

Abstract
Atopic dermatitis is a skin condition resulting in a skin rash from exposure to environmental factors. Skin biopsies taken from patients suffering from atopic dermatitis were micro-dissected and analyzed using a microchip-based immunoaffinity CE system for the presence of CXCL1, CXCL5 and CXCL8 and CCL1, CCL3 and CCL5 chemokines. Disposable immunoaffinity disks with immobilized antibodies were used to capture the CXC and CC chemokines from the homogenized skin samples. The captured analytes were then labeled with AlexaFluor 633, eluted from the disk and separated by CE. The labeled chemokines were identified and quantified by laser induced fluorescence. The total analysis time was less than 40 min, including the biopsy microdissection, pre-analysis preparation of the sample and the ICE-CHIP analysis, which took less than 10 min with inter- and intra-assay CV's below 6.4%. Microchip-based immunoaffinity CE could distinguish between normal skin biopsies and those with inflammation. Patients with neutrophil cellular infiltrates by histopathology showed increased concentrations of CXCL1, CXCL5 and CXCL8 while increases of CCL1, CCL3 and CCL5 corresponded to the patient group demonstrating monocytic and T-lymphocyte infiltration by histopathology. This system demonstrates the ability to identify and quantify immunochemical analytes in frozen sections taken from clinical histopathology samples.
Keywords: Atopic dermatitis, Biopsy, Chemokines, Chip-based CE, Immunoaffinity
1. Introduction
Atopic dermatitis (AD) occurs when the skin of a patient has an abnormal reaction. AD is the most common and relapsing allergic disease of the skin [1]. The irritants can include food or environmental allergens, such as pollen or animal dander [2], and can leave the patient susceptible to bacterial infections. AD is characterized by eczematous changes in the epidermis [3]. AD usually presents in children and continues into adulthood, and while there is no cure, it can be effectively managed with treatment.
Histopathological examination of inflammation-associated dermal lesions provides valuable information for determining clinical diagnosis of the disease. However, such examinations assess the cellular composition of the lesion at the time of the biopsy and cannot predict the prognosis of the disease. The infiltration of the inflammatory cells is under the control of a number of factors including cytokines and especially a subgroup known as chemokines. Immunochemical assessment of these low molecular weight cytokines (8–10 kDa) could prove to be a better predictor of the future cellular events taking place within the lesion; infiltrating neutrophils leading to clinical inflammation, while infiltrating T-cells indicate allergic reactions with prolonged associated inflammation.
Chemokines are chemoattractants inducing the migration and activation of selected types of leukocytes into the lesion [4,5], thus perpetuating the inflammatory process. Several chemokines have been found to participate in leukocyte recruitment to the sites of inflammation in AD [6]. When T helper type 2 (Th2) cells are activated, the production of a proinflammatory cytokine and chemokine pattern sustains the persistence of inflammation [7]. AD-associated chemokines have been extensively studied in both adults and children and include cutaneous T-cell attracting cytokine (CTACK), macrophage-derived chemokine (MDC), thymus and activation-regulated chemokine (TARC) and leukotriene E4, although there are other biochemical markers that have not been well studied in this complex disease [8]. To date, none of these studies measure the chemokine concentrations in vivo.
Chemokines are divided into several families but of importance in this work are the CXC chemokines (IL-8, GRO alpha, ENA-78) which are reported to attract and activate neutrophils [9,10] and the CC chemokines (MIP-1 alpha, RANTES, I-309) which attract and activate monocytes, dendritic cells, T-lymphocytes, natural killer cells, B-lymphocytes, basophils and eosinophils [11,12]. Defining the concentrations of these two groups of chemokines, within the lesion itself, would greatly help in (a) determining the potential progression of the disease and (b) help determine the status in vivo. Such knowledge would greatly aid in the treatment regime used to control the disease.
Microdissection of inflammatory lesions, within the biopsy, and consequent immunochemical analysis of the resident chemokines could prove to be a superior approach to determining the prognosis of the disease. Our group has previously used this approach to define pro-inflammatory cytokines within defined histological areas of skin lesions [13]. Further, we have developed a chip-based immunoaffinity capillary electrophoresis (ICE) system to measure the recovered cytokines within the lesion [13]. Chip-based ICE exploits the selectivity of antibodies coupled to the high resolving power of CE in a microfluidic device. This method affords the analysis of complex mixtures of defined analytes within small samples such as histopathology tissue sections. In this communication, we have applied our chip ICE system to the measurement of chemokines within histologically defined lesions in AD. Current immunological thought indicates that the presence of certain early-onset chemokines may be of greater importance in diagnosis than the latter presence of pro-inflammatory cytokines [1,3,6].
2. Experimental section
2.1. Materials
Recombinant human chemokines (growth regulating oncogen alpha - CXCL1/GRO-α, cat # 275-GR-010/CF; epithelial cell-derived neutrophil-activating peptide 78 - CXCL5/ENA-78, cat # 254-XB-025/CF; interleukin-8 - CXCL8/IL-8, cat # 618-IL-010/CF; CCL1/I-309, cat # 272-I-010/CF; macrophage inflammatory protein 1 alpha - CCL3/MIP-1α, cat # 270-LD-010/CF; regulated upon activation, normal T cell expressed and secreted - CCL5/RANTES, cat # 278-RN-101/CF) and their specific reactive antibodies (anti-CXL1 cat # AF-272; anti-CXC-L5, cat # AF-254; anti-CXCL8, cat # AF-208-NA; anti-CCL1, cat # AF-275; anti-CCL3, cat # AF-270; anti-CCL-5 cat # AF-278) were purchased from R & D Systems (Minneapolis, MN, USA). All of these reagents were reconstituted to stock solutions of 1 μg/mL in 0.1 M phosphate buffer, pH 7.4 and their specificity and cross-reactivity tested by Western Blot against all six chemokines. Only antibodies demonstrating specificity for only the analyte to which they were raised and no cross-reactivity with the other analytes of interest were used. Antibody F(ab)′2 digestion kits (cat # 44688), Tris(2-carboxyethyl)-phosphine hydrochloride (TCEP.HCl, cat # 20490) disulfide reducing agent, bis-maleimidoethane (BMOE, cat # 22323) sulfhydryl cross-linking agent, 3-aminopropyltriethoxysilane and sulfosuccinimidyl 4-[N-maleimidomethyl] cyclohexane-1-carboxylate (SMCC, cat # 22122) were purchased from Pierce Biotechnology (Rockford, IL, USA). All other chemicals were purchased from Acros Chemicals (Fisher Scientific, Pittsburgh, PA, USA). Immediately prior to use, all solutions were passed through 0.2 μm Millex-GS filters (Millipore, Bedford, MA, USA, cat # SGL0250S) to remove particulate impurities.
2.2. Digestion of the chemokine capture antibodies
The anti-chemokine antibodies were enzymatically digested as previously described [13,14]. Briefly, the antibodies were reduced to F(ab)′2 fragments using the Pierce IgG F(ab)′2 digestion kit according to the manufacturers instructions. The fragments were recovered and further reduced to Fab fragments by incubation with 200 mM TCEP. Finally, a mixture of equal volumes (2 ng) of the six digested anti-chemokine antibodies was stored at 4 °C until immobilized on the immunoaffinity disks.
2.3. Preparation of the immunoaffinity insert
The digested antibody fragments were immobilized on an immunoaffinity insert as previously described [13,14]. Briefly, the insert was prepared by chemically modifying AP40 glass fiber filters (Millipore). 2-mm disks were punched from the filter and soaked in 10% v/v aqueous 3-aminopropyltriethoxysilane for 10 min, heating to 100 °C for 60 min and then cooled to room temperature. This process was repeated four times using fresh silane solution. The disks were cleaned in 10 mM hydrochloric acid by incubating them for 60 min at 100 °C, cooled, washed twice in distilled water and finally incubated in a solution of 1 μg/mL SMCC dissolved in 50 mM carbonate buffer, pH 9.0 for 2 h at room temp. Prior to Fab immobilization, the disks were washed 5 times in formamide and then placed into a 1 μg/mL solution of BMOE. A 1 μg/mL solution of the Fab mixture dissolved in 200 mM TCEP (to prevent reformation of the F(ab)′2 complex) was added and the disks incubated on a rotary mixer overnight at 4 °C. Finally, the disks were extensively washed in 0.1 M phosphate buffer, pH 7.4.
Disk performance was tested using the procedure outlined in Section 2.8. The LOD of the disk was determined by running dilutions of a chemokine standard (a mixture containing 100 pg/mL of each of the chemokines) through the system until no further signal could be detected. Saturation was determined in a similar manner by applying increasing concentrations of a chemokine mixture to the immunoaffinity disk until no further increase could be detected. In these studies a new disk was used for each analysis, although previous studies had shown that the disks were re-usable [14]. Additionally, concentrations of 10 pg, 100 pg and 500 pg of each chemokine were added to a saline extract of normal tissue and run throughout the system as described in Section 2.8. This was performed to examine the recovery efficiency of the system.
2.4. Assembly of the lCE-CHIP
The lCE-CHIP was fashioned around a commercially available T3530 CE chip (Micronit Microfluidics BV, Enschede, The Netherlands). This chip was made from low-fluorescence borosilicate glass 45-mm long × 15-mm wide with a double T cruciform channel etched into the main body of the chip. The channels were 50-μm wide and 20-μm deep terminating in 2-mm ports and a separation channel length of 35 mm. The ports were numbered clockwise 1–4 from the left hand side of the chip. The immunoaffinity disk was placed into port 2 and connections for the electrodes and the fluid lines were via a poly-ether-ether-ketone (PEEK) top plate with 4 drilled and tapped holes corresponding to the positions of the four ports.
2.5. Instrumentation
ICE analysis was performed on a modified Micralyne μTK microfluidic electrophoresis system (Micralyne, Edmonton, Alberta, Canada) as previously described [13,14]. The instrument consisted of an electrophoresis chamber and an electronics controller equipped with four dual-channel 6 kV power boards, a power interlock and a 16-bit data acquisition board. The electrophoresis chamber was equipped with eight platinum electrodes and an integrated 635 nm, 8 mW red diode laser. Signal detection was achieved by an epi-illumination microscope equipped with a Hamamatsu H5773-03 photomultiplier tube coupled to the data acquisition board. In these analyses, the instrument was equipped with four electrodes plus a chip stage designed to accept the T3530 CE chip. Fluid handling was achieved by a Harvard “11” syringe pump (Harvard Apparatus, Holliston, MA, USA) connected via an automatic injection valve (Upchurch Scientific) fitted with a calibrated 200-nL loop to port 2 of the chip. This connection was achieved by placing 20 mm i.d., 360 mm o.d. PEEK tubing (Upchurch Scientific) into port 1, alongside the electrode using a modified F-121H Nano-port nut (Upchurch Scientific, Oak Harbor, WA, USA). This connection was used for both sample injection, immunoaffinity port washing and introduction of the laser dye. Control of the entire system was achieved by a PC, running Microsoft Windows XP with a compiled LabView interface (National Instruments, Austin, TX, USA). To facilitate washing another tubing/electrode plug was placed into port 4 and electrode plugs were placed into ports 1 and 3 of the PEEK cover plate [14].
2.6. Patient samples
Patients with active atopic dermatitis to metals (nickel and brass) were seen at the Allergy Clinic of the George Washington University Hospital, Washington, DC, USA. A single skin biopsy was taken from each patient and control subject. The biopsy was divided in two and half processed for routine histopathology while the other half was donated to this study. All patient biopsies were taken during episodes of atopic dermatitis. Histopathological examination, with immunocytochemical verification of the infiltrating cell types was performed by the Pathology Department and divided the patients into two groups, namely 20 patients with neutrophilic infiltrations and perivascular inflammation (Group 1) and 22 patients with monocytic and lymphocytic infiltration (Group 2). Biopsies were also obtained from a group of 30 non-allergic subjects (Group 3). All subjects were 25–40 years of age and consented to use of the samples for this study. No name indicators were assigned to any samples as required by the hospital institutional review board, however, numerical identifiers where assigned to each sample in order to compare the ICE-Chip results with the histopathology findings.
2.7. Micro-dissection of patient biopsies
Fresh-frozen samples were used in this study as previous experience had led us to believe that formalin-fixed tissue could serious impair the availability of the analytes of interest to immunoaffinity capture [14]. All samples were micro-dissected as previously described [13,14]. Briefly, a single 6 μm frozen section was taken from each biopsy and air-dried on a chilled glass microscope slide. Once dry, the section was stained with a 0.01% aqueous solution of cotton blue to aid in morphological identification. Using microscopic examination, tissue areas containing either cellular-infiltration or normal tissue were dissected using an Eppendorf Microdissector (Eppendorf North America, Westbury, NY, USA), shown in Fig. 1A, equipped with an oscillating micro-chisel,(shown in Fig. 1B). The chisel was moved around the area of interest to leave an island of tissue surrounded by a circle of clear glass. The isolated tissue island was dissolved by covering it with 50-μL of warm (22 °C) 100 mM phosphate buffer, pH 7.4, containing 0.1% v/v Brij 35 detergent and 1 mM of leupeptin, using the Micro-dissector's micropipette. The buffer was recovered with the micropipette and clarified by centrifugation in a hematocrit centrifuge.
Fig. 1.
(A) An Eppendorf Microdissector (Eppendorf North America, Westbury, NY, USA), equipped with, (B) an oscillating micro-chisel.
In order to be able to compare the concentrations of the different analytes in the recovered samples, the samples were “normalized” to a standard protein content to enable comparison of recovered analytes between the different study groups. Results were then expressed as pg/μg extracted protein The total protein content of each sample was measured by direct spectrophotometry at A280/260 nm using a NanoDrop ND-100 microspectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) using a percent extinction coefficient with units of (g/100 ml−1 cm−1) as recommended by the Pierce Biotechnology technical bulletin #6 [15]. The samples were adjusted to a A280 nm value that was equivalent to a protein concentration of 1 μg/mL in 100 mM phosphate buffer/0.1%Brij 35, pH 7.4 prior to analysis.
2.8. ICE measurement of in situ chemokines
ICE-CHIP analysis of each sample was performed in the Micralyne system using the LabView interface sequence to control the injection, washing, labeling and separation of the sample. Prior to each analysis, the chip was flushed with 100 mM phosphate/0.1% Brij 35 running buffer, pH 7.4. The pump was programmed to inject a 200-nL sample into the immunoaffinity port at a flow rate of 400-μL/min. The sample was allowed to react for 2 min, during which the immunoaffinity port bound the analytes of interest. The pump then flushed the immunoaffinity port with 500-μL of phosphate buffer to remove all of the non-bound materials; these waste materials passing out through port 2. 0.2 mL of a 5 mM solution of AlexaFluor 633 laser dye (Molecular Probes/Invitrogen, Eugene, OR, USA) was pumped into port 1, and allowed to incubate for 2 min, thus labeling only the bound analytes. The port was again flushed with phosphate buffer to remove the unbound dye before introducing 0.2 μL of 100 mM phosphate buffer/HCl, pH 1.0 into port 1 for 1 min. A potential of 1.5 kV was introduced between ports 4 and 2 for 20 s to inject the released analytes into the separation channel. The current was grounded and a potential of 4 kV was set between ports 1 and 3 to separate the analytes, which were detected by the on-line detector set 32-mm from the injection site. All of the assays were performed at room temperature and the concentrations of each separated peak calculated from a calibration curve constructed from known amounts of each analyte run under identical separation conditions.
Inter- and intra-assay coefficients of variance (CV) were calculated from data obtained by applying a test mixture containing 100 pg of each chemokine on five separate days and by running five repetitive assays on the same day. Additionally, the same analysis was performed on a sample from a normal subject to check the assay CVs at low concentrations in a real sample.
3. Results
3.1. Characteristics of the system
The total time taken to perform a complete analysis was 35–40 min; including 20–25 min taken for microdissection, recovery and pre-analysis preparation of the sample. Chemokine ICE-CHIP analysis was 8 min during which all six chemokines were captured, labeled, recovered, separated and measured. Fig. 2 illustrates a typical electropherogram produced by the chip, which represents the separation and measurement of a mixture containing 100 pg/mL of each chemokine. Additionally, there is always a small free dye peak, which elutes as the last peak; this represents that an excess of dye, in relationship to the amount of bound analytes, is always injected. Protein analysis performed on the immunoaffinity disk demonstrated that ca. 10 ng of antibody mixture could be reliably immobilized on a 2-mm disk. Dilution studies indicated that the LOD for each chemokine was 0.2 pg/mL (2.5 × 10−14 M) for CXCL1, 0.3 pg/mL (3.6 × 10−14 M) for CXCL5, 0.2 pg/mL (2.5 × 10−14 M) for CXCL8, 0.4 pg/mL (4.7 × 10−14 M) for CCL1, 0.3 pg/mL (4.0 × 10−14 M) for CCL3 and 0.4 pg/mL (5.1 × 10−14 M) for CCL5. Saturation studies indicated that the immunoaffinity disk was capable for binding 1.7 ng of CXCL1, 1.6 ng of CXCL5, 1.9 ng of CXCL8, 1.4 ng of CCL1, 1.6 ng of CCL3 and 1.5 ng of CCL5, respectively. Recovery of all six chemokines from spiked normal human skin tissue extracts, containing various concentrations of each chemokine are summarized in Table 1. The inter- and intra-assay CVs are summarized in Table 2.
Fig. 2.
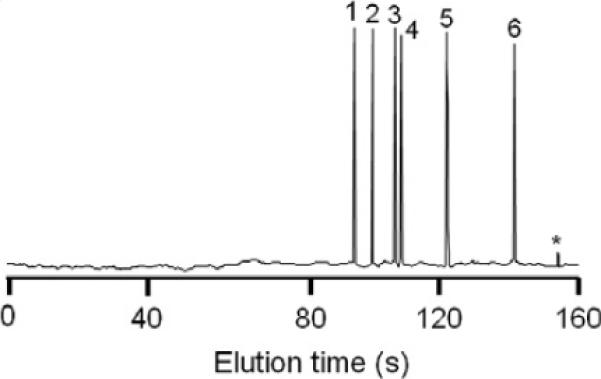
Typical ICE-CHIP electropherogram illustrating the electrophoretic pattern of the six chemokines, eluted in the following order: 1. CXCL8, 2. CCL5, 3. CXCL1, 4. CXCL5, 5. CCL3, 6. CCL1, *free dye.
Table 1.
Chemokine recovery from normal human tissue extract.
| Chemokine | Amount spiked (pg) | Amount recovered (pg) |
|---|---|---|
| CXCL1 | 10 | 9.6 |
| 100 | 95.8 | |
| 500 | 488.9 | |
| CXCL5 | 10 | 9.3 |
| 100 | 95.7 | |
| 500 | 497.2 | |
| CXCL8 | 10 | 9.7 |
| 100 | 97.6 | |
| 500 | 498.1 | |
| CCL1 | 10 | 9.3 |
| 100 | 96.4 | |
| 500 | 497.5 | |
| CCL3 | 10 | 9.3 |
| 100 | 98.1 | |
| 500 | 496.9 | |
| CCL5 | 10 | 9.5 |
| 100 | 98.9 | |
| 500 | 497.7 |
Table 2.
Inter- and intra-assay CV's for each chemokine in normal human tissue extract and in a sample from a normal subject.
| Chemokine | Inter-assay CV | Intra-assay CV |
|---|---|---|
| Tissue extract | ||
| CXCL1 | 6.2 | 5.8 |
| CXCL5 | 4.7 | 4.1 |
| CXCL8 | 4.3 | 3.6 |
| CCL1 | 5.5 | 5.1 |
| CCL3 | 6.4 | 5.6 |
| CCL5 | 5.1 | 4.8 |
| Normal subject sample | ||
| CXCL1 | 9.4 | 8.6 |
| CXCL5 | 8.2 | 6.6 |
| CXCL8 | 6.5 | 5.9 |
| CCL1 | 7.8 | 8.2 |
| CCL3 | 10.1 | 8.5 |
| CCL5 | 9.7 | 6.8 |
3.2. Measurement of chemokines in patient and control groups
The ability of the lCE-CHIP to distinguish not only samples taken from normal controls and patients but also between different cellular events within the patient groups 1 and 2 is illustrated on Fig. 3. Normal concentrations (Group 3) of all six chemokines are shown in Fig. 3A and were found to be less than 10 pg/μg extracted protein for CXCL5, CCL1, CCL3 and less than 20 pg/μg extracted protein for CXCL1, CXCL8 and CCL5. Analysis of the patient group demonstrated two distinct patterns which corresponded with the cellular infiltrates found by routine histopathology. The group 1 patients demonstrating increased concentrations of CXCL1, CXCL5 and CXCL8 corresponded to the subset of patients with clearly demonstrable neutrophil cellular infiltrates by histopathology (Fig. 3B) while the other patients (Group 2) showed increased concentrations of CCL1, CCL3 and CCL5 (Fig. 3C). This latter group corresponded to the patient group demonstrating monocytic and T-lymphocyte infiltration by histopathology with immunocytochemical verification. Table 3 summarizes the concentrations of the six chemokines in the three subject groups.
Fig. 3.
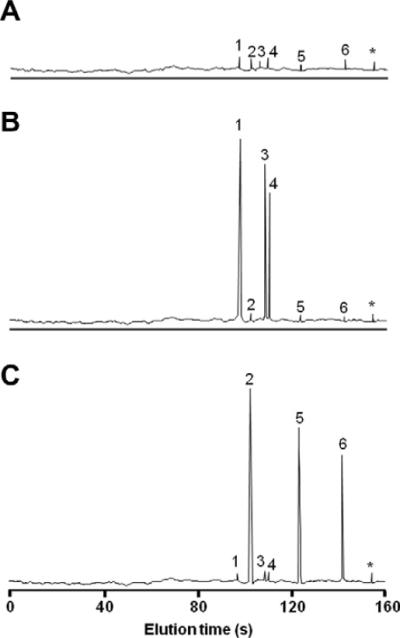
Electropherograms of the chemokine profiles of the three subject groups. (A) Normal subjects (Group 3); (B) Patients with neutrophil infiltrations (Group 1); (C) Patients with monocyte and lymphocyte infiltrations (Group 2). Chemokines eluted in the following order: 1. CXCL8, 2. CCL5, 3. CXCL1, 4. CXCL5, 5. CCL3, 6. CCL1, *free dye.
Table 3.
Chemokine concentrations in the three subject groups.
| Chemokine | Group 1 | Group 2 | Group 3 |
|---|---|---|---|
|
|
|||
| (n = 20) | (n = 22) | (n = 30) | |
| CXCL1 | 168.4 ± 33.9 | 28.2 ± 3.6 | 18.4 ± 6.6 |
| CXCL5 | 104.5 ± 21.3 | 20.6 ± 5.5 | 7.3 ± 2.5 |
| CXCL8 | 381.6 ± 43.1 | 14.8 ± 3.4 | 19.6 ± 8.1 |
| CCL1 | 11.5 ± 4.2 | 85.9 ± 28.4 | 6.2 ± 2.5 |
| CCL3 | 16.4 ± 5.1 | 287.6 ± 35.5 | 8.2 ± 2.3 |
| CCL5 | 11.7 ± 3.4 | 340.4 ± 41.7 | 16.5 ± 7.2 |
All values are the mean ± SEM.
ICE analysis could not only differentiate the two histopathologically distinct patient groups but also provide information on chemokine concentrations in different parts of the biopsy. Chemokine concentrations were greatest within the actual lesion itself with a gradual reduction at sites taken from areas up to 15 mm away from the lesion epicenter. In the patients with neutrophil infiltrations, concentrations of CXCL8 within the lesion were found to be 381.6 ± 43.1 pg/μg of extracted proteins, which diminished to 61.3 pg/μg extracted protein in samples taken 15 mm from the lesion. In samples taken from the second patient group, lesion concentrations of CCL3 and CCL5 were shown to be 287.6 ± 35.5 pg/μg extracted protein and 340.4 ± 41.7 pg/μg extracted protein, respectively. These patients also demonstrated a similar lessening of the resident chemokines at sites peripheral to the lesion itself (Fig. 4). These findings indicate that the chemokines are being produced locally and indicates the importance of measuring chemokines in situ. Further, the data demonstrates that measurement of concentrations of chemokines taken from the periphery of the biopsy and away from the lesion may provide better baseline measurements than comparisons to analytes measured in normal tissues. It must be borne in mind that the analytes being measured are only a minute fraction of the extracted protein and as such must be compared to each other in the groups tested. Although ICE is reasonably sensitive, it relies on the selectivity of the immobilized antibodies to capture all of the analytes present in the sample. Previous analyses have shown that a 50 times excess of immobilized antibody captures greater than 95% of the total analyte in the sample [13,14]. However, no internal standard is included in the system, so the overall concentrations of each analyte must be compared with the concentrations in the control or other group and not taken as an absolute value.
Fig. 4.
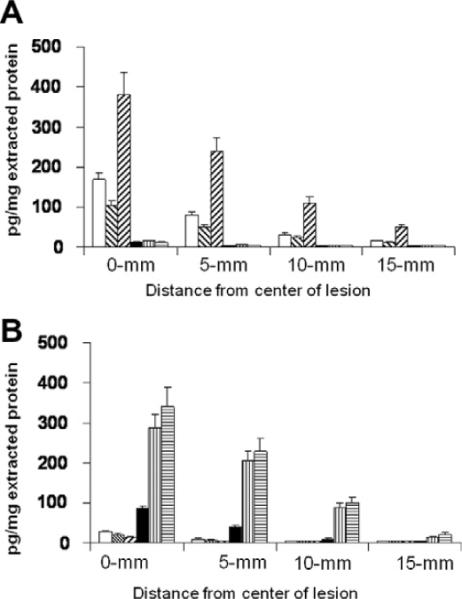
Chemokine concentrations within the biopsy tissue of the two patient groups (A) Patients with neutrophil infiltrations (Group 1); (B) Patients with monocyte and lymphocyte infiltrations (Group 2).
4. Discussion
An escalation in the popularity of chip-based devices and their application to problems in biomedical research has occurred over the past few years [16–22]. A major attraction of these devices is their ability to handle minute samples coupled with decreased reagent consumption. Further, in many cases, extremely short analysis times may be achieved, thus providing the potential for faster diagnosis. Microfluidic devices are also capable of analyzing multiple analytes, within the same sample, utilizing samples as low as 200 nL in size [13], especially when CE is employed as the analytical tool. CE analysis can be greatly enhanced by coupling the separation technology with one or several high sensitivity detectors such as LIF detectors or mass spectrometers. Chip-based CE can also be enhanced by incorporating pre-separation devices such as affinity or immunoaffinity cartridges to the CE system to produce affinity (ACE) [23–28] or ICE systems [13,19,29–32], the procedure described in these studies. The advantage of adding a pre-separation cartridge is that the affinity or immunoaffinity ligands act not only as selective capture devices, but also as pre-analytical concentrators [19,33–35]. When handling complex biological matrices, pre-analytical immunoaffinity extraction can greatly enhance the efficiency of the separation procedure by actively selecting and extracting the analytes of interest from the biological matrix. In the present study, an immunoaffinity port selected and extracted the analytes of interest from a complex tissue homogenate. Depending on the specificity of the capture antibody and its presence in excess, there is a reasonable degree of assurance that the majority of the desired analyte will be successfully extracted and released into the analytical section of the CE.
Immunoaffinity extraction followed by CE separation and analysis holds several advantages over conventional plate-based immunoassays. ICE requires minute quantities of sample (200 nL versus 50–100 μL). Additionally, the immunoaffinity ligands can be regenerated and used for up to 200 times, thus reducing assay costs and assay variability [36]. The use of a replaceable immunoaffinity disk further helps to reduce assay costs and extends the working life of the costly glass chip. In comparison with conventional immunoassays such as the enzyme-linked immunosorbent assay (ELISA), ICE can be used to analyze multiple analytes in the same sample [13,22,37], whereas ELISA usually measures a single analyte. However, future advances in ELISA hold the potential for multi-analyte analysis [38]. ICE can also be considered a two-dimension assay as opposed to the single dimension of most immunoassays as it uses immunoaffinity selectivity and capture in one dimension and electrophoretic separation and analysis in the second dimension. The combination of these two techniques reducing the incidence of non-specific reactions; the electrophoretic phase separating the immunoaffinity bound materials and checking on selectivity [33,36].
ICE and chip-based ICE are versatile and can be applied to the analysis of a multitude and different analytes. It has one limiting factor; the availability of a specific antibody capable of selecting the analyte of interest. However, this has not stopped ICE being used to analyze a large number of important biomedical analytes [39] including inflammatory biomarkers found in the cerebrospinal fluid of head injury patients [40] and for the measurement of hormones in human body fluids [37]. Additionally, ICE has been used to detect and measure a number of drugs including the anti-inflammatory drug, naproxene [19,41].
In the studies reported in this communication, ICE was used to detect and measure six important mediators of inflammatory skin lesions. The ICE system was able to distinguish between two clinically important types of allergic skin lesions and correlated well with the histopathological classifications of the lesion. The ability to measure the immunochemical mediators within the lesion areas of the biopsy added important information to the pathological findings. Definition of the chemokines present and their relative concentrations enables clinicians to better understand the in vivo situation and thus provide suitable therapy. Whereas the classical histopathology coupled with immunocytochemistry can identify the infiltrating cells, it is unable to measure the intensity of the chemoattractive molecules causing such infiltration. Ice measurements can not only identify the types of infiltrating cells but can provide information on the intensity of the reaction. Such information is vital for the clinician to be able to surmise if the lesion is ongoing or static; situations that effect treatment. ICE in such cases is a adjunct assay, providing valuable additional information thus supplementing the histopathological findings. Although the pre-analysis time is quite long (microdissection and sample preparation) the actual analysis of six chemokines is quite rapid (ca. 8 min).
The advent of large arrays of commercially available antibodies widens the potential applications for immunoaffinity CE especially in areas where small or precious samples are the only source of materials such as in newborn screening and analysis of archival tissues. Microdissection opens many additional areas where ICE may be applied, especially in forensic and small animal model studies.
5. Conclusion
Analyses that use small patient samples and yield results quickly are being sought in all fields of medicine. The chip-based ICE method presented in this paper could analyze small tissue samples and identify and quantify six chemokines from a single sample in less than 10 min. The low reagent and sample consumption, decreased analysis time and increased sensitivity make this method an attractive alternative to other conventional assays. As the technology continues to improve, this method could be easily adapted to a point-of-care setting providing timely results to both clinicians and their patients.
Acknowledgment
This work was supported by the Intramural Program of the National Institutes of Health, Bethesda, MD, USA.
Footnotes
The authors have declared no conflict of interest.
References
- [1].Yamanaka K-I, Mizutani H. Curr. Prob. Dermatol. 2011;41:80–92. doi: 10.1159/000323299. [DOI] [PubMed] [Google Scholar]
- [2].Agrawal R, Wisniewski JA, Woodfolk JA. Curr. Prob. Dermatol. 2011;41:112–124. doi: 10.1159/000323305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Komine M. J. Pharmacol. Sci. 2009;110:260–264. doi: 10.1254/jphs.09r06fm. [DOI] [PubMed] [Google Scholar]
- [4].Ransohoff RM. Immunity. 2009;31:711–721. doi: 10.1016/j.immuni.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Surmi BK, Hasty AH. Vascul. Pharmacol. 2010;52:27–36. doi: 10.1016/j.vph.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Avgerinou G, Goules AV, Stavropoulos PG, Katsambas AD. Int. J. Dermatol. 2008;47:219–224. doi: 10.1111/j.1365-4632.2008.03471.x. [DOI] [PubMed] [Google Scholar]
- [7].Incorvaia C, Frati F, Verna N, D'Alo S, Motolese A, Pucci S. Clin. Exp. Immunol. 2008;153:27–29. doi: 10.1111/j.1365-2249.2008.03718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hon K.-l.E., Ching G.K.-w., Wong K.-y., Leung T.-f., Leung AK. J. Natl. Med. Assoc. 2008;100:500–505. doi: 10.1016/s0027-9684(15)31296-7. [DOI] [PubMed] [Google Scholar]
- [9].Williams MA, Cave CM, Quaid G, Solomkin JS. Arch. Surg. 1999;134:1360–1366. doi: 10.1001/archsurg.134.12.1360. [DOI] [PubMed] [Google Scholar]
- [10].Saetta M, Baraldo S, Zuin R. Am. J. Respir. Crit. Care Med. 2003;168:911–913. doi: 10.1164/rccm.2308002. [DOI] [PubMed] [Google Scholar]
- [11].Ward SG, Bacon K, Westwick J. Immunity. 1998;9:1–11. doi: 10.1016/s1074-7613(00)80583-x. [DOI] [PubMed] [Google Scholar]
- [12].Fischereder M. Acta Physiol. Hung. 2007;94:67–81. doi: 10.1556/APhysiol.94.2007.1-2.7. [DOI] [PubMed] [Google Scholar]
- [13].Phillips TM, Wellner EF. Electrophoresis. 2007;28:3041–3048. doi: 10.1002/elps.200700193. [DOI] [PubMed] [Google Scholar]
- [14].Phillips TM, Wellner EF. Electrophoresis. 2009;30:2307–2312. doi: 10.1002/elps.200900095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pierce Biotechnology, Tech Tip 6. Thermo Scientific; Rockford, IL: 2008. [Google Scholar]
- [16].Chiem N, Harrison DJ. Anal. Chem. 1997;69:373–378. doi: 10.1021/ac9606620. [DOI] [PubMed] [Google Scholar]
- [17].Regnier FE, He B, Lin S. J. Trends Biotechnol. 1999;17:101–106. doi: 10.1016/s0167-7799(98)01294-3. [DOI] [PubMed] [Google Scholar]
- [18].Sato K, Tokeshi M, Kimura H, Kitamori T. Anal. Chem. 2001;73:1213–1218. doi: 10.1021/ac000991z. [DOI] [PubMed] [Google Scholar]
- [19].Guzman NA. Electrophoresis. 2003;24:3718–3727. doi: 10.1002/elps.200305647. [DOI] [PubMed] [Google Scholar]
- [20].Haes AJ, Terray A, Collins GE. Anal. Chem. 2006;78:8412–8420. doi: 10.1021/ac061057s. [DOI] [PubMed] [Google Scholar]
- [21].Demianova Z, Shimmo M, Poysa E, Franssila S, Baumann M. Electrophoresis. 2007;28:422–428. doi: 10.1002/elps.200600334. [DOI] [PubMed] [Google Scholar]
- [22].Phillips TM, Wellner E. J. Chromatogr. A. 2006;1111:106–111. doi: 10.1016/j.chroma.2006.01.102. [DOI] [PubMed] [Google Scholar]
- [23].Sloat AL, Roper MG, Lin X, Ferrance JP, Landers JP, Colyer CL. Electrophoresis. 2008;29:3446–3455. doi: 10.1002/elps.200700808. [DOI] [PubMed] [Google Scholar]
- [24].Yang W, Sun X, Pan T, Woolley AT. Electrophoresis. 2008;29:3429–3435. doi: 10.1002/elps.200700704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hou C, Herr AE. Electrophoresis. 2008;29:3306–3319. doi: 10.1002/elps.200800244. [DOI] [PubMed] [Google Scholar]
- [26].Montes RE, Hanrahan G, Gomez FA. Electrophoresis. 2008;29:3325–3332. doi: 10.1002/elps.200700693. [DOI] [PubMed] [Google Scholar]
- [27].Heegaard NH, Schou C, Ostergaard J. Methods Mol. Biol. 2008;421:303–338. doi: 10.1007/978-1-59745-582-4_21. [DOI] [PubMed] [Google Scholar]
- [28].Danel C, Azaroual N, Brunel A, Lannoy D, Vermeersch G, Odou P, Vaccher C. J. Chromatogr. A. 2008;1215:85–193. doi: 10.1016/j.chroma.2008.10.094. [DOI] [PubMed] [Google Scholar]
- [29].Guzman NA. Anal. Bioanal. Chem. 2004;378:37–39. doi: 10.1007/s00216-003-2326-y. [DOI] [PubMed] [Google Scholar]
- [30].Delaunay-Bertoncini N, Hennion MC. J. Pharm. Biomed. Anal. 2004;34:717–736. doi: 10.1016/S0731-7085(03)00559-4. [DOI] [PubMed] [Google Scholar]
- [31].Benavente F, Hernandez E, Guzman NA, Sanz-Nebot V, Barbosa J. Anal. Bioanal. Chem. 2007;387:2633–2639. doi: 10.1007/s00216-007-1119-0. [DOI] [PubMed] [Google Scholar]
- [32].Miksa B, Chinnappan R, Dang NC, Reppert M, Matter B, Tretyakova N, Grubor NM, et al. Chem. Res. Toxicol. 2007;20:1192–1199. doi: 10.1021/tx7001096. [DOI] [PubMed] [Google Scholar]
- [33].Guzman NA, Blanc T, Phillips TM. Electrophoresis. 2008;29:3259–3278. doi: 10.1002/elps.200800058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Guzman NA, Phillips TM. Anal. Chem. 2005;77:61A–67A. doi: 10.1021/ac053325c. [DOI] [PubMed] [Google Scholar]
- [35].Guzman NA, Stubbs RJ, Phillips TM. Drug Discov. Today: Technol. 2006;3:29–37. doi: 10.1016/j.ddtec.2006.03.009. [DOI] [PubMed] [Google Scholar]
- [36].Guzman NA, Phillips TM. Electrophoresis. 2011;32:1565–1578. doi: 10.1002/elps.201000700. [DOI] [PubMed] [Google Scholar]
- [37].Wellner EF, Kalish H. Electrophoresis. 2008;29:3477–3483. doi: 10.1002/elps.200700785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Adrian J, Pinacho DG, Granier B, Diserens JM, Sánchez-Baeza F, Marco MP. Anal. Bioanal. Chem. 2008;391:1703–1712. doi: 10.1007/s00216-008-2106-9. [DOI] [PubMed] [Google Scholar]
- [39].Amundsen LK, Siren H. Electrophoresis. 2007;28:99–113. doi: 10.1002/elps.200500962. [DOI] [PubMed] [Google Scholar]
- [40].Phillips TM. Electrophoresis. 2004;25:1652–1659. doi: 10.1002/elps.200305873. [DOI] [PubMed] [Google Scholar]
- [41].Phillips TM, Wellner EF. Biomed. Chromatogr. 2006;20:662–667. doi: 10.1002/bmc.673. [DOI] [PubMed] [Google Scholar]