

Abstract
Studies from a single laboratory have shown that in rodent models of permanent stroke, administration of the sulfonylurea glibenclamide (Glib) is highly effective in reducing edema, mortality, and lesion volume. The Stroke Therapy Academic Industry Roundtable (STAIR) recommends that new acute treatments for ischemic stroke to be replicated across different laboratories. Accordingly, we examined the effect of low-dose Glib in a permanent suture occlusion model of stroke. Male Sprague-Dawley rats underwent permanent middle cerebral artery occlusion (pMCAO) followed by an initial intraperitoneal injection of Glib (10 μg/kg) and the start of a constant infusion (200 ng/h) via miniosmotic pump at the onset of ischemia. Functional deficits were assessed by Neurological Severity Score (NSS) and grip-strength meter at 24 and 48 h after pMCAO. Glib-treated rats showed a significant reduction in infarct volume, lower NSS, and less hemispheric swelling compared to vehicle. Grip strength was decreased significantly in pMCAO rats compared to shams and significantly improved by treatment with Glib. Taken together, these data indicate that Glib has strong neuroprotective effects following ischemic stroke and may warrant further testing in future clinical trials for human stroke.
1. Introduction
Stroke has recently been downgraded from the third to the fourth leading cause of death in the United States, partly because of better acute ischemic stroke prevention and management among the elderly. However, despite these advances, therapeutic options for brain ischemic stroke remain limited, and there has been a rise in midlife stroke cases [1]. Currently, the only interventions to improve functional recovery after stroke are rehabilitative strategies, which have had limited success, and genetically engineered tissue plasminogen activators (tPAs), which are still the only medications approved by the FDA and come with serious medical risks and limitations. Thus, the problem of finding a safe and effective acute neuroprotective treatment after stroke remains urgent.
Posttreatment with Glibenclamide (Glib), a second-generation sulfonylurea, has been shown to reduce edema and mortality in brain injury models [2–5]. Several studies have found that Glib exerts beneficial effects by blocking the K-ATP channel [6] and/or SUR1 [2, 4]. Glib has also been shown to suppress neutrophil migration and chemotaxis following acute inflammatory response by blocking the K-ATP channel [7, 8] and to ameliorate damage caused by renal and intestinal ischemic reperfusion (IR) injury [7, 9]. Glib treatment also produces antioxidant effects independent of its K-ATP blocking activity [10, 11].
Previous studies have shown that in rodent models of severe ischemic stroke, administration of Glib is effective in reducing edema, mortality, and lesion volume [2, 4]. However, these findings have not yet been replicated in a permanent stroke model or motor function tests. Recently, Abdallah et al. [12] have shown Glib's neuroprotective efficacy in a global IR model via modulating oxidative stress and inflammatory mediators.
Numerous drug failures in clinical trials during the late 1990s resulted in the Stroke Therapy Academic Industrial Roundtable (STAIR) guidelines [13, 14], but these are often overlooked in the experimental literature. One STAIR recommendation calls for replicating findings in multiple laboratories and using focal permanent middle cerebral artery occlusion (pMCAO) models to better simulate the typical human stroke injury without reperfusion. The present study assessed whether Glib would show neuroprotective efficacy on hemispheric swelling, cerebral infarction, and functional deficits induced by pMCAO in the rat. Focal brain ischemia (pMCAO) was mechanically induced in Sprague-Dawley (SD) rats.
2. Materials and Methods
2.1. Subjects
Twenty-five male SD rats, approximately 90 days of age (300–350 g) at the time of surgery, were randomly assigned to a treatment or control group (IACUC, Emory University protocol 251–2008). Approved protocols meeting NIH guidelines require that investigators take all necessary steps to minimize the number of animals needed for statistical analysis and any unnecessary pain and discomfort caused by the experimental procedures. All animals were housed in a temperature-, humidity-, and light-controlled environment, placed under a 12 : 12 h reverse light : dark cycle, and handled daily for at least 5 days prior to surgery.
2.2. Permanent Focal Ischemia Model
Rats underwent pMCAO by intraluminal filament and suture method to occlude the right MCA. The animals were separated into three groups: sham-operated, vehicle-treated control (Sham, n = 7); pMCAO vehicle-treated (pMCAO, n = 9); pMCAO Glib-treated (pMCAO + Glib, n = 8). One rat was excluded because of inadequate occlusion. Criteria for inclusion of rats from the study group were based on laser-Doppler flowmetry (LDF) measurement of cerebral blood flow (CBF). LDF is an established, practical, and reliable system for monitoring changes in CBF due to induction of focal cerebral ischemia [15]. To ensure relative uniformity of the ischemic insult, animals with mean ischemic CBF > 30% (measured over a 5-minute period after occlusion) of baseline CBF were excluded from the cohort. This procedure resulted in more edema and more uniform infarcts, reducing experimental variability.
Anesthesia was induced by inhalation of 5% isoflurane (in a N2/O2 70%/30% mixture) and maintained by inhalation of 2% isoflurane. Using a SurgiVet pulse oximeter (model V3304; Waukesha, WI, USA), blood SpO2 was monitored and maintained at levels ≥ 90%. Body temperature was monitored throughout surgery via rectal probe and maintained at 37°C using a homeothermic heat lamp temperature controller (Harvard Apparatus, South Natick, MA, USA) by shutting off the lamp when rectal temperature exceeded 37°C. A small incision was made in the skin overlying the temporalis muscle and the laser-Doppler probe (Moor Instruments, Wilmington, DE, USA) was then positioned on the superior portion of the temporal bone (6 mm lateral and 2 mm posterior from bregma). Focal cerebral ischemia was induced by occlusion of the right MCA as previously described [16] with minor modifications. A midline incision was made on the ventral surface of the neck, and the right common carotid arteries were isolated and ligated with a 6.0 silk suture (Ethicon, San Angelo, TX, USA). The internal carotid artery and the pterygopalatine artery were temporarily occluded using a microvascular clip. A 4-0 silicon-coated monofilament 0.41 mm along its distal coating (4-5 mm) length (Doccol Co., Albuquerque, NM, USA) was introduced into the internal carotid artery through the incision in the external carotid artery. The filament was carefully advanced until the drop in blood flow was noticed. Relative CBF was monitored for 5 min following occlusion before the wound was sutured and the rats allowed to recover from the anesthesia.
2.3. Treatment
The rats subjected to MCAO and incurring ischemic insult less than 30% of baseline CBF measured for the 5 min period of occlusion were randomly assigned to receive Glib or vehicle treatment. Treatment included a loading dose of drug or vehicle plus delivery through a mini-osmotic pump (Alzet, Cupertino, CA, USA) subcutaneously implanted during the surgery. MCAO rats received an initial intraperitoneal injection of one bolus/loading dose of Glib (10 μg/kg) 5 min after occlusion plus the start of a constant subcutaneous infusion (200 ng/h) at the onset of ischemia and lasting for 48 h as described earlier [3]. All treatments were blinded and coded.
2.4. Analysis of Infarct Volume
The rats were killed for histology at 48 h after occlusion with an overdose (75 mg/kg) of Nembutal sodium solution and their brains extracted and evaluated for hemispheric swelling and infarct volume. The brains were carefully removed and placed in chilled saline, and then sliced into 7 serial coronal sections of 2 mm thickness using a rat brain matrix (Harvard Apparatus) starting at 3 mm posterior to the anterior pole. After sectioning, the slices were stained with 2% 2, 3, 5-triphenyltetrazolium chloride (TTC; Sigma Aldrich, St. Louis, MO, USA) in saline and kept for 15 min at 37°C. Both hemispheres of each stained coronal section were scanned using a high-resolution scanner (Epson Perfection 2400 Photo), and then evaluated by digital image analysis (Image Pro System, Media Cybernetics, Silver Spring, MD, USA). Unstained areas (pale color) were defined as ischemic lesions. The areas of infarction and total areas of both hemispheres were calculated for each brain slice. An edema index was calculated by dividing the total volume of the left hemisphere by the total volume of the right hemisphere. The actual infarct volume adjusted for edema was calculated by dividing the infarct volume by the edema index. Infarct volumes are expressed as percentage of contralateral hemisphere. These techniques have been used repeatedly in the literature to measure and evaluate stroke outcome in experimental preparations [17, 18].
2.5. Hemispheric Swelling
To assess the extent of cerebral hemisphere edema, the volumes of both hemispheres were calculated in arbitrary units (pixels) from the sum of coronal slice areas using Image J software after scanning with an optical scanner. Brain edema area was expressed as a percentage of the normal areas in the contralateral, unaffected hemisphere. The extent of swelling was calculated using the equation: Extent of edema = (volume of ipsilateral hemisphere − volume of contralateral hemisphere)/volume of contralateral hemisphere.
2.6. Neurological Severity Score
The neurological severity score (NSS) for severity of deficit was based on a scale of 0–8 (0, no neurological deficit; 1, left forelimb flexion when suspended by the tail, or failure to extend the right forepaw fully; 2, left shoulder adduction when suspended by the tail; 3, reduced resistance to lateral push toward the left side; 4, spontaneous movements in all directions with circling to the left exhibited only if pulled by the tail; 5, circle or walk spontaneously only to the left; 6, walk only when stimulated; 7, no response to stimulation; 8, stroke-related death) [19].
2.7. Assessment of Grip Strength
Forelimb grip strength in rats was determined before ischemia and 24 and 48 h after surgery using a grip-strength meter (Columbus Instruments, Columbus, OH, USA). We used an electronic digital force gauge that measured the peak force exerted by the action of the animal while gripping the sensor bar. While being drawn back along a straight line leading away from the sensor, the animal released its grip at some point and the gauge then recorded the maximum force attained at the time of release. The digital reading (in Newtons) of three successive trials was obtained for each rat, averaged, and used for data analysis.
2.8. Statistical Analysis
Mean ischemic CBF and hemispheric swelling were analyzed by one-way ANOVA. Student's t-test was used to compare lesion volume; a nonparametric Kruskal-Wallis rank test was used to compare NSS. Grip strength was expressed as a percentage of presurgery values for each rat and analyzed by ANOVA for repeated measures. Percent difference between pre- and postsurgical scores for each animal was used to control for individual differences in grip strength. All results were expressed as mean ± SE. The criterion for statistical significance was set at P < 0.05. The calculations were obtained using SPSS 19.0 software.
3. Results
3.1. Infarct Volume
Glib-treated rats showed a significant (P < 0.05) reduction in the volume of the infarct (% contralateral hemisphere) (42.79 ± 6.016.59%) compared to vehicle controls (57.44 ± 2.15) (Figure 1). Blood flow was measured over the ipsilateral parietal cortex. Mean relative CBF observed (5 min after MCAO) during occlusion in rats treated with Glib (76.83% ± 1.38%) was not statistically different from that observed in vehicle-treated animals (78.45% ± 1.84%).
Figure 1.
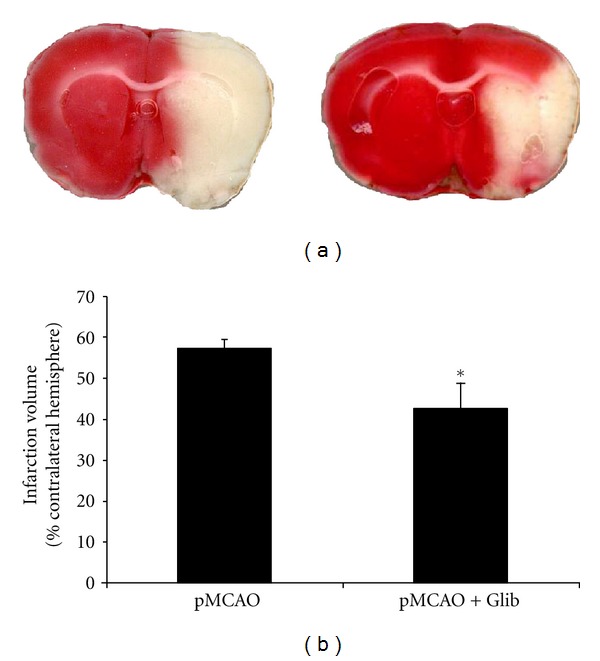
Glib reduces infarct volume. (a) TTC-stained coronal sections from representative animals given either vehicle or Glib, in brains harvested 48 h after occlusion. Infarcts are shown as pale (unstained) regions involving the striatum and overlying cortex. The infarct area in Glib-treated animals is substantially reduced.(b) Infarct volumes after 48 h of occlusion. Compared to vehicle alone, Glib significantly reduced hemispheric infarct volumes (% of contralateral structure). The data are represented as mean ± SE; P < 0.05; *significant difference compared to MCAO + Vehicle.
3.2. Hemispheric Swelling
Figure 2 graphs the effects of Glib on hemispheric swelling. Treatments with Glib significantly decreased hemispheric swelling (>50%) compared to the vehicle-treated group. Hemispheric swelling in Glib-treated rats was 12.16 ± 2.84% compared with 23.54 ± 2.5% in vehicle-treated animals.
Figure 2.
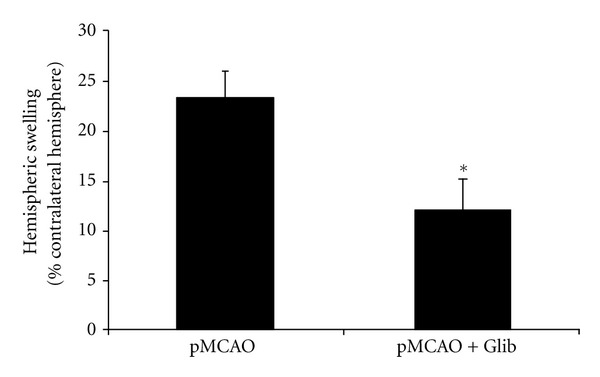
Glib attenuates hemispheric swelling in a rat model of pMCAO. The extent of swelling was calculated using the equation: Extent of edema = (volume of ipsilateral hemisphere − volume of contralateral hemisphere)/volume of contralateral hemisphere. Treatment with Glib significantly decreased hemispheric swelling compared to the vehicle-treated group at 48 h. P < 0.05; *significant difference compared to MCAO + Vehicle. Values are expressed as mean ± SE.
3.3. Neurological Severity Score
Repeated measures ANOVA on NSS showed significant group (F(1,13) = 5.17, P = 0.041) effects. Treatments with Glib after pMCAO significantly (P < 0.05) decreased the NSS (4.00 ± 0.92) compared to the vehicle-treated group (6.5 ± 0.42) at 2 days after occlusion (Figure 3). At 1 day after occlusion we observed no significant decrease in NSS in Glib-treated (4.43 ± 0.72) versus vehicle-treated animals (6.0 ± 0.50).
Figure 3.
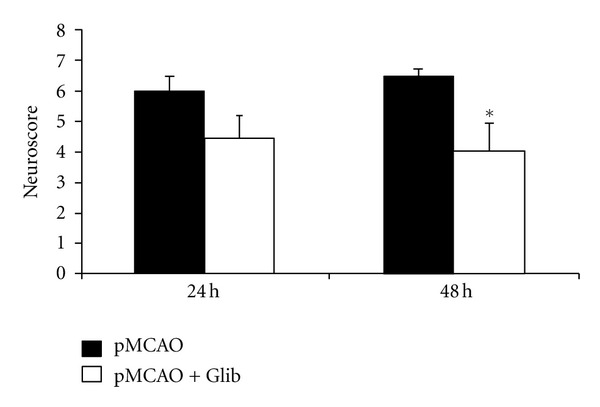
Glib improves neurological functions in a rat model of pMCAO. Treatments with Glib significantly decreased the NSS compared to the vehicle-treated group at 48 h after occlusion. P < 0.05; *significant difference compared to MCAO + Vehicle. Values are expressed as mean ± SE.
3.4. Grip-Strength Test
Grip-strength score (in Newtons (N)) is expressed as a percentage of presurgery control value. Treatments with Glib improved grip-strength deficits resulting from pMCAO (Figure 4). A repeated measures ANOVA showed significant group (F(2,21) = 14.14, P < 0.05) and time (F(1,63) = 8.10, P < 0.05) effects. Grip strength was decreased significantly (P < 0.05) in rats subjected to pMCAO (62.59 ± 3.63 and 40.83 ± 10.64 at 24 and 48 h after occlusion, resp.) compared to shams (98.03 ± 4.70 and 98.71 ± 2.88 N at 24 and 48 h after surgery, resp.), but was significantly (P < 0.05) improved with Glib treatment (85.48 ± 7.09 N and 60.63 ± 10.97 at 24 and 48 h).
Figure 4.
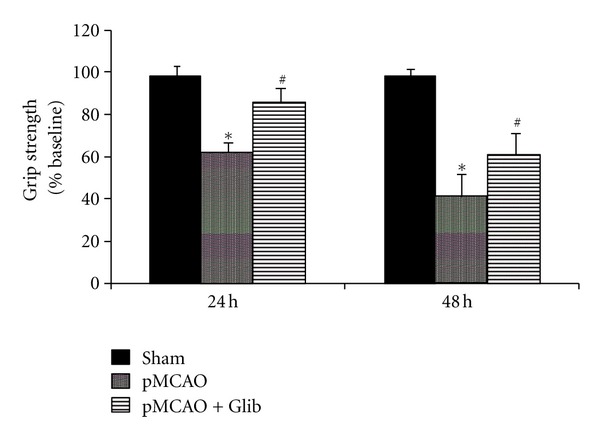
Glib improves grip strength in a rat model of pMCAO. The rats subjected to pMCAO + vehicle showed significantly (P < 0.05) lower grip-strength scores at both time points compared to sham + vehicle rats. Glib-treated rats showed significantly (P < 0.05) improved average scores at both time points (24 and 48 h after injury) compared to pMCAO + vehicle-treated animals. P < 0.05; *significant difference compared to Sham + Vehicle, #significant difference compared to pMCAO + Vehicle. Values are expressed as mean ± SE.
3.5. Physiological Variables
Using a SurgiVet pulse oximeter, blood SpO2 was monitored and maintained at levels greater than or equal to 90%. Body temperature was monitored throughout surgery by rectal probe and maintained at 37°C using a homeothermic heat lamp temperature controller (Harvard Apparatus) by shutting off the heat lamp when the rectal temperature exceeded 37°C. Laser-Doppler measurement of relative CBF was measured over the ipsilateral parietal cortex for 5 minutes before and after occlusion and expressed as a percentage drop from baseline. We observed no significant differences between groups in CBF during 5 minutes post-occlusion, pre- surgery glucose, or hemoglobin (Hb), suggesting that the relative ischemic insult, glucose, and Hb were equivalent for all groups (Table 1).
Table 1.
Physiological variables: Using a SurgiVet pulse oximeter, blood SPO2 was monitored and maintained at levels greater than or equal to 90%. Body temperature was monitored throughout surgery by rectal probe and maintained at 37°C using a homeothermic heat lamp temperature controller.
| Weight | % occlusion | Glucose | Hb | |
|---|---|---|---|---|
| Sham (n = 7) | 330.28 ± 14.37 | 203.85 ± 19.54 | 14.7 ± 0.72 | |
| pMCAO (n = 8) | 332.87 ± 13.85 | 78.45 ± 5.19 | 192.13 ± 21.84 | 15.14 ± 0.52 |
| pMCAO + Glib (n = 7) | 326.85 ± 18.08 | 76.83 ± 3.64 | 205.14 ± 29.13 | 15.11 ± 1.08 |
4. Discussion
Our study shows that continuous low-dose Glib administration after stroke significantly reduces cerebral infarct volume, hemispheric swelling, and functional deficits as assessed by NSS and grip-strength testing in adult male rats at 24 and 48 h after pMCAO. To date, only one previous study administered Glib to permanently occluded subjects [4]. In the present study, we administered a bolus dose of Glib (10 μg/kg) 5 min after occlusion followed by a constant subcutaneous infusion of 200 ng/h. This treatment regimen has been shown to reduce lesion volume after cerebral ischemia [2, 4].
Malignant brain edema resulting from a massive middle cerebral stroke contributes to the substantial mortality in patients [20]. Glib has been shown to reduce cerebral edema, infarct volume, and mortality by 50% in rodent models of stroke [2]. Although there are still unknowns about Glib's effects, some of the mechanisms underlying its edema-attenuating effects have been established. Glib blocks the sulfonylurea receptor 1 (SUR 1), which is thought to play a role in the formation and resolution of cerebral edema [2].
The mechanism of Glib may include direct cytoprotection as well as effects on regional CBF. Recently, it was shown that peripherally administered, radiolabeled Glib can reach the brain, and this uptake is increased in ischemia following pMCAO-induced blood-brain barrier breakdown [21]. The observation that there is preferential uptake of peripherally administered Glib into the ischemic brain but without significant accumulation in the brain [22] is important, given that some investigators maintain that reperfusion is required to achieve adequate compound penetration to observe any efficacy. The finding that Glib is neuroprotective in permanent focal ischemia suggests that reperfusion may not be essential. Since stroke produces systemic effects, the drug could also work via peripheral as well as central mechanisms. This is particularly relevant from a clinical perspective, since the majority of human stroke victims suffer from permanent occlusion [23–25].
To assess acute functional outcome, we chose to use a grip-strength meter. Recent studies indicate that grip strength is a sensitive index for the assessment of motor impairment induced by ischemia in the rat [26] and one of the few behavioral tests that yield objective quantitative data. To the best of our knowledge, previous studies have not examined the attenuation of motor deficits by Glib after a stroke. In the present study, we found that Glib treatment substantially improved behavioral deficits caused by pMCAO at 24 and 48 h after surgery and decreased the NSS compared to the vehicle-treated group at 48 h after occlusion, consistent with earlier observations.
Studies have shown that Glib also suppresses the inflammatory response, including the expression of proinflammatory cytokines, notably IL-10 [12], via blockade of K-ATP channels suppresses neutrophil migration and chemotaxis and indirectly reduces the proinflammatory cytokine TNF-α released by the cells [7–9]. Taken together, these findings suggest that some of the therapeutic benefits of Glib administration can be attributed to its regulation of inflammatory proteins, which prevents the increase in immune cell invasion and thereby reduces cerebral edema. It is likely that administration of Glib, by reducing the actions of these proinflammatory cytokines after ischemic reperfusion injury, results in a smaller cerebral infarction and less hemispheric swelling.
Inflammation can also result in activation of signaling pathways that can induce apoptosis [27]. Glib's inhibitory effect on apoptosis has been reported in other injury models. Caspase-3 activation has been described as a major cause of apoptotic processes after central nervous system (CNS) injury [28, 29]. Recently, Simard et al. [4] showed that Glib decreased cell death after subarachnoid hemorrhage by reducing caspase-3 activity. It has been suggested that Glib-induced inhibition of the SUR1-regulated NC(Ca-ATP) channel is involved in preventing activation of endothelial caspase-3 [5]. It is possible that Glib interrupts neuronal apoptosis occurring after ischemic episodes, and this would lead to a smaller infarct size and thus a better clinical outcome.
The present study looked only at Glib's acute effects when administered at the onset of ischemic stroke in relatively young adult subjects. For translational research, it will be important to perform a therapeutic time window study and replicate the findings in middle-aged and senescent subjects, where stroke is more prevalent. Ischemic stroke maturation occurs over several days. Therefore, in future preclinical studies, it will be important to compare the effects of Glib on long-term morphological and functional outcomes after stroke. Here, we can conclude that low-dose Glib has significant beneficial effects on hemispheric swelling, lesion volume, and functional outcome measures immediately following ischemic stroke. Taken together, our data and previous reports indicate that Glib is beneficial and warrants further testing in future clinical trials for human stroke.
Conflict of Interests
The authors declare no conflict of interests.
Acknowledgments
The authors thank Leslie McCann for her editorial assistance. This work was supported by AHA SDG Grant 11SDG5430002 to IS and research funds subcontracted from the Department of Neurology, University of Maryland, MD, USA.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation. 2012;125(1):e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simard JM, Chen M, Tarasov KV, et al. Newly expressed SUR1-regulated NCCa-ATP channel mediates cerebral edema after ischemic stroke. Nature Medicine. 2006;12(4):433–440. doi: 10.1038/nm1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simard JM, Tsymbalyuk N, Tsymbalyuk O, Ivanova S, Yurovsky V, Gerzanich V. Glibenclamide is superior to decompressive craniectomy in a rat model of malignant stroke. Stroke. 2010;41(3):531–537. doi: 10.1161/STROKEAHA.109.572644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simard JM, Geng Z, Kyoon Woo S, et al. Glibenclamide reduces inflammation, vasogenic edema, and caspase-3 activation after subarachnoid hemorrhage. Journal of Cerebral Blood Flow and Metabolism. 2009;29(2):317–330. doi: 10.1038/jcbfm.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou Y, Fathali N, Lekic T, Tang J, Zhang JH. Glibenclamide improves neurological function in neonatal hypoxia-ischemia in rats. Brain Research. 2009;1270:131–139. doi: 10.1016/j.brainres.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nistico R, Piccirilli S, Sebastianelli L, Nisticò G, Bernardi G, Mercuri NB. The blockade of K(+)-ATP channels has neuroprotective effects in an in vitro model of brain ischemia. International Review of Neurobiology. 2007;82:383–395. doi: 10.1016/S0074-7742(07)82021-6. [DOI] [PubMed] [Google Scholar]
- 7.Pompermayer K, Amaral FA, Fagundes CT, et al. Effects of the treatment with glibenclamide, an ATP-sensitive potassium channel blocker, on intestinal ischemia and reperfusion injury. European Journal of Pharmacology. 2007;556(1–3):215–222. doi: 10.1016/j.ejphar.2006.10.065. [DOI] [PubMed] [Google Scholar]
- 8.Da Silva-Santos JE, Santos-Silva MC, De Queiroz Cunha F, Assreuy J. The role of ATP-sensitive potassium channels in neutrophil migration and plasma exudation. Journal of Pharmacology and Experimental Therapeutics. 2002;300(3):946–951. doi: 10.1124/jpet.300.3.946. [DOI] [PubMed] [Google Scholar]
- 9.Pompermayer K, Souza DG, Lara GG, et al. The ATP-sensitive potassium channel blocker glibenclamide prevents renal ischemia/reperfusion injury in rats. Kidney International. 2005;67(5):1785–1796. doi: 10.1111/j.1523-1755.2005.00276.x. [DOI] [PubMed] [Google Scholar]
- 10.Al-Azzam SI, Abdul-Razzak KK, Jaradat MW. The nephroprotective effects of pioglitazone and glibenclamide against gentamicin-induced nephrotoxicity in rats: a comparative study. Journal of Chemotherapy. 2010;22(2):88–91. doi: 10.1179/joc.2010.22.2.88. [DOI] [PubMed] [Google Scholar]
- 11.Nazaroglu NK, Sepici-Dincel A, Altan N. The effects of sulfonylurea glyburide on superoxide dismutase, catalase, and glutathione peroxidase activities in the brain tissue of streptozotocin-induced diabetic rat. Journal of Diabetes and its Complications. 2009;23(3):209–213. doi: 10.1016/j.jdiacomp.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Abdallah DM, Nassar NN, Abd-El-Salam RM. Glibenclamide ameliorates ischemia-reperfusion injury via modulating oxidative stress and inflammatory mediators in the rat hippocampus. Brain Research. 2011;1385:257–262. doi: 10.1016/j.brainres.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 13.Fisher M. Recommendations for advancing development of acute stroke therapies: stroke Therapy Academic Industry Roundtable 3. Stroke. 2003;34(6):1539–1546. doi: 10.1161/01.STR.0000072983.64326.53. [DOI] [PubMed] [Google Scholar]
- 14.Fisher M, Feuerstein G, Howells DW, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40(6):2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dirnagl U, Kaplan B, Jacewicz M, Pulsinelli W. Continuous measurement of cerebral cortical blood flow by Laser-Doppler flowmetry in a rat stroke model. Journal of Cerebral Blood Flow and Metabolism. 1989;9(5):589–596. doi: 10.1038/jcbfm.1989.84. [DOI] [PubMed] [Google Scholar]
- 16.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 17.Bederson JB, Pitts LH, Germano SM. Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke. 1986;17(6):1304–1308. doi: 10.1161/01.str.17.6.1304. [DOI] [PubMed] [Google Scholar]
- 18.Belayev L, Busto R, Zhao W, Fernandez G, Ginsberg MD. Middle cerebral artery occlusion in the mouse by intraluminal suture coated with poly-L-lysine: neurological and histological validation. Brain Research. 1999;833(2):181–190. doi: 10.1016/s0006-8993(99)01528-0. [DOI] [PubMed] [Google Scholar]
- 19.Xia CF, Smith RS, Shen B, et al. Postischemic brain injury is exacerbated in mice lacking the kinin B2 receptor. Hypertension. 2006;47(4):752–761. doi: 10.1161/01.HYP.0000214867.35632.0e. [DOI] [PubMed] [Google Scholar]
- 20.Ayata C, Ropper AH. Ischaemic brain oedema. Journal of Clinical Neuroscience. 2002;9(2):113–124. doi: 10.1054/jocn.2001.1031. [DOI] [PubMed] [Google Scholar]
- 21.Ortega FJ, Gimeno-Bayon J, Espinosa-Parrilla JF, et al. ATP-dependent potassium channel blockade strengthens microglial neuroprotection after hypoxia-ischemia in rats. Experimental Neurology. 2012;235(1):282–296. doi: 10.1016/j.expneurol.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 22.Tomiyama Y, Brian JE, Jr., Todd MM. Cerebral blood flow during hemodilution and hypoxia in rats: role of ATP-sensitive potassium channels. Stroke. 1999;30(9):1942–1948. doi: 10.1161/01.str.30.9.1942. [DOI] [PubMed] [Google Scholar]
- 23.Kassem-Moussa H, Graffagnino C. Nonocclusion and spontaneous recanalization rates in acute ischemic stroke: a review of cerebral angiography studies. Archives of Neurology. 2002;59(12):1870–1873. doi: 10.1001/archneur.59.12.1870. [DOI] [PubMed] [Google Scholar]
- 24.Pantano P, Caramia F, Bozzao L, Dieler C, Von Kummer R. Delayed increase in infarct volume after cerebral ischemia: correlations with thrombolytic treatment and clinical outcome. Stroke. 1999;30(3):502–507. doi: 10.1161/01.str.30.3.502. [DOI] [PubMed] [Google Scholar]
- 25.Hacke W, Brott T, Caplan L, et al. Thrombolysis in acute ischemic stroke: controlled trials and clinical experience. Neurology. 1999;53(7, supplement 4):S3–S14. [PubMed] [Google Scholar]
- 26.Ishrat T, Sayeed I, Atif F, Stein DG. Effects of progesterone administration on infarct volume and functional deficits following permanent focal cerebral ischemia in rats. Brain Research. 2009;1257:94–101. doi: 10.1016/j.brainres.2008.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eltzschig HK, Eckle T. Ischemia and reperfusion—from mechanism to translation. Nature Medicine. 2011;17(11):1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broughton BRS, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40(5):e331–e339. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- 29.Yakovlev AG, Faden AI. Caspase-dependent apoptotic pathways in CNS injury. Molecular Neurobiology. 2001;24(1–3):131–144. doi: 10.1385/MN:24:1-3:131. [DOI] [PubMed] [Google Scholar]