, Wataya Y, Santi DV. Mechanism-based inhibition of thymidylate synthase by 5-(trifluoromethyl)-2′-deoxyuridine 5′-monophosphate. Biochemistry. 1994;3:15086–15094. doi: 10.1021/bi00254a018. [DOI] [PubMed] [Google Scholar]
- 5.Umeda M, Heidelberger C. Comparative studies of fluorinated pyrimidines with various cell lines. Cancer Res. 1968;28:2529–2538. [PubMed] [Google Scholar]
- 6.Murakami Y, Kazuno H, Emura T, Tsujimoto H, Suzuki N, Fukushima M. Different mechanisms of acquired resistance to fluorinated pyrimidines in human colorectal cancer cells. Int J Oncol. 2000;17:277–283. doi: 10.3892/ijo.17.2.277. [DOI] [PubMed] [Google Scholar]
- 7.Emura T, Nakagawa F, Fujioka A, Ohshimo H, Yokogawa T, Okabe H, Kitazato K. An optimal dosing schedule for a novel combination antimetabolite, TAS-102, based on its intracellular metabolism and its incorporation into DNA. Int J Mol Med. 2004;13:249–255. [PubMed] [Google Scholar]
- 8.Emura T, Suzuki N, Yamaguchi M, Ohshimo H, Fukushima M. A novel combination antimetabolite, TAS-102, exhibits antitumor activity in FU-resistant human cancer cells through a mechanism involving FTD incorporation in DNA. Int J Oncol. 2004;25:571–578. [PubMed] [Google Scholar]
- 9.Suzuki N, Emura T, Fukushima M. Mode of action of trifluorothymidine (TFT) against DNA replication and repair enzymes. Int J Oncol. doi: 10.3892/ijo.2011.1003. (In press) [DOI] [PubMed] [Google Scholar]
- 10.Nakamura J, Walger VE, Upton PB, Chiang SY, Kow YW, Swenberg JA. Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Cancer Res. 1998;58:222–225. [PubMed] [Google Scholar]
- 11.Nishikawa T, Munshi A, Story MD, Ismail S, Stevens C, Chada S, Meyn RE. Adenoviral-mediated mda-7 expression suppresses DNA repair capacity and radiosensitizes non-small-cell lung cancer cells. Oncogene. 2004;16:7125–7131. doi: 10.1038/sj.onc.1207917. [DOI] [PubMed] [Google Scholar]
- 12.Hartman KY, Heidelberger C. Studies on fluorinated pyrimidines: XIII. Inhibition of thymidylate synthase. J Biol Chem. 1961;236:3006–3013. [PubMed] [Google Scholar]
- 13.Santi DV, McHenry CS. 5-Fluoro-2′-deoxyuridinylate: covalent complex with thymidylate synthase. Proc Natl Acad Sci USA. 1972;69:1855–1857. doi: 10.1073/pnas.69.7.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glazer RI, Lloyd LS. Association of cell lethality with incorporation of 5-fluorouracil and 5-fluorouridine into nuclear RNA in human colon carcinoma cells in culture. Mol Pharmacol. 1982;21:468–473. [PubMed] [Google Scholar]
- 15.Greenhalgh DA, Parish JH. Effect of 5-fluorouracil on cytotoxicity and RNA metabolism in human colonic carcinoma cells. Cancer Chemother Pharmacol. 1989;25:37–44. doi: 10.1007/BF00694336. [DOI] [PubMed] [Google Scholar]
- 16.Rogers WI, Hartman AC, Palm PE, Okstein C, Kensler CJ. The fate of 5-trifluoromethyl-2′-deoxyuridine in monkeys, dogs, mice, and tumor-bearing mice. Cancer Res. 1969;29:953–961. [PubMed] [Google Scholar]
- 17.Dexter DL, Wolberg WH, Ansfield FJ, Helson L, Heidelberger C. The clinical pharmacology of 5-trifluoromethyl-2′-deoxyuridine. Cancer Res. 1972;32:247–53. [PubMed] [Google Scholar]
- 18.Suzuki N, Fukushima M. Simple and rapid enzymatic method for the synthesis of single-strand oligonucleotides containing trifluorothymidine. Nucleosides Nucleotides Nucleic Acids. 2010;29:896–904. doi: 10.1080/15257770.2010.535803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bijnsdorp IV, Kruyt FA, Gokoel S, Fukushima M, Peters GJ. Synergistic interaction between trifluorothymidine and docetaxel is sequence dependent. Cancer Sci. 2008;99:2302–2308. doi: 10.1111/j.1349-7006.2008.00963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Backus HH, Pinedo HM, Wouters D, Kuiper CM, Jansen G, van Groeningen CJ, Peters GJ. Differences in the induction of DNA damage, cell cycle arrest, and cell death by 5-fluorouracil and antifolates. Oncol Res. 2000;12:231–239. doi: 10.3727/096504001108747729. [DOI] [PubMed] [Google Scholar]
- 21.Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003;17:615–628. doi: 10.1101/gad.1067403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morrison C, Sonoda E, Takao N, Shinohara A, Yamamoto K, Takeda S. The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J. 2000;19:463–471. doi: 10.1093/emboj/19.3.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, Goodarzi AA, Krempler A, Jeggo PA, Löbrich M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009;28:3413–3427. doi: 10.1038/emboj.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008;9:297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]