

Abstract
We report the systematic rational design and synthesis of new monovalent Smac mimetics that bind preferentially to the BIR2 domain of the anti-apoptotic protein XIAP. Characterization of compounds in vitro (including 9i; ML101) led to the determination of key structural requirements for BIR2 binding affinity. Compounds 9h and 9j sensitized TRAIL-resistant breast cancer cells to apoptotic cell death, highlighting the value of these probe compounds as tools to investigate the biology of XIAP.
Programmed cell death plays an essential role in development, typically occurring in animal species by apoptosis.1 Defects in apoptosis are associated with many diseases characterized by either insufficient cell death (e.g. cancer) or excessive cell death (e.g. degenerative diseases).2,3 The inhibitor of apoptosis proteins (IAPs) contain ~70 amino acid motifs termed baculovirus IAP repeat (BIR) domains.4,5 These BIR domains are primarily responsible for the anti-apoptotic activity of IAPs due to their ability to bind and inhibit distinct caspases, cysteine-aspartyl proteases that are critical for the initiation and execution phases of apoptosis.6 Human X-linked inhibitor of apoptosis protein (XIAP) is the most potent caspase inhibitor in the IAP family.7 XIAP contains three BIR domains (designated 1, 2, and 3) which exhibit specificity for different caspases. A short linker peptide in the BIR2 region of XIAP mediates the interaction with the effector caspases-3 and -7, whereas the third BIR domain (BIR3) targets the initiator caspase-9.8,9,10 All apoptotic signaling, triggered via either the intrinsic or the extrinsic pathway, converges on caspases-3 or -7 and this points to the importance of developing novel chemical entities with preferential affinity for the BIR2 domain of XIAP.
In the intrinsic cell death pathway, apoptotic signaling is regulated by the mitochondrial protein Smac, an endogenous dimeric proapoptotic antagonist of XIAP. The release of Smac from the intermembrane space of the mitochondria into the cytosol perpetuates the apoptotic signal by competing with caspases for binding to XIAP.11 The four hydrophobic amino acids Ala-Val-Pro-Ile (AVPI) at the N-terminus of mature Smac bind to the surface groove on the BIR3 domain of XIAP, removing caspase-9 inhibition, and bind with lower affinity to the BIR2 domain, alleviating inhibition by caspase-3 and -7.12 Up-regulation of XIAP expression in tumors causes resistance to current chemotherapeutic agents, and thus inhibition of the protein-protein interaction between XIAP and caspases-3, -7 and -9 represents a promising approach for the treatment of cancer.13
In the past few years substantial efforts have focused on small molecule Smac mimetics that target the BIR3 domain of XIAP.14 Conversely, there have been only a few reports on the design and synthesis of compounds that effect inhibition by binding to the BIR2 domain of XIAP. Examples include bivalent dimers, macrocyclic peptides, polyphenylureas and the natural product delaquinium.15 While these compound classes exhibit interesting properties, including cellular activity, their utility as potential drug leads is limited by high molecular weight, low potency, poor solubility or other disadvantageous physicochemical properties. The present study was performed within the framework of the Molecular Libraries Probe Production Centers Network (MLPCN; http://mli.nih.gov/mli/mlpcn/) with the goal of developing novel BIR2-selective probes. Herein we report the rational design and SAR of low molecular weight tripeptide derivatives that bind to the BIR2 domain of XIAP with high affinity. We further demonstrate that these small molecule probes are effective tools for investigating the biology of XIAP in cells.
Monovalent Smac mimetics based on the AVPI tetrapeptide have previously been shown to inhibit the interaction of XIAP with caspase-9 by binding to the XIAP BIR3 domain. For our studies we used compounds exemplified by the structures shown in Fig. 1 as a starting point and employed a rational design approach to investigate the structural requirements for XIAP BIR2 vs. BIR3 domain potency.16
Figure 1.

Examples of monovalent tripeptide XIAP BIR3 inhibitors.
At the outset we developed a putative binding model based on the structures of 1a and 1b (Fig. 2). Our approach consisted of retaining the common structural features of 1a and 1b, namely the N-methyl alanine moiety at the P1 position and the proline residue at P3, while investigating the effects of varying the P2 position and the C-terminal substituent. Thus, our preliminary synthetic efforts were focused on: (1) investigating the optimal R2 and R4 substituents; and (2) studies to determine the effect of amino acid stereochemistry at the P2 and P3 positions on the potency of inhibitors at the BIR2 and BIR3 domains of XIAP.
Figure 2.

Initial tripeptide binding model.
The general procedure for the synthesis of analogues is summarized in Scheme 1. Condensation of amino acid derivative 2 with proline derivative 3, followed by acidic cleavage of the N-terminal Boc protecting group afforded dipeptide 4. Condensation of 4 with alanine derivative 5 followed by hydrogenolysis of the benzyl ester afforded tripeptide acid 6. Finally, condensation of 6 with the appropriate amine followed by removal of the Boc group provided the target XIAP inhibitors 7.17
Scheme 1.

General synthetic route for the synthesis of small molecule XIAP inhibitors. Reagents and conditions: (a) EDC, HOBt, N-methylmorpholine, DMF (b) TFA, CH2Cl2 (c) H2, Pd/C, MeOH.
All of the Smac mimetics synthesized were evaluated for their ability to bind the BIR2 or BIR3 domains of XIAP by employing fluorescence-polarization (FP) competition assays.18 The results are expressed as competitive inhibition constants (Ki), derived from the corresponding IC50 values by application of a mathematical equation developed by Wang and coworkers.19
In the initial phase of SAR studies we investigated the effects of different substituents at the P2 position on BIR2 and BIR3 potency. The results are summarized in Table 1. The previously reported compound 1a16a was synthesized and tested as a benchmark against which the new analogues were compared. Incorporation of cyclohexyl, phenyl, benzyl or ethyl as the R2 substituent provided compounds 8a–d which exhibited moderate affinity for BIR2 and good BIR3 binding affinity. A valine residue at the P2 position (8e) improved potency against both BIR2 and BIR3. Compounds 8f–8h that incorporate an amino acid with unnatural D stereochemistry were essentially inactive, demonstrating the importance of employing amino acids with natural L stereochemistry at P1, P2 and P3. Other R2 substitutents such as iso-butyl, sec-butyl or propylamide (compounds 8i–8k) did not improve on the BIR2 potency exhibited by 8e with Val at P2.
Table 1.
Binding affinities to BIR2 or BIR3 domains of XIAP for analogues 8a–8k
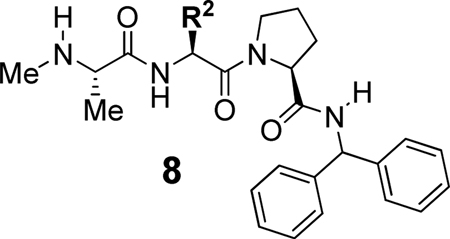 | |||
|---|---|---|---|
| Cmpds. | R2 | BIR2 Ki, µMa |
BIR3 Ki, µMa |
| 1a | iso-propyl | 1.36 (± 0.02) | 0.25 (± 0.03) |
| 8a | cyclohexyl | 3.58 (± 0.11) | 0.66 (± 0.11) |
| 8b | phenyl | 8.06 (± 0.28) | 0.66 (± 0.06) |
| 8c | benzyl | 9.07 (± 3.24) | 1.05 (± 0.31) |
| 8d | ethyl | 3.12 (± 0.69) | 0.70 (± 0.01) |
| 8e | iso-propyl | 1.84 (± 0.96) | 0.44 (± 0.13) |
| 8f | iso-propylb | > 56 | > 39 |
| 8g | iso-propylc | > 56 | > 39 |
| 8h | iso-propyld | > 56 | 29.88 (±0.17) |
| 8i | iso-butyl | 1.67 (± 0.11) | 0.35 (± 0.01) |
| 8j | sec-butyl | 4.41 (± 0.31) | 0.80 (± 0.01) |
| 8k | propylamide | 2.33 (± 0.45) | 0.65 (± 0.02) |
Values were determined in duplicate and reported as the Ki ±, standard deviation which is given in parentheses.
D valine at P2.
D proline at P3.
D alanine at P1.
We next investigated the effect of varying the C-terminal substituent (R4) on BIR2 and BIR3 potency and selectivity. For this phase the P1–P2–P3 tripeptide was retained as NMeAla-Val-Pro and the C-terminal was varied to determine the optimal R4 substituent. Compounds 9a–9j were synthesized and their BIR2 and BIR3 potencies determined in the FP assays (Table 2). The approximate BIR2:BIR3 selectivity for each compound is shown as a ratio in the last column of Table 2.
Table 2.
Binding affinities to BIR2 or BIR3 domains of XIAP for selected tripeptide analogues 9a–9j
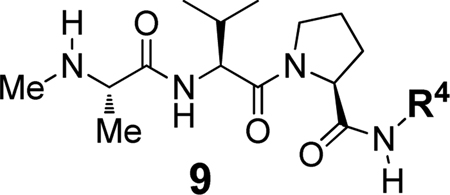 | ||||
|---|---|---|---|---|
| Cmpds. | R4 | BIR2 Ki, µMa |
BIR3 Ki, µMa |
BIR2 vs BIR3 Selectivity |
| 9a | 3.36 (± 0.96) | 0.59 (± 0.03) | 1 :6 | |
| 9b | 9.70 (± 1.32) | 5.62 (± 0.44) | 1 : 2 | |
| 9c | 10.93 (± 0.96) | 8.18 (± 0.45) | 1 : 1 | |
| 9d | 20.07 (± 1.71) | 26.15 (± 0.96) | 1 : 1 | |
| 9e | 2.01 (± 0.16) | 0.12 (± 0.02) | 1 : 17 | |
| 9f | 1.37 (± 0.11) | 0.055 (± 0.011) | 1 : 25 | |
| 9g | 4.97 (± 0.04) | 1.40 (± 0.22) | 1 : 4 | |
| 9h | 0.44 (± 0.02) | 1.51 (± 0.13) | 3 : 1 | |
| 9i | 1.91 (± 0.35) | 12.71 (± 0.81) | 7 : 1 | |
| 9j | 0.39 (± 0.17) | 1.08 (± 0.06) | 3 : 1 | |
Values are determined in duplicate and reported ± standard deviation which is given in parentheses.
As shown in Table 1, the benzylamine derivative 9a exhibited similar BIR2 and BIR3 potency (Ki = 3.36 µM and 0.59 µM) to the benzhydryl derivative 8e, while the isomeric pyridinylmethanamine derivatives 9b–9d were significantly less potent than 9a at both BIR2 and BIR3. However, despite the loss of potency, the BIR2 vs BIR3 selectivity ratio improved to 1:1 for 9c and 9d. The effect of a tetrahydronaphthyl-1-amine R4 substituent was investigated in compounds 9e–9g by analogy to compound 1b. As expected based on previous studies, these compounds exhibited good potency vs. BIR3, with 9f, containing the R-tetrahydronaphthyl-1-amine isomer, being especially potent against BIR3 (Ki = 0.055 µM). A breakthrough came when the tetrahydronaphthyl substituent was replaced with a 1-naphthyl group, as in 9h. Compound 9h displayed a significant potency boost for BIR2 (BIR2 Ki = 0.44 µM) and was 3-fold selective (BIR3 Ki = 1.51 µM). Interestingly, the quinolin-5-amine derivative 9i was less potent (BIR2 Ki = 1.91 µM; BIR3 Ki = 12.71 µM) but showed improved selectivity for BIR2 vs. BIR3 (7:1), suggesting that heteroatom incorporation may favor BIR2 selectivity. A second breakthrough occurred in the form of the phenylhydrazine derivative 9j. Thus, compound 9j was found to be potent (BIR2 Ki = 0.39 µM; BIR3 Ki = 1.08 µM) and three-fold selective for BIR2 vs. BIR3.
Based on the promising selectivity for BIR2 displayed by the quinoline derivative 9i we synthesized a small library of tripeptide derivatives with heteroaromatic (quinoline, isoquinoline or indole) C-terminal substituents. The structures and data for these analogues are shown in Table 3. Several of these compounds, including 9k–n, showed good selectivity for BIR2 although with lower overall potency at both BIR2 and BIR3. Other analogues, such as 9p–r, exhibited good potency but were not as selective as 9i. However, incorporation of an indole moiety at the C-terminal position provided a compound (9t20) that was both 7-fold selective for BIR2 and retained potency (BIR2 Ki = 0.74 µM).
Table 3.
Binding affinities to BIR2 or BIR3 domains of XIAP for selected heterocyclic analogues 9k–9t.
| Compds | R4 | BIR2 Ki, µMa |
BIR3 Ki, µMa |
BIR2 vs BIR3 selectivity ratio |
|---|---|---|---|---|
| 9k b | 7.20 (± 1.06) | 56.13 (± 10.73) | 8 : 1 | |
| 9l | 6.07 (± 0.08) | 37.06 (± 7.88) | 6 : 1 | |
| 9m | 4.94 (± 0.15) | 13.78 (± 0.64) | 3 : 1 | |
| 9n | 2.55 (± 0.13) | 16.21 (± 1.22) | 6 : 1 | |
| 9o | 1.40 (± 0.10) | 4.07 (± 0.07) | 3 : 1 | |
| 9p | 1.29 (± 0.05) | 1.50 (± 0.15) | 1 : 1 | |
| 9q | 1.29 (± 0.01) | 0.54 (± 0.01) | 1 : 2 | |
| 9r | 0.99 (± 0.08) | 0.42 (± 0.03) | 1 : 2 | |
| 9s | 4.75 (± 0.32) | 6.10 (± 0.10) | 1 : 1 | |
| 9t | 0.74 (± 0.08) | 5.5 (± 0.09) | 7 : 1 |
Values are determined in duplicate ± standard deviation which is given in parentheses.
In this case the residue employed at P2 was Abu instead of Val.
Structural modeling of 9t on the BIR2 domain was performed based on binding modes observed in BIR3/SMAC and BIR2 complexes8,12 and suggests a basis for the properties of the compound (Fig 3). Thus, compound 9t was first manually docked into the Smac-binding pocket of BIR2 based on available BIR3-Smac and BIR2-Caspase-3 structures followed by energetic minimization.21 The model suggests that BIR2 residues Lys 206 and Gln 197 contribute to the binding of the indole moiety of 9t which can readily be accommodated in the P4 pocket. In this binding mode the aliphatic stem of Lys 206 is poised to assist binding of the aromatic ring structure, while Gln 197 is positioned to form a hydrogen bond with the indole nitrogen. Comparison to the equivalent surface on BIR3 indicates that in BIR3 Lys 206 is replaced by a Glycine and Gln 197 by a Lysine residue. The resulting pocket is unlikely to accommodate the indole as favorably, accounting for the increased BIR2 selectivity of compound 9t.
Figure 3.

Model of compound 9t (magenta) bound to BIR2 (grey) with residues Lys 206 and Gln 197 depicted in yellow.
To assess the activity of the compounds in a relevant cellular context their ability to sensitize TRAIL-resistant cells to apoptosis was investigated. Thus, MDA-MB-231 breast adenocarcinoma cells were incubated with the respective compounds as described for 4 h before adding varying concentrations of TRAIL for 20 h, at which time cell viability was assessed.22 Both 9h (BIR2 selective) and 9f (BIR3 selective) showed strong TRAIL sensitizing activity (Fig. 4). Compound 9j (BIR2 selective) showed a similar trend but was slightly less potent at the concentration used. An LD50 value could not be achieved in MDA-MB-231 cells with TRAIL alone but values of 12.2 ng/mL (0.68 nM), 11.3 ng/mL (0.63 nM) and 24.6 ng/mL (1.37 nM) were obtained with 5 µM 9h, 9f, and 9j, respectively. Most interestingly, these compounds showed little to no toxicity as single agents (data not shown) but rather sensitized the cells to TRAIL-induced apoptosis. Thus these agents are cell permeable TRAIL sensitizing agents that may serve as potential lead compounds in the search for clinically relevant drugs to circumvent TRAIL-resistant cancers.
Figure 4.

Apoptosis in TRAIL-resistant cells sensitized with compounds 9f, 9hor 9j.
In summary, using a rational design approach we have systematically optimized small molecule monovalent Smac mimetics that inhibit XIAP by binding preferentially to the BIR2 domain. Compounds 9h, 9j, and 9t bind to the BIR2 domain with submicromolar affinities and are 3- to 7-fold selective for BIR2 over BIR3. In addition, the XIAP inhibitory potency observed in vitro translates into cell killing activity in breast cancer cells, suggesting that these compounds are potentially useful tools for probing apoptosis signaling pathways in cells. Further optimization of these compounds towards the discovery of potent and selective drug-like analogues is currently in progress.
Acknowledgements
This work was supported by NIH grant HG005033 and R01AA017238 to S.J.R. The authors thank Andrey Bobkov for expert technical assistance. M.G.L. acknowledges Fundación Ramón Areces for a postdoctoral fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Riedl SJ, Salvesen GS. Nat. Rev. Mol. Cell. Bio. 2007;8:405. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- 2.Lowe SW, Lin AW. Carcinogenesis. 2000;21:485. doi: 10.1093/carcin/21.3.485. [DOI] [PubMed] [Google Scholar]
- 3.Okouchi M, Ekshyyan O, Maracine M, Aw TY. Antioxid. Redox. Signal. 2007;9:1059. doi: 10.1089/ars.2007.1511. [DOI] [PubMed] [Google Scholar]
- 4.Deveraux QL, Reed JC. Genes Dev. 1999;1:239. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 5.LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG. Oncogene. 2008;27:6252. doi: 10.1038/onc.2008.302. [DOI] [PubMed] [Google Scholar]
- 6.Pop C, Salvesen GS. J. Biol. Chem. 2009;284:21777. doi: 10.1074/jbc.R800084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schimmer AD, Dalili S, Batey RA, Riedl SJ. Cell Death Differ. 2006;13:179. doi: 10.1038/sj.cdd.4401826. [DOI] [PubMed] [Google Scholar]
- 8.Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC, Salvesen GS. Cell. 2001;104:791. doi: 10.1016/s0092-8674(01)00274-4. [DOI] [PubMed] [Google Scholar]
- 9.(a) Chai J, Shiozaki E, Srinivasula SM, Wu Q, Dataa P, Alnemri ES, Shi Y. Cell. 2001;104:769. doi: 10.1016/s0092-8674(01)00272-0. [DOI] [PubMed] [Google Scholar]; (b) Huang Y, Park YC, Rich RL, Segal D, Myszka DG, Wu H. Cell. 2001;104:781. [PubMed] [Google Scholar]
- 10.Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, Lee R-A, Robbins PD, Fernandes-Alnemri T, Shi Y, Alnemri ES. Nature. 2001;410:112. doi: 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- 11.Shiozaki EN, Shi Y. Trends Biochem. Sci. 2004;39:486. doi: 10.1016/j.tibs.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 12.(a) Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T, Herrmann J, Wu JC, Fesik SW. Nature. 2000;408:1004. doi: 10.1038/35050006. [DOI] [PubMed] [Google Scholar]; (b) Wu G, Chai J, Suber TL, Wu J-W, Du C, Wang X, Shi Y. Nature. 2000;408:1008. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 13.Dean EJ, Ranson M, Blackhall F, Dive C. Expert Opin. Ther. Targets. 2007;11:1459. doi: 10.1517/14728222.11.11.1459. [DOI] [PubMed] [Google Scholar]
- 14.(a) Sun H, Nikolovska-Coleska Z, Yang C-Y, Qian D, Lu J, Qiu S, Bai L, Peng Y, Cai Q, Wang S. Acc. Chem. Res. 2008;41:1264. doi: 10.1021/ar8000553. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mannhold R, Fulda S, Carosati E. Drug Discov. Today. 2010;15:210. doi: 10.1016/j.drudis.2010.01.003. [DOI] [PubMed] [Google Scholar]; (c) Flygare JA, Fairbrother WJ. Expert Opin. Ther. Pat. 2010;20:251. doi: 10.1517/13543770903567077. [DOI] [PubMed] [Google Scholar]
- 15.(a) Sun H, Liu L, Lu J, Bai L, Li X, Nikolovska-Coleska Z, McEachern D, Yang CY, Qiu S, Yi H, Sun D, Wang S. J. Med. Chem. 2011;54:3306. doi: 10.1021/jm101651b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sun H, Liu L, Lu J, Qiu S, Yang CY, Yi H, Wang S. Bioorg. Med. Chem. Lett. 2010;20:3043. doi: 10.1016/j.bmcl.2010.03.114. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schimmer AD, Welsh K, Pinilla C, Wang Z, Krajewska M, Bonneau MJ, Pedersen IM, Kitada S, Scott FL, Bailly-Maitre B, Glinsky G, Scudiero D, Sausville E, Salvesen G, Nefzi A, Ostresh JM, Houghten RA, Reed JC. Cancer Cell. 2004;5:25. doi: 10.1016/s1535-6108(03)00332-5. [DOI] [PubMed] [Google Scholar]; (d) Orzaez M, Gortat A, Sancho M, Carbajo RJ, Pineda-Lucena A, Palacios-Rodriguez Y, Perez-Paya E. Apoptosis. 2011;16:460. doi: 10.1007/s10495-011-0582-4. [DOI] [PubMed] [Google Scholar]
- 16.(a) Sun H, Nikolovska-Coleska Z, Chen J, Yang C-Y, Tomita Y, Pan H, Yoshioka Y, Krajewski K, Roller PP, Wang S. Bioorg. Med. Chem. Lett. 2005;15:793. doi: 10.1016/j.bmcl.2004.11.008. [DOI] [PubMed] [Google Scholar]; (b) Oost TK, Sun C, Armstrong RC, Al-Assaad A-S, Betz SF, Deckwerth TL, Ding H, Elmore SW, Meadows RP, Olejniczak ET, Oleksijew A, Oltersdorf T, Rosenberg SH, Shoemaker AR, Tomaselli KJ, Zou H, Fesik SW. J. Med. Chem. 2004;47:4417. doi: 10.1021/jm040037k. [DOI] [PubMed] [Google Scholar]
- 17.All synthesized compounds were purified by either medium pressure silica gel chromatography or preparative HPLC using a C18 column. Compound purity was determined using analytical HPLC/MS and NMR (1H and 13C). The yields for the first two steps ranged from 70 – 90% while the yields for the last step ranged from 20 – 90%. The purity of each final product was greater than 95% as determined by HPLC/MS analysis.
- 18.Fluorescence polarization assays were run in 20 µL volume in 384 well format with black plates. The assay solution was 25 mM Hepes at pH 7.5/1 mM tris(2-carboxyethyl)phosphine/0.005% Tween 20 and 20 nM AVPIAQK-rhodamine. XIAP BIR2 was present at 1 µM or XIAP BIR3 at 0.2 µM. Compound was present within a range of 100 µM to 6.1 nM using two fold dilutions starting at 100 µM generating a 16 point curve. Plates were read on an Analyst HT in fluorescence polarization mode with excitation at 530 nm, emission at 580 nm and a dichroic mirror at 565 nm. Resulting data in mP was fit in GraphPad Prism 5 with a non-linear regression using a sigmodial curve with variable slope.
- 19.Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, Roller PP, Krajewski K, Saito N, Stuckey J, Wang S. Anal. Biochem. 2004;332:261. doi: 10.1016/j.ab.2004.05.055. [DOI] [PubMed] [Google Scholar]
- 20.Spectroscopic data for selected compound 9t: 1H NMR (400 MHz, D2O) δ 7.52 (d, J= 1.8 Hz, 1H), 7.37 (d, J= 8.5 Hz, 1H), 7.28 (d, J= 3.1 Hz, 1H), 7.01 (dd, J= 9.1, 2.4 Hz, 1H), 6.43 (d, J= 3.1 Hz, 1H), 4.44 (d, J= 7.3 Hz, 1H), 4.39 (m, 1H), 3.83 (m, 2H), 3.65 (m, 1H), 2.56 (s, 3H), 2.05 (m, 2H), 1.94 (m, 2H), 1.38 (d, J= 6.7 Hz, 3H), 0.91 (d, J= 6.7 Hz, 3H), 0.83 (d, J= 6.7 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 169.6, 169.3, 132.6, 131.1, 127.4, 125.9, 114.5, 111.1, 110.4, 100.9, 60.2, 56.4, 55.9, 47.3, 38.2, 31.3, 30.0, 29.5, 24.7, 19.0, 18.2, 16.3. MS m/z= 414 (M+H).
- 21.The specific details of model-generation to illustrate the putative binding mode of compound 9t to XIAP BIR2 are as follows. The experimental basis for the approach was the prototypical structure of a Smac peptide (AVPI) bound to XIAP BIR312, and our previous crystal structure showing a “Smac-like” binding of the N-terminus of caspase-3 from a crystallographic neighbor to the Smac binding groove on XIAP BIR2 as described in ref. 8. The latter still represents the only experimental structural data available for Smac-like binding by the XIAP BIR2 domain. The BIR domains of both structures were overlaid using fitting algorithms from COOT and Pymol, which resulted in an overlay of the P1–P3 residues of bound SMAC (AVP) and caspase-3 (SGV) of excellent fidelity, and allowed us to place the SMAC sequence AVPI into the BIR2 SMAC binding groove. We utilized the close structural similarity of compound 9t and AVPI to model compound 9t onto BIR2 with backbone architecture and sidechain-like moieties of compound 9t in similar orientations to AVP, while the sidechain bearing the P4 indole was placed in the orientation observed for the Ile residue of AVPI. This procedure was followed by a final energetic and geometrical minimization step using Phenix23Comparison of this model with AVPI and the BIR3 SMAC binding groove pointed to a generally very similar putative binding of AVPI/compound 9t between XIAP BIR2 and 3 but revealed a different binding pocket composition in P4 as described in the text and illustrated in Fig. 3
- 22.Cell viability was assessed using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega Corp., Madison, WI). Briefly, cells were seeded at 5000 cells/well in 100 µL complete medium and allowed to attach overnight. The medium was aspirated and replaced with fresh media containing the specified drug at the concentrations described before re-incubation of the cells at 37°C for 4 h. TRAIL was then added to the desired final concentration and the cells again incubated at 37°C for 20 h. The plates were removed to room temperature for 10 mins before addition of one half volume (50 µL) of freshly prepared CellTiterGlo reagent. The plates were gently shaken to ensure complete cellular lysis before luminescence measurement on a Biotek Synergy 2 plate reader. All experiments were carried out in at least triplicate, at least three times. MDA-MB-231 cells were maintained in DMEM with 10% (v/v) FBS, and penicillin/streptomycin/L-Glutamine (Omega Scientific Inc, Tarzana, CA).
- 23.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta. Crystallogr. D Bio.l Crystallogr. 2010;66:213. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]