

Abstract
Host species switches by bacterial pathogens leading to new endemic infections are important evolutionary events that are difficult to reconstruct over the long term. We investigated the host switching of Staphylococcus aureus over a long evolutionary timeframe by developing Bayesian phylogenetic methods to account for uncertainty about past host associations and using estimates of evolutionary rates from serially sampled whole-genome data. Results suggest multiple jumps back and forth between human and bovids with the first switch from humans to bovids taking place around 5500 BP, coinciding with the expansion of cattle domestication throughout the Old World. The first switch to poultry is estimated at around 275 BP, long after domestication but still preceding large-scale commercial farming. These results are consistent with a central role for anthropogenic change in the emergence of new endemic diseases.
Keywords: Bayesian phylogenetics, molecular clocks, bacterial evolution, host switching
1. Introduction
Staphylococcus aureus is a leading cause of hospital- and community-associated human infections. It has also adapted to other hosts, including wild birds and livestock, causing substantial losses to the dairy and poultry industries [1,2]. Genetic analyses have demonstrated that livestock-associated strains evolved adaptively following human-to-animal host jumps, leading to endemic clones that are largely host-restricted [2–4]. Some of these strains may also act as zoonoses for human infection [5]. A better understanding of these host jumps, and their ecological context, is necessary in order to design ways to prevent the emergence of new pathogens.
Here, we examine the long-term evolutionary history of S. aureus, using a panel of strains that represents the breadth of the species’ diversity. To deal with the challenges of these data, we introduce two methodological innovations. First, to add a temporal scale to our phylogeny, we estimate rates of substitution from whole-genome sequences of S. aureus with known dates of isolation in the recent past [6–8]. We use these estimates to constrain rates at sites least likely to be affected by weak purifying selection (which can decrease rates over the longer term [9]), while also allowing for rate variation across the tree. Second, we adapt a method from Bayesian phylogeography [10], to infer uncertain associations with ancestral hosts. Together, our method estimates topology, substitution rates, divergence dates, ancestral host states and number of host switches in a single simultaneous analysis.
2. Material and methods
To represent the global diversity of S. aureus, we selected 123 genotypes of multilocus sequence type data [11] (electronic supplementary material, table S1). Human strains comprised nasal carriage, common epidemic hospital and community-associated strains, as well as four divergent sequence types (STs) of clonal complex (CC) 75 [12]. We also include the major clones specific to bovid hosts (cattle, sheep and goats) and avian hosts (poultry, reared and wild birds). Despite no formal evidence of recombination (electronic supplementary material, methods), we conservatively excluded two genes with unusual patterns of conservation (electronic supplementary material, figure S1), leaving an alignment of 2265 bp from five genes. Although our data represent the global diversity of S. aureus, standard tests do not provide evidence of sequence saturation (electronic supplementary material, methods).
Bayesian phylogenetic analyses were performed in Beast v. 1.7.1 [6] using Beagle [13]. We used the HKY85 + Γ substitution model, and the uncorrelated lognormal model of changes in substitution rate [6], and partitioned our alignment by codon position [14].
To infer a temporal scale, we estimated the rate of nucleotide substitution using published whole-genome data from ST239, sampled over a 20-year period [7]. A rate estimate obtained from these data was used to inform a prior applying only to third codon positions. Such sites are largely synonymous, and so less affected by weak purifying selection that is effective over longer timescales (see electronic supplementary material, methods). To reconstruct host switching, we applied a phylogeographical model [10], replacing geographical locations with host types (electronic supplementary material, methods). Each global strain was classified as human, avian or bovid, and hosts of ancestral strains were inferred jointly with the other parameters. We did not distinguish between strains isolated from the bovid genera Bos, Ovis and Capra because they group closely within the tree and some strains are found in all three genera. For any given tree in the Markov chain Monte Carlo (MCMC) sample, the minimum date of the earliest jump from human to livestock corresponds to the oldest node with a livestock host state.
For our data, prior information strongly suggests that the ancestral host state was human, namely, the pseudogenization in animal strains of proteins involved in human colonization or pathogenesis [3,4] and the presence of related outgroup strains in indigenous human communities [12] and New World monkeys (Staphylococcus simiae) [15]. Accordingly, we extended published methods to allow us to constrain the root state in our analyses (electronic supplementary material, methods).
3. Results and discussion
Figure 1 shows the maximum clade credibility tree of our global sample of S. aureus. The four strains from CC75 form a well-supported outgroup to the other strains, with an estimated root age of 69 738 BP (24 678–142 433).
Figure 1.

Maximum clade credibility time tree. Node labels are posterior support values more than 80%. CC75 strains are not shown.
For the livestock-associated strains, figure 1 indicates five human-to-bovid jumps (table 1), which agrees well with estimates from the complete posterior sample (median 5.12 jumps; 95% Bayesian credible interval (BCI): 4.98–7.08), and from reconstructions using alternative methods implemented in Mesquite [16] (electronic supplementary material, methods). Each of the groups contains Bos, Ovis and Capra hosts with the exception of CC97 and ST126/694 identified only in cattle (Bos). The bounds on the ages of the jumps range from 0 to 9000 BP, but all occurred post-domestication [17,18], suggesting intimate contact between humans and animals as a principal driver of transmission and subsequent spread of S. aureus in domesticated animals. Incorporating uncertainty in ancient host associations, and the relative ages of the different jumps, we estimate the first transmission of S. aureus from human to bovids at 5512 years ago (BCI: 3656–9007), which corresponds well to the period of expansion of agriculture throughout the Old World—the Neolithic revolution [17] (figure 2a). The influence of domestication on human diseases in the agricultural age is well established [23], but this is the first dating study to imply its role in the emergence of animal diseases.
Table 1.
Dates (BP) of the inferred jumps between human (H), bovid (B) or avian (A) hosts. Dates are median posterior estimates with Bayesian credible intervals (BCIs) of the most common recent ancestor of the clade. Large groups of strain types (ST) are given as clonal complexes (CC) [2].
| direction | strains | date of host switch |
|
|---|---|---|---|
| median | 95% BCI | ||
| H to B | CC97 | 1832 | (879, 3108) |
| H to B | CC133/CC425 | 3113 | (1183, 6113) |
| H to B | CC151/CC130 | 5429 | (3082, 8981) |
| H to B | ST126/694 | 83 | (0, 379) |
| H to B | ST521/688 | 1134 | (192, 2760) |
| H to A | ST385/692 | 273 | (23, 786) |
| B to H | ST59/966/754 | 456 | (62, 1189) |
Figure 2.
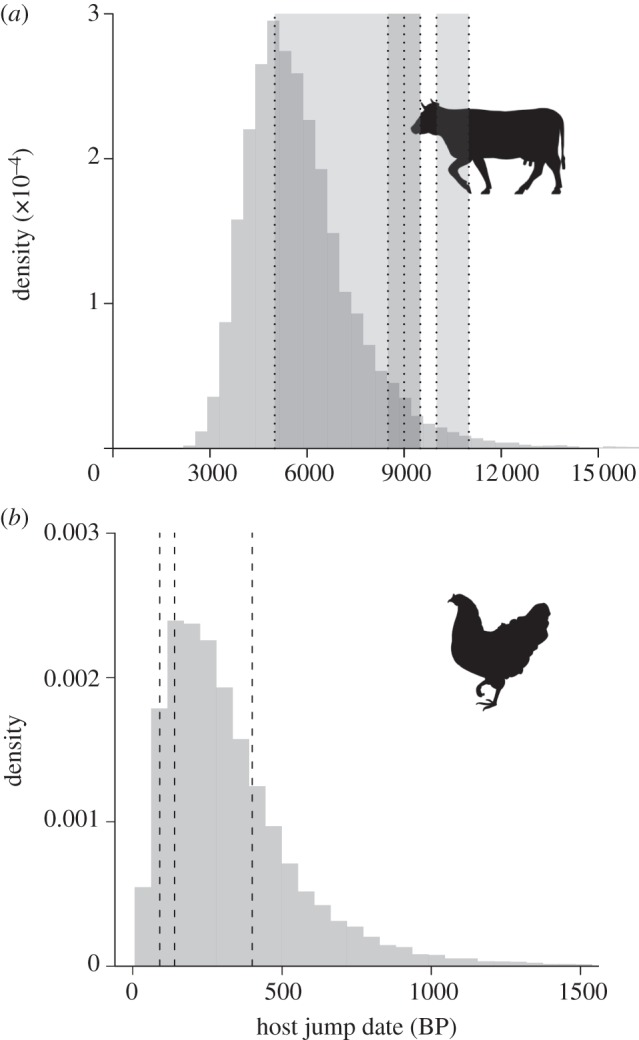
(a) The estimated date of the first host jump of Staphylococcus aureus from humans to bovids. Shaded areas represent the first evidence of independent management of sheep, goat and taurine cattle herds (10 000–11 000 BP); morphological responses to domestication (9500–9000 BP) [17,18]; expansion of bovid herds throughout the Fertile Crescent (8500–9500 BP) [18,19]; and bovid introduction to Europe (5000–9000 BP) [17]. (b) First host jump from humans to avian hosts, with dashed lines representing the arrival of poultry in America (400 BP) [20]; the expansion of flocks in North America (140 BP) [21,22]; and the first factory farms (90 BP) [21].
We also estimate that two bovid strains have subsequently jumped back into humans (median 2.06; BCI: 1.98–2.16). The first putative back-jump, ST25, presumably switched host very recently; ST25 is unique in lacking mecA-mediated resistance to methicillin but still exhibiting borderline oxacillin resistance. It has been suggested that the administering of antibiotics during non-lactating periods as a prophylaxis for mastitis in cattle could have selected for this borderline resistance phenotype [24]. The other bovid-to-human jump, involving ST59, occurred around 500 BP (table 1). The ST59 clone is a major epidemic clone in Southeast Asia and many strains demonstrate mecA-mediated resistance. However, the date of the host switch predates the introduction of methicillin and so the resistance phenotype cannot be due to farm management practices. These instances demonstrate the benefit of understanding historical host associations.
In contrast to bovids, the two strain types from poultry cluster together with approximately 100 per cent probability (figure 1), and so our data indicate a single human-to-avian jump that led to the establishment of an endemic clone (median 1.03; BCI: 0.97–1.21). We estimate that the first transmission occurred around 274 years ago (BCI: 75–785), well after the domestication of the modern-day chicken (about 8000 years ago; figure 2b) [21,25]. The recent age for this host jump is unexpected given the wide range of domesticated and wild avian hosts that are infected with these strain types [4]. This may imply more stringent host species-dependent barriers for transmission from humans to birds than to cattle (although our data preclude examination of very recent host jumps that have not led to endemic strains, such as the jump of an ST5 strain around 40 years ago [4]). With current data, we cannot predict if the initial host was a wild or domesticated bird, but we speculate that the higher frequency of interactions between humans and domesticated birds may have provided the opportunities for the initial host jump. During the period of the inferred jump, poultry was still farmed by small-holdings [21] but the poultry industry started to expand considerably about 140 years ago [21,22] presumably contributing to the dissemination of S. aureus among flocks.
4. Summary
We have employed novel Bayesian methods to investigate the frequency and timing of S. aureus host switching events that led to the emergence of global livestock pathogens. A correlation of the first human-to-bovid host jump with the spread of domestication in the Old World is consistent with increased opportunities for human to animal transmission. However, it is currently unclear whether our results are general, i.e. whether other pathogens associated with both humans and bovids (e.g. Streptococcus agalactiae) underwent host jumps around the time of the spread of domestication. This is due to the absence, for most bacterial pathogens, of datasets that yield reliable molecular timescales. However, with the increased use of next-generation sequencing of serially sampled and ancient data [7,26], this is certain to change in the near future, and our approach could be used to infer the ecological context of host shifts in a comparative framework.
Acknowledgements
We thank Michael Stanhope for S. simiae sequences, Francois Balloux and Andrew Leigh Brown for support, two anonymous reviewers for comments and the MRC and BBSRC (L.W. and J.R.F.), the NSF and NIH (M.A.S.) and the NESCent (M.A.S., A.R. and P.L.) for funding. Initial research received funding from the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 278433 and ERC grant agreement no. 260864.
References
- 1.Miles H., Lesser W., Sears P. 1992. The economic implications of bioengineered mastitis control. J. Dairy Sci. 75, 596–605 10.3168/jds.S0022-0302(92)77797-2 (doi:10.3168/jds.S0022-0302(92)77797-2) [DOI] [PubMed] [Google Scholar]
- 2.Smyth D. S., Feil E. J., Meaney W. J., Hartigan P. J., Tollersrud T., Fitzgerald J. R., Enright M. C., Smyth C. J. 2009. Molecular genetic typing reveals further insights into the diversity of animal-associated Staphylococcus aureus. J. Med. Microbiol. 58, 1343–1353 10.1099/jmm.0.009837-0 (doi:10.1099/jmm.0.009837-0) [DOI] [PubMed] [Google Scholar]
- 3.Guinane C. M., et al. 2010. Evolutionary genomics of Staphylococcus aureus reveals insights into the origin and molecular basis of ruminant host adaptation. Genome Biol. Evol. 2, 454–466 10.1093/gbe/evq031 (doi:10.1093/gbe/evq031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lowder B. V., et al. 2009. Recent human-to-poultry host jump, adaptation, and pandemic spread of Staphylococcus aureus. Proc. Natl Acad. Sci. USA 106, 19 545–19 550 10.1073/pnas.0909285106 (doi:10.1073/pnas.0909285106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.García-Álvarez L., et al. In press Meticillin-resistant Staphylococcus aureus with a novel mecA homologue in human and bovine populations in the UK and Denmark: a descriptive study. Lancet Infect. Dis. 11, 595–603 10.1016/S1473-3099(11)70126-8 (doi:10.1016/S1473-3099(11)70126-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drummond A. J., Suchard M. A., Xie D., Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. (doi:10.1093/molbev/mss075) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harris S. R., et al. 2010. Evolution of MRSA during hospital transmission and Intercontinental spread. Science 327, 469–474 10.1126/science.1182395 (doi:10.1126/science.1182395) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gray R. R., Tatem A. J., Johnson J. A., Alekseyenko A. V., Pybus O. G., Suchard M. A., Salemi M. 2010. Testing spatiotemporal hypothesis of bacterial evolution using methicillin-resistant Staphylococcus aureus ST239 genome-wide data within a Bayesian framework. Mol. Biol. Evol. 28, 1593–1603 10.1093/molbev/msq319 (doi:10.1093/molbev/msq319) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hasegawa M., Cao Y., Yang Z. 1998. Preponderance of slightly deleterious polymorphism in mitochondrial DNA: nonsynonymous/synonymous rate ratio is much higher within species than between species. Mol. Biol. Evol. 15, 1499–1505 10.1093/oxfordjournals.molbev.a025877 (doi:10.1093/oxfordjournals.molbev.a025877) [DOI] [PubMed] [Google Scholar]
- 10.Lemey P., Rambaut A., Drummond A. J., Suchard M. A., Fraser C. 2009. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 5, e1000520. 10.1371/journal.pcbi.1000520 (doi:10.1371/journal.pcbi.1000520) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Enright M. C., et al. 2000. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J. Clin. Microbiol. 38, 1008–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng J. S., Holt D. C., Lilliebridge R. A., Stephens A. J., Huygens F., Tong S. Y. C., Currie B. J., Giffard P. M. 2009. A phylogenetically distinct Staphylococcus aureus lineage prevalent among Indigenous communities in Northern Australia. J. Clin. Microbiol. 47, 2295–2300 10.1128/JCM.00122-09 (doi:10.1128/JCM.00122-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suchard M. A., Rambaut A. 2009. Many-core algorithms for statistical phylogenetics. Bioinformatics 25, 1370–1376 10.1093/bioinformatics/btp244 (doi:10.1093/bioinformatics/btp244) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shapiro B., Rambaut A., Drummond A. J. 2006. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol. Biol. Evol. 23, 7–9 10.1093/molbev/msj021 (doi:10.1093/molbev/msj021) [DOI] [PubMed] [Google Scholar]
- 15.Pantucek R., et al. 2005. Staphylococcus simiae sp. nov., isolated from South American squirrel monkeys. Int. J. Syst. Evol. Microbiol. 55, 1953–1958 10.1099/ijs.0.63590-0 (doi:10.1099/ijs.0.63590-0) [DOI] [PubMed] [Google Scholar]
- 16.Maddison W. P., Maddison D. R. 2011. Mesquite: a modular system for evolutionary analysis. Version 2.75 (http://mesquiteproject.org)
- 17.Zeder M. A. 2008. Domestication and early agriculture in the Mediterranean Basin: origins, diffusion, and impact. Proc. Natl Acad. Sci. USA 105, 11 597–11 604 10.1073/pnas.0801317105 (doi:10.1073/pnas.0801317105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeder M. 2009. The neolithic macro-(r)evolution: macroevolutionary theory and the study of culture change. J. Archaeol. Res. 17, 1–63 10.1007/s10814-008-9025-3 (doi:10.1007/s10814-008-9025-3) [DOI] [Google Scholar]
- 19.Horwitz L. K., Ducos P. 1998. An investigation into the origins of domestic sheep in the southern Levant. In Archaeozoology of the near east III (eds Buitenhuis H., Bartosiewicz L., Choyke A. M.), pp. 80–95 Groningen, The Netherlands: ARC Publications; (no. 18) [Google Scholar]
- 20.Gongora J., et al. 2008. Indo-European and Asian origins for Chilean and Pacific chickens revealed by mtDNA. Proc. Natl Acad. Sci. USA 105, 10 308–10 313 10.1073/pnas.0801991105 (doi:10.1073/pnas.0801991105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crawford R. D. 1990. Poultry breeding and genetics. Amsterdam, The Netherlands: Elsevier [Google Scholar]
- 22.Gillespie J. R., Flanders F. B. 2010. Modern livestock and poultry production. Clifton Park, NY: Delmar Cengage Learning [Google Scholar]
- 23.Diamond J. 2002. Evolution, consequences and future of plant and animal domestication. Nature 418, 700–707 10.1038/nature01019 (doi:10.1038/nature01019) [DOI] [PubMed] [Google Scholar]
- 24.Nadarajah J., Lee M. J. S., Louie L., Jacob L., Simor A. E., Louie M., McGavin M. J. 2006. Identification of different clonal complexes and diverse amino acid substitutions in penicillin-binding protein 2 (PBP2) associated with borderline oxacillin resistance in Canadian Staphylococcus aureus isolates. J. Med. Microbiol. 55, 1675–1683 10.1099/jmm.0.46700-0 (doi:10.1099/jmm.0.46700-0) [DOI] [PubMed] [Google Scholar]
- 25.West B., Zhou B.-X. 1988. Did chickens go North? New evidence for domestication. J. Archaeol. Sci. 15, 515–533 10.1016/0305-4403(88)90080-5 (doi:10.1016/0305-4403(88)90080-5) [DOI] [Google Scholar]
- 26.Bos K. I., et al. 2011. A draft genome of Yersinia pestis from victims of the Black Death. Nature 478, 506–510 10.1038/nature10549 (doi:10.1038/nature10549) [DOI] [PMC free article] [PubMed] [Google Scholar]