

Abstract
Angiostatin blocks tumor angiogenesis in vivo, almost certainly through its demonstrated ability to block endothelial cell migration and proliferation. Although the mechanism of angiostatin action remains unknown, identification of F1-FO ATP synthase as the major angiostatin-binding site on the endothelial cell surface suggests that ATP metabolism may play a role in the angiostatin response. Previous studies noting the presence of F1 ATP synthase subunits on endothelial cells and certain cancer cells did not determine whether this enzyme was functional in ATP synthesis. We now demonstrate that all components of the F1 ATP synthase catalytic core are present on the endothelial cell surface, where they colocalize into discrete punctate structures. The surface-associated enzyme is active in ATP synthesis as shown by dual-label TLC and bioluminescence assays. Both ATP synthase and ATPase activities of the enzyme are inhibited by angiostatin as well as by antibodies directed against the α- and β-subunits of ATP synthase in cell-based and biochemical assays. Our data suggest that angiostatin inhibits vascularization by suppression of endothelial-surface ATP metabolism, which, in turn, may regulate vascular physiology by established mechanisms. We now have shown that antibodies directed against subunits of ATP synthase exhibit endothelial cell-inhibitory activities comparable to that of angiostatin, indicating that these antibodies function as angiostatin mimetics.
Tumor expansion beyond a prevascular size requires generation of new blood vessels, termed angiogenesis. As the tumor expands, vascular remodeling and continued angiogenesis is essential to support this growth (1–3). Inhibition of angiogenesis not only arrests tumor growth, but also results in tumor shrinkage to a prevascular size (4, 5). The regulation of angiogenesis is a complex process involving enzymatic and signal-transduction cascades that function in fibrinolysis, matrix remodeling, inflammation, hemodynamic control of oxygenation, and growth regulation (6, 7). The activation of these pathways produces many molecules with either proangiogenic or antiangiogenic properties. Folkman and colleagues (3, 8, 9) have hypothesized that the local balance between these various positive and negative factors determines the net tendency toward angiogenesis or angiostasis.
Critical to this angiogenic “set point” is the behavior of the cancer itself. Proangiogenic signals elaborated by tumor cells set into motion a series of highly regulated endothelial responses. The fibrinolytic system of plasminogen, its activators, and inhibitors plays a central role in the interface events that occur between endothelial and tumor cells (10). Plasmin is important enzymatically through its ability to activate matrix metalloproteinases (MMPs), which enable tumor cells to proteolytically degrade basement membrane barriers and metastasize (11, 12). Plasminogen and its activators bind to tumor cell membrane receptors, where plasmin is generated and signal transduction is triggered, promoting MMP-9 secretion (13, 14). However, further proteolytic cleavage of plasmin by both host and tumor cell proteinases produces additional fragments lacking proteolytic activity, some of which inhibit angiogenesis (15–17). These fragments, termed angiostatins, are among the most potent antiangiogenic substances currently known. All angiostatins have in common the presence of one or more of the five kringle domains contained within plasminogen. The form used in the present study consists of human kringles 1–3, which inhibit proliferation and migration of cultured endothelial cells (18). Suppression of primary and metastatic tumor growth occurs in mice injected with purified angiostatin (15, 19, 20), with evidence of increased tumor cell apoptosis (21).
We previously identified a candidate angiostatin receptor, endothelial cell surface-associated F1-FO ATP synthase (hereafter referred to as ATP synthase), based on affinity purification of this enzyme's α- and β-subunits from human umbilical vein endothelial cell (HUVEC) plasma membranes over an angiostatin-Sepharose column (18). Supporting the identity of this receptor was the observation that polyclonal antibodies against the α-subunit of ATP synthase blocked angiostatin binding to HUVEC as well as its ability to inhibit endothelial cell proliferation. This initial study, however, did not demonstrate an intact, enzymatically active, surface-associated ATP synthase complex, nor did it assess whether angiostatin could block activity of this putative complex. In the current study, we demonstrate conclusively that all components forming the core catalytic complex of F1 ATP synthase are present on the external endothelial cell surface. Moreover, we show that this enzyme is catalytically competent in the synthesis of ATP on the surface of endothelial cells. Finally, we show that both angiostatin and antibodies against the α- and β-subunits of ATP synthase inhibit activity of the surface-associated enzyme and the purified enzyme. Taken together, these data strongly support our original hypothesis that ATP synthase is the primary target for angiostatin on the surface of endothelial cells and further suggest that cell surface ATP metabolism is likely to play a central role in the endothelial cell response to angiostatin. The ability of angiostatin or antibodies to disrupt ATP production may render endothelial cells more vulnerable to hypoxic challenge in the microenvironment of a growing tumor and additionally may alter ATP-mediated signal transduction at the endothelial cell surface.
Materials and Methods
Confocal Microscopy.
HUVECs were plated in microvascular endothelial cell growth medium (EGM-MV; Clonetics, San Diego) medium at 150,000 cells/ml on glass coverslips and allowed to adhere overnight. Cells were incubated at 4°C and washed with PBS before fixation in 2% paraformaldehyde solution. A control slide was permeabilized in 100% ethanol for 5 min at room temperature before fixation. All coverslips were incubated in 5% goat serum in Dulbecco's PBS, pH 7.4 (Life Technologies, Gaithersburg, MD) for 15 min and washed before coincubation with murine monoclonal anti-α ATP synthase (Molecular Probes) (1:100) and rabbit polyclonal antibody raised against the recombinant human β-subunit of ATP synthase (1:500) diluted in staining buffer (1% BSA/0.02% Tween 20/0.005 M EDTA/1% goat serum/1× PBS, pH 7.4). To verify surface staining, cells were incubated with murine monoclonal anti-CD31 (PharMingen). All cells were washed three times and incubated for 1 h in the dark at 4°C with goat anti-mouse IgG AF488 F(ab′)2 (Molecular Probes) (1:100), goat anti-rabbit IgG AF546 F(ab′)2 (Molecular Probes) (1:200), or goat anti-mouse IgG1 AF633 (1:100) in staining buffer. After the final washes, cells were visualized by using a Zeiss LSM-410 (Switzerland) confocal microscope at ×630.
ELISA-Binding Studies.
Binding studies were performed with purified F1 subunit of bovine ATP synthase (20 μg/ml) passively adsorbed onto polyvinylchloride microtiter 96-well, flat-bottom plates (Dynex Technologies, Chantilly, VA) as described (22). Briefly, plates were coated with protein in 50 μl of 0.1 M Na2CO3, pH 9.6, and incubated for 2 h at 37°C. Nonspecific sites were blocked by incubating with PBS, pH 7.0, containing 1% BSA for 30 min at room temperature. Binding studies were performed with increasing amounts of angiostatin (0–0.5 mg/ml) added in a 50 μl final volume for 1 h at 37°C. Plates were washed (0.1% Tween 20/PBS, pH 7.0) and incubated with anti-human angiostatin antibody (goat IgG) (R & D Systems) (1:200 dilution) overnight at 4°C. Plates were washed and incubated with anti-goat IgG peroxidase conjugate (Sigma) (1:7,500 dilution) for 1 h at 37°C. Plates were washed, 150 μl of OPD substrate was added to each well, and the reaction was stopped with 25 μl of H2SO4 (25%) before monitoring the absorbance at λ = 492 nm in a Molecular Devices SpectraMax Plus-384 plate reader. Control studies were performed in the absence of angiostatin to detect any nonspecific binding of secondary proteins. Isotype-specific controls were performed by using a goat antifibronectin antibody (Sigma).
Purification of Bovine Heart F1 ATP Synthase and F1 Activity Assay.
Fresh bovine heart mitochondria were obtained as described (23) and sonicated to yield submitochondrial particles (24). The F1 portion was separated from membrane-bound FO by chloroform extraction. The aqueous layer was centrifuged at 105,000 × g to remove particulate matter before purifying over an S300 gel-filtration column. The purified F1 ATP synthase only exhibits activity in the reverse reaction (ATPase), because the forward reaction (ATP synthase) requires the F1-FO holoenzyme correctly assembled into a membrane across which a proton gradient exists. ATPase activity was measured spectrophotometrically by monitoring at λ = 340 nm by coupling the production of ADP to the oxidation of NADH via pyruvate kinase and lactate dehydrogenase reactions as described (25).
Cell Surface ATP Assay.
Quiescent, confluent HUVECs in 24-well plates were washed and equilibrated into DMEM/F-12 (HAM) 1:1 medium (Life Technologies) containing 10 mM potassium phosphate. Cells were treated with 1 μM angiostatin or 0.1 mg/ml oligomycin for 1 h at 37°C. All cells then were incubated with 0.1 μCi 32Pi and 0.1 μCi (50 μM) [2,8-3H]-ADP (NEN) for 1 min. Supernatants were removed and centrifuged before assaying for ATP production by TLC or firefly luciferase assay.
ATP Generation by Bioluminescent Luciferase Assay.
Aliquots (50 μl) of cellular supernatants from cell surface ATP assays were analyzed by using the ATP bioluminescence assay kit (Sigma). In this firefly luciferin–luciferase reaction, only ATP is readily detected because the enzymatic reaction of firefly luciferase to oxidize luciferin is specific for ATP relative to all other nucleotides. Samples were injected with the ATP assay mixture, and recordings were made in a LuminoskanRS (Labsystems, Franklin, MA) over a 10-s period. The response in a given sample or standard was quantified as area under the peak of the response and averaged for duplicate determinations. Data are expressed as picomoles of ATP produced per cell based on standards determined under the same conditions with each experiment. Angiostatin was prepared as described (26). Angiostatin did not interfere with the luciferin–luciferase assay, indicating that the effects of angiostatin are not an assay artifact (data not shown).
Dual-Label Radioactive TLC of ATP.
Supernatants were obtained as described above. Cell pellets were obtained after washing wells with 1.0 ml of medium (as described above) and lysing with 1 M NaOH (100 μl). Aliquots of supernatant (3 μl) and cell pellet (10 μl) were applied to microcrystalline polyethyleneimine-cellulose plates (Analtech) along with an authentic [γ-32P]ATP standard (0.025 μCi). Plates were developed in 1.4 M LiCl for a distance of 15 cm (27). Dried spots containing [32P]ATP, [3H]ATP, and [3H]ADP were detected by sodium iodide and phosphoimaging on a Storm 850 (Molecular Dynamics). Areas corresponding in Rf value to cochromatographed authentic [32P]ATP standard were scraped off the plate, and their radioactivity was determined in a liquid scintillation analyzer (Packard). No ATP was detectable by TLC in the [3H]ADP preparation used for all experiments.
Cell Proliferation Assay.
HUVECs were plated at a density of 5,000 cells per well in medium depleted of FCS overnight to allow the cells to become quiescent. Fresh medium containing 5% FCS, 10 ng/ml basic fibroblast growth factor, and 3 ng/ml vascular endothelial growth factor were added to the wells along with angiostatin (1.0 μM), antibody directed against the recombinant human α-subunit of ATP synthase (1:10 dilution), antibody directed against the recombinant human β-subunit of ATP synthase (1:10 dilution), preimmune serum (1:10 dilution), or cycloheximide (10 μg/ml). Cell density was measured after 24 h by using the CyQUANT Cell Proliferation Assay Kit (Molecular Probes) in a fluorometric plate reader (Molecular Devices). The absorbance values used to calculate the percentage of proliferation of the cells ranged from 1.24 or 1.00 for treated and 2.57 for untreated.
Results
α- and β-Subunits of ATP Synthase Colocalize on the Surface of HUVECs.
Our previous studies demonstrated the presence of the α-subunit of ATP synthase on the surface of endothelial cells by immunofluorescence and flow cytometry (18). We now demonstrate extensive colocalization of the α- and β-subunits of ATP synthase on the endothelial cell surface by using confocal microscopy with a mAb specific for the α-subunit of ATP synthase and affinity-purified antibodies generated against the recombinant β-subunit of ATP synthase (Fig. 1C). The immunofluorescence occurs in two distinctive patterns. First, there are numerous fine punctate structures distributed over the entire cell surface, except where the bulging nucleus displaces the plasma membrane from the optical section. Second, each cell displays one or more irregular clusters of punctate structures, suggesting an organized distribution on the cell surface. The cells were fixed in situ before the addition of antibodies to eliminate antibody-capping artifacts. Parallel studies of immunolocalization between the α- and γ-subunits of ATP synthase showed virtually identical patterns of colocalization (data not shown).
Figure 1.
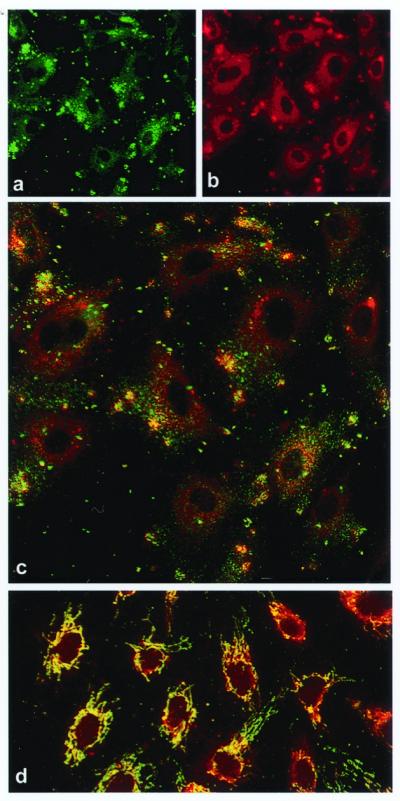
Colocalization of the α- and β-subunits of ATP synthase on the surface of HUVECs by immunostaining and confocal microscopy. (a) Nonpermeabilized HUVECs immunostained with a murine mAb specific for the α-subunit of ATP synthase. (b) The same cells immunostained with a rabbit polyclonal antiserum specific for the β-subunit of ATP synthase. (c) Composite colocalization images obtained by digital overlays of the above images. (d) Colocalization image obtained from cells permeabilized with ethanol (100%). Representative images are shown; n = 26.
Fluorescence in all images was determined to be cell surface-associated by three criteria. (i) Permeabilized cells (Fig. 1D) produced a dramatically different pattern characteristic of mitochondrial staining of ATP synthase. Endothelial cell mitochondria are characteristically tubular and reticular in pattern, with a perinuclear concentration as seen in Fig. 1D. (ii) Confocal optical sectioning along the z axis confirmed an apical concentration of antigen distribution characteristic of surface staining (Fig. 2 A–D, discussed below). (iii) Costaining with a known endothelial cell surface marker, CD31, produced overlapping distributions in z axis optical sections, as would be expected for surface localization (Fig. 2C). Flow cytometry of primary HUVECs also was performed with cells determined to be nonpermeabilized by dye exclusion. These cells demonstrated cell surface expression of the α-subunit of ATP synthase (data not shown), confirming that the staining of ATP synthase that we detect is not due to permeabilization but rather to the presence of this enzyme on the cell surface.
Figure 2.

Surface localization of the α-subunits of ATP synthase and CD31 on nonpermeabilized HUVECs by immunostaining and confocal microscopy. (A–D) Confocal optical sections were taken along the z axis every 1.5 μm. Each section is ≈0.6 μm in thickness. A series of z sections from a representative field is shown, starting with the basal surface in A and ending with the apical surface in D. (E) The same section shown in C; fluorescence from red channel only. (F) The same section shown in C; fluorescence from green channel only; n = 3.
The cell surface localization was investigated further by confocal microscopy comparing staining with CD31, an established endothelial cell surface marker, and the α-subunit of ATP synthase. Fig. 2 E and F demonstrates CD31 and α-ATP synthase staining, respectively, and Fig. 2C shows the overlay of these two images. The image in Fig. 2C was taken at approximately the midpoint between the basal and apical surfaces of cultured endothelial cells and clearly shows a marginal distribution of the punctate structures containing ATP synthase along the periphery of the cells. Fig. 2 A–D shows the same field of view in optical sections along the z axis, with A starting near the basal aspect and D ending near the apical aspect of the cultured cells. It is important to note that the apical surface would be equivalent to the luminal surface if these cells were within the vasculature. Although confocal microscopy emphasizes structures within the optical plane of sectioning, structures that are above or below the plane of section also may be visualized if their staining is particularly intense. However, structures that are outside the confocal plane will display fuzzy margins, whereas structures within the focal plane will show sharp margins. This effect is seen with the α-ATP synthase staining. Examination of the sharp, green spots demonstrates that essentially all α-ATP synthase staining is confined to the cellular margins in subapical sections of nonpermeabilized cells (Fig. 2 A–C). Moreover, α-ATP synthase exhibits a greater intensity of staining in the apical section (Fig. 2D), in which the confocal plane grazes the majority of the exposed apical surface, as would be expected for a surface-localized marker.
Angiostatin Binds to Bovine F1 ATP Synthase.
Human angiostatin bound to purified bovine F1 ATP synthase passively adsorbed onto microtiter wells in a concentration-dependent manner (Fig. 3). Human and bovine ATP synthase are highly homologous, differing only by eight aa residues in the mature α-chains (SWISS-PROT accession nos. P25705 and P19483, respectively) and six residues in the mature β-chains (SWISS-PROT accession nos. P06576 and P00829, respectively). Similar results were obtained with purified bovine F1 ATP synthase binding to immobilized angiostatin (data not shown). Microtiter wells coated with decreasing concentrations of purified bovine F1 ATP synthase also showed concentration-dependent binding of angiostatin (data not shown). Background binding of angiostatin to BSA-coated wells gave values comparable to the baseline seen in Fig. 3 (compare 0 μg/ml with 0.2 μg/ml), indicating that angiostatin requires F1 ATP synthase for efficient binding. Apparent dissociation constants [Kd(app)] were determined from double-reciprocal plots of the binding data as shown in Fig. 3 Inset. The Kd(app) for angiostatin binding to purified F1 ATP synthase is 14.1 nM. Although this suggests a 10-fold-higher affinity than our previously determined value for binding of angiostatin to the cell surface form of ATP synthase (18), it must be recognized that the holoenzyme associates with numerous other proteins on the cell surface and, thus, may be sterically hindered to a significant degree. In contrast, the purified F1 subcomplex would not be subjected to steric hindrance by associated proteins and would be expected to exhibit an increased affinity for its ligands.
Figure 3.

Binding of angiostatin to purified bovine F1 ATP synthase. ELISA was used to determine concentration-dependent binding of angiostatin to a constant amount of F1 ATP synthase. Each well was coated with 1 μg of F1 ATP synthase before addition of decreasing amounts of angiostatin. Control lane (−) shows binding of secondary antibody only (n = 6). (Inset) Apparent dissociation constants [Kd(app)] were determined from double-reciprocal plots of the binding data. Angiostatin bound to bovine F1 ATP synthase with a Kd(app) of 14.1 nM.
Angiostatin Inhibits Purified Bovine F1 ATP Synthase Activity.
Although the F1-FO ATP synthase holoenzyme efficiently catalyzes both the forward ATP synthase reaction and the reverse ATP hydrolysis reaction, the purified F1 ATP synthase subcomplex catalyzes only the reverse reaction. The ATP hydrolytic activity of the purified bovine F1 ATP synthase was measured by using a coupled enzymatic assay in which production of ADP is linked to oxidation of NADH via pyruvate kinase and lactate dehydrogenase (28). Thus, a decrease in the absorbance measured at λ = 340 nm indicates ATPase activity. Angiostatin completely inhibited purified ATPase activity, similar to a known F1 inhibitor, NaN3 (Fig. 4). Polyclonal antibodies directed against the recombinant α-, β-, and γ-subunits of ATP synthase also were tested. Both α- and β-subunit-specific ATP synthase polyclonal antibodies abolished ATPase activity. In contrast, the γ-subunit-specific ATP synthase antibody had no effect (data not shown). In addition, a commercial mAb specific for the α-subunit of ATP synthase (Molecular Probes) also inhibited activity. Preimmune, unrelated polyclonal, and unrelated mAbs used as controls had no effect on activity.
Figure 4.

Inhibition of purified F1 ATP synthase by angiostatin. Purified F1 ATP synthase activity was measured spectrophotometrically at λ = 340 nm by coupling the production of ADP to the oxidation of NADH via the pyruvate kinase and lactate dehydrogenase reaction, in which a decrease in the absorbance at λ = 340 nm indicates active protein ( ) (28). Angiostatin (10 μM) completely inhibited purified F1 ATP synthase activity (◊), comparable to a known F1 ATP synthase inhibitor, NaN3 (2%) (⧫) and an enzyme-free control (▴). Polyclonal antibodies directed against the recombinant α-subunit of ATP synthase (500 μg/ml) (▾) and β-subunit ATP synthase (700 μg/ml) (▿) abolished ATPase activity. A mAb to the α-subunit of ATP synthase (25 μg/ml) also inhibited activity (■). Control antibodies had no effect on activity (○, □). Representative data are shown; n = 3.
Surface-Associated ATP Synthase Is Catalytically Competent.
The presence of the α-, β-, and γ-subunits of ATP synthase on the endothelial cell surface suggests that the entire catalytic complex is present. To test for functional activity, we incubated HUVECs with [3H]ADP, [32Pi], and unlabeled phosphate (100 mM) and followed ATP production by analysis of supernatants on microcrystalline polyethyleneimine-cellulose TLC plates. ATP generation was detected within the first 15 s and reached maximal levels by 1 min. The possibility of ATP release from intracellular pools was discounted because the cells tested were intact, as evident by Trypan blue exclusion and lack of lactate dehydrogenase release (data not shown). Moreover, the ratio of [3H]ATP/[32P]ATP remained constant at 1.56 ± 0.04 over the time measured, thus demonstrating that the ADP and Pi substrates used to form ATP were derived exclusively from the external medium. No labeled product was detectable within the cell pellets (data not shown), again confirming that the ATP was synthesized on the cell surface.
To determine whether angiostatin inhibits ATP synthesis on the endothelial cell surface, we measured ATP production in the extracellular medium by using a bioluminescence assay. This assay is highly specific for ATP, to the exclusion of all other nucleotides. ATP concentrations significantly more than basal levels were detected in the extracellular milieu, indicating that de novo ATP synthesis occurred on the cell surface (Table 1). Release of intracellular ATP pools was excluded by the low level of ATP measured in the absence of ADP. Angiostatin inhibited ATP synthesis in a dose-dependent manner (Fig. 5). ATP synthesis was inhibited 81% by 1 μM angiostatin as shown in Table 1. Polyclonal antibodies against either the α- or β-subunits of ATP synthase inhibited cell surface production of ATP by 65% and 57%, respectively. In contrast, preimmune serum showed no inhibition. Oligomycin, a known inhibitor of the FO subcomplex, inhibited ATP synthesis by 84% under these conditions, confirming that the major activity assayed was a result of ATP synthase.
Table 1.
Inhibition of ATP generation on the surface of HUVECs as measured by bioluminescent luciferase assay
| Treatment applied to cultured HUVEC in the presence of 50 μM ADP | Percent inhibition ± SEM |
|---|---|
| Medium alone | 0 |
| Angiostatin (1 μM) | 81.0 ± 6.0 |
| Polyclonal α-ATP synthase (1.0 mg/ml) | 64.8 ± 3.2 |
| Polyclonal β-ATP synthase (0.5 mg/ml) | 56.8 ± 5.8 |
| Preimmune serum (1.0 mg/ml) | 0 ± 7.6 |
| Oligomycin (50 μg/ml) | 83.5 ± 4.5 |
Basal ATP levels in the assay medium (in the absence of ADP) produced bioluminescence signals equivalent to approximately 4% of the uninhibited levels in the presence of ADP. n = 3.
Figure 5.

Inhibition of ATP generation by angiostatin on the surface of HUVECs as measured by bioluminescent luciferase assay. ATP generation on the surface of HUVECs was inhibited in a dose-dependent manner in the presence of increasing concentrations of angiostatin. Representative data are shown; n = 3.
Inhibition of HUVEC Proliferation in the Presence of Angiostatin and Anti-β-ATP Synthase Antibodies.
We previously showed that an antibody raised against the α-subunit of ATP synthase abrogated the antiproliferative effects of angiostatin in an endothelial cell-proliferation assay. In the present study, polyclonal antibodies raised against the β-subunit of ATP synthase inhibited endothelial cell proliferation 1.5- to 2-fold more effectively than angiostatin (Table 2). The antiproliferative effect of the β-subunit-specific antibody approached that of cycloheximide (80.9% and 100%, respectively). However, this effect is unlikely to represent a toxic response because the cellular morphology of the anti-β-subunit ATP synthase-treated cells was unchanged from that of untreated cells, in contrast to the obvious rounded morphology caused by cycloheximide (data not shown). The preimmune serum exhibited no effect on proliferation. These data suggest that antibodies directed against the β-subunit of ATP synthase act as angiostatin mimetics in both biochemical and cell-based assays of ATP synthase function.
Table 2.
Inhibition of HUVEC proliferation in the presence of angiostatin and anti-β-ATP synthase antibody as measured by CyQUANT
| Percent inhibition ± SEM | |
|---|---|
| Medium only | 0 ± 1.034 |
| Angiostatin (1 μM) | 57.2 ± 1.16 |
| Cycloheximide (10 μg/ml) | 100 ± 7.0 |
| Polyclonal β-ATP synthase (100 μg/ml) | 80.9 ± 15.9 |
| Preimmune serum (100 μg/ml) | 0 ± 1.7 |
n = 3.
Discussion
We previously demonstrated by affinity chromatography that the α- and β-subunits of ATP synthase constitute the major endothelial cell binding site for angiostatin (18). Our initial study used a polyclonal antiserum raised against the α-subunit of ATP synthase to detect cell surface subunits and to block binding and endothelial cell-inhibitory activity of angiostatin. In that study, we did not investigate whether the cell surface form of ATP synthase was functional or whether other ATP synthase subunits also were present on the cell surface. We now extend our initial study to show that ATP synthase is catalytically active on the endothelial cell surface and that angiostatin-mediated inhibition of this activity correlates with inhibition of proliferation. Moreover, we demonstrate that certain antibodies directed against the α- and β-subunits of ATP synthase also inhibit enzymatic activity and endothelial cell proliferation. These antibodies, therefore, appear to constitute functional mimetics of angiostatin.
In addition to angiostatin, there are known inhibitors of ATP synthase that exhibit antitumor effects including piceatannol (25) and resveratrol (29). Resveratrol inhibits the development of 7,12-dimethylbenz[a]anthracene-induced preneoplastic lesions and tumor growth (30). Piceatannol was shown to inhibit tumor cell growth by inhibition of protein–tyrosine kinases (25). That both of these antitumor compounds inhibit ATP synthase suggests a relationship between the endothelial cell antiproliferative effects of angiostatin and cell surface-associated ATP synthesis.
The generally accepted concept that ATP synthesis is strictly an intracellular process now appears questionable. It is well established that both nucleosides and nucleotides act as extracellular-signaling molecules (31, 32). Indeed, the extracellular role of nucleotides in regulating multicellular communication is widely conserved in eukaryotic evolution (33). Extracellular receptors for ATP exist as both ion channels (P2X) and G protein-coupled receptors (P2Y) that are ubiquitous to all mammalian tissues (32). A recent study demonstrates that ATP release by physical or chemical means contributes to the set point of cellular-signaling pathways coupled to P2Y receptors (34). In the cardiovascular system, adenosine released from myocytes maintains blood flow to ischemic areas of the heart (35), and ATP may play a role in the moment-to-moment regulation of cardiac blood flow in nonpathological states (36). Endothelial cells release ATP in response to stimuli such as shear stress and vasoactive agonists including ATP (37, 38).
The availability of significant amounts of ATP on the surface of endothelial cells may provide several mechanisms to promote endothelial cell proliferation and increases in tumor blood flow. We originally hypothesized that ATP generated on the cell surface is transported into the cell, providing a source of energy for endothelial cells in the tumor microenvironment, where vascular pO2 levels can be as low as 12 ± 3 mmHg in tumor central vessels (39). A functional test of this hypothesis remains to be performed, and the present study does not address this issue. An alternate hypothesis is that surface-generated ATP is available to act through the P2Y receptor to activate Ca2+-dependent-signaling cascades, which increase DNA synthesis (32). This would tend to promote endothelial cell proliferation in the tumor environment. Our finding that angiostatin suppresses the generation of cell surface-associated ATP suggests that this antiangiogenesis factor might regulate one or more mechanisms that promote tumor vascularization.
Our immunolocalization data show that the α-, β-, and γ-subunits of ATP synthase are present on the endothelial cell surface, where they colocalize extensively. These subunits represent the minimal components required for efficient ATPase catalytic activity and strongly suggest that the cell surface form of ATP synthase is similar to the mitochondrial form. Moreover, cell surface ATP synthase is present in discrete foci, indicating structural organization of the enzyme complex. We also have detected numerous other mitochondrial matrix enzymes that colocalize with ATP synthase into these discrete foci on the endothelial cell surface, but mitochondrial outer-membrane markers were not detected (data not shown). These findings suggest that a highly complex energy-production apparatus exists on the endothelial cell surface.
Although the mechanism of angiostatin action on endothelial cells remains largely unknown, our data strongly suggest that angiostatin operates through inhibition of the enzymatic activity of cell surface ATP synthase. Data supporting this interpretation include concordance of inhibitory activities of angiostatin, known small molecule inhibitors of ATP synthase, and antibodies specific for ATP synthase subunits in binding and biochemical assays by using purified bovine enzyme. There are also similar effects of angiostatin and subunit-specific antibodies in the inhibition of ATP production on the surface of cultured human endothelial cells. It should be noted that existing small molecule inhibitors cannot be used in this assay because they also inhibit the mitochondrial form of ATP synthase, whereas neither angiostatin nor the subunit-specific antibodies cross the cell membrane. Finally, angiostatin and anti-β-subunit antibodies show similar ability to inhibit proliferation of cultured human endothelial cells.
Collectively, these data suggest a steric hindrance model of ATP synthase inhibition by angiostatin. ATP synthase couples proton flux across a membrane to rotation of the γ-subunit, which, in turn, induces cyclical conformational changes in the catalytic β-subunits (40). These conformational changes, required for conversion of ADP to ATP, can be blocked by any compound that either inhibits rotation of the γ-subunit or locks the conformation of the α- or β-subunits. It is well established that binding of specific antibodies can lock the conformation of flexible antigens. Thus, we propose that both angiostatin and certain antibodies to the α- or β-subunits of ATP synthase inhibit the enzyme by blocking conformational changes of the enzyme complex required for ATP synthesis or hydrolysis. This observation provides a great deal of hope that functional mimetics of angiostatin can be derived by developing mAbs against the α- or β-subunits of ATP synthase. Such antibodies likely would be far easier to produce and administer than angiostatin and may offer significant advances in antiangiogenic therapy of cancer and other proliferative diseases.
Acknowledgments
We thank Steve Conlon for confocal image preparation.
Abbreviation
- HUVEC
human umbilical vein endothelial cell
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Folkman J. In: Cancer Medicine. Holland J F, Frei E, Bast R, Kufe D, Morton D, Weichselbaum R, editors. Vol. 1. Baltimore: Williams & Wilkins; 1997. pp. 181–204. [Google Scholar]
- 2.Gullino P M. J Natl Cancer Inst. 1978;61:639–643. [PubMed] [Google Scholar]
- 3.Hanahan D, Folkman J. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 4.Folkman J, Watson K, Ingber D, Hanahan D. Nature (London) 1989;339:58–61. doi: 10.1038/339058a0. [DOI] [PubMed] [Google Scholar]
- 5.Folkman J. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 6.Kerbel R S. Carcinogenesis. 2000;21:505–515. doi: 10.1093/carcin/21.3.505. [DOI] [PubMed] [Google Scholar]
- 7.Carmeliet P. Nature (London) 1999;401:657–658. doi: 10.1038/44304. [DOI] [PubMed] [Google Scholar]
- 8.Bouck N, Stellmach V, Hsu S C. Adv Cancer Res. 1996;69:135–174. doi: 10.1016/s0065-230x(08)60862-3. [DOI] [PubMed] [Google Scholar]
- 9.Hanahan D, Weinberg R A. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 10.Folkman J. J Natl Cancer Inst. 2000;92:94–95. doi: 10.1093/jnci/92.2.94. [DOI] [PubMed] [Google Scholar]
- 11.Liotta L A. Sci Am. 1992;266:54–63. doi: 10.1038/scientificamerican0292-54. [DOI] [PubMed] [Google Scholar]
- 12.DeClerck Y A, Laug W E. Enzyme Protein. 1996;49:72–84. doi: 10.1159/000468617. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez-Gronow M, Gawdi G, Pizzo S V. J Biol Chem. 1994;269:4360–4366. [PubMed] [Google Scholar]
- 14.Gonzalez-Gronow M, Grenett H E, Weber M R, Gawdi G, Pizzo S V. Biochem J. 2001;355:397–407. doi: 10.1042/0264-6021:3550397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Reilly M S, Holmgren L, Shing Y, Chen C, Rosenthal R A, Moses M, Lane W S, Cao Y, Sage E H, Folkman J. Cell. 1994;79:315–328. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 16.Gately S, Twardowski P, Stack M S, Patrick M, Boggio L, Cundiff D L, Schnaper H W, Madison L, Volpert O, Bouck N, et al. Cancer Res. 1996;56:4887–4890. [PubMed] [Google Scholar]
- 17.Gately S, Twardowski P, Stack M S, Cundiff D L, Grella D, Castellino F J, Enghild J, Kwaan H C, Lee F, Kramer R A, et al. Proc Natl Acad Sci USA. 1997;94:10868–10872. doi: 10.1073/pnas.94.20.10868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moser T L, Stack M S, Asplin I, Enghild J J, Hojrup P, Everitt L, Hubchak S, Schnaper H W, Pizzo S V. Proc Natl Acad Sci USA. 1999;96:2811–2816. doi: 10.1073/pnas.96.6.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Reilly M S, Holmgren L, Chen C, Folkman J. Nat Med. 1996;2:689–692. doi: 10.1038/nm0696-689. [DOI] [PubMed] [Google Scholar]
- 20.Drixler T A, Rinkes I H, Ritchie E D, van Vroonhoven T J, Gebbink M F, Voest E E. Cancer Res. 2000;60:1761–1765. [PubMed] [Google Scholar]
- 21.Claesson-Welsh L, Welsh M, Ito N, Anand-Apte B, Soker S, Zetter B, O'Reilly M, Folkman J. Proc Natl Acad Sci USA. 1998;95:5579–5583. doi: 10.1073/pnas.95.10.5579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moser T L, Enghild J J, Pizzo S V, Stack M S. J Biol Chem. 1993;268:18917–18923. [PubMed] [Google Scholar]
- 23.Johnson D, Lardy H. Methods Enzymol. 1967;10:94–96. [Google Scholar]
- 24.Li S G, Gui L L, Lin Z H, Wan Z L, Chang W R, Liang D C. Biochem Mol Biol Int. 1996;40:479–486. doi: 10.1080/15216549600201043. [DOI] [PubMed] [Google Scholar]
- 25.Zheng J, Ramirez V D. Biochem Biophys Res Commun. 1999;261:499–503. doi: 10.1006/bbrc.1999.1063. [DOI] [PubMed] [Google Scholar]
- 26.Sottrup-Jensen L, Claeys H, Zajdel M, Petersen T E, Magnusson S. Progress in Chemical Fibrinolysis and Thrombolysis. New York: Raven; 1978. [Google Scholar]
- 27.Kirchner J G. Techniques of Chemistry in Thin Layer Chromatography. New York: Wiley; 1978. [Google Scholar]
- 28.Zheng J, Ramirez V D. Eur J Pharmacol. 1999;368:95–102. doi: 10.1016/s0014-2999(99)00012-6. [DOI] [PubMed] [Google Scholar]
- 29.Zheng J, Ramirez V D. Br J Pharmacol. 2000;130:1115–1123. doi: 10.1038/sj.bjp.0703397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jang M, Cai L, Udeani G O, Slowing K V, Thomas C F, Beecher C W, Fong H H, Farnsworth N R, Kinghorn A D, Mehta R G, et al. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 31.Hoyle C H V, Burnstock G. Adenosine in the Nervous System. London: Academic; 1991. [Google Scholar]
- 32.Ralevic V, Burnstock G. Pharmacol Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- 33.Soderbom F, Loomis W F. Trends Microbiol. 1998;6:402–406. doi: 10.1016/s0966-842x(98)01348-1. [DOI] [PubMed] [Google Scholar]
- 34.Ostrom R S, Gregorian C, Insel P A. J Biol Chem. 2000;275:11735–11739. doi: 10.1074/jbc.275.16.11735. [DOI] [PubMed] [Google Scholar]
- 35.Berne R M. Methods in Pharmacology. New York: Plenum; 1985. [Google Scholar]
- 36.Buxton I L O, Cheek D J. Adenosine and Adenine Nucleotides: From Molecular Biology to Integrative Physiology. New York: Kluwer; 1995. [Google Scholar]
- 37.Yang S, Cheek D J, Westfall D P, Buxton I L. Circ Res. 1994;74:401–407. doi: 10.1161/01.res.74.3.401. [DOI] [PubMed] [Google Scholar]
- 38.Bodin P, Burnstock G. J Cardiovasc Pharmacol. 1996;27:872–875. doi: 10.1097/00005344-199606000-00015. [DOI] [PubMed] [Google Scholar]
- 39.Dewhirst M W, Ong E T, Klitzman B, Secomb T W, Vinuya R Z, Dodge R, Brizel D, Gross J F. Radiat Res. 1992;130:171–182. [PubMed] [Google Scholar]
- 40.Boyer P D. Annu Rev Biochem. 1997;66:717–749. doi: 10.1146/annurev.biochem.66.1.717. [DOI] [PubMed] [Google Scholar]