

Abstract
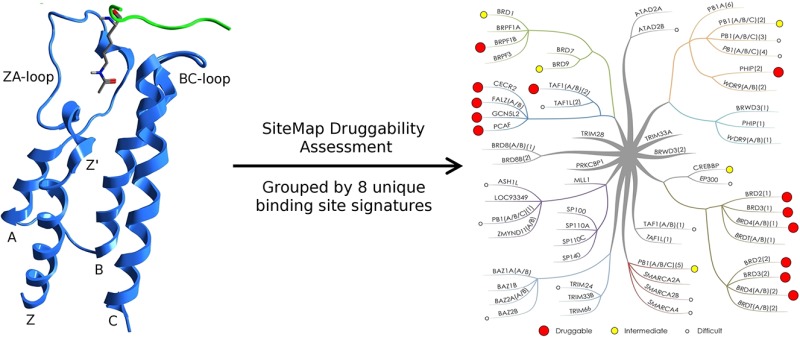
Bromodomains are readers of the epigenetic code that specifically bind acetyl-lysine containing recognition sites on proteins. Recently the BET family of bromodomains has been demonstrated to be druggable through the discovery of potent inhibitors, sparking an interest in protein–protein interaction inhibitors that directly target gene transcription. Here, we assess the druggability of diverse members of the bromodomain family using SiteMap and show that there are significant differences in predicted druggability. Furthermore, we trace these differences in druggability back to unique amino acid signatures in the bromodomain acetyl-lysine binding sites. These signatures were then used to generate a new classification of the bromodomain family, visualized as a classification tree. This represents the first analysis of this type for the bromodomain family and can prove useful in the discovery of inhibitors, particularly for anticipating screening hit rates, identifying inhibitors that can be explored for lead hopping approaches, and selecting proteins for selectivity screening.
Introduction
Epigenetic targets are increasingly explored in the field of drug discovery. Proteins of this target class are classified into readers, writers, and erasers of marks on histones or other nuclear proteins and DNA.1 The complex combinations of these posttranslational marks regulate gene expression and have been termed the “histone code”.2
Bromodomains represent one of the readers of these marks, specifically recognizing acetyl-lysine (KAc) through an architecturally conserved interaction module.3 Sixty-one unique bromodomains have been identified from the human genome,4 each containing a conserved tertiary structure as described by Mutjaba et al.5 This tertiary structure is an “atypical left-handed four-helix bundle”, with the hydrophobic KAc binding site at one end formed between the Z′ short helix, the ZA loop, and the BC loop (Figure 1A). This binding site is primarily hydrophobic, with the carbonyl oxygen of the acetyl-group forming two hydrogen bonds, one to a donor from either asparagine or threonine and the other to a conserved water molecule at the base of the pocket (Figure 1B,C).
Figure 1.

(A) Conserved protein fold of bromodomains comprising the four canonical helices αZ, αA, αB, and αC. (B) Surface representation of a typical KAc binding site. (C) Typical binding of KAc to bromodomain. All illustrated by FALZ (PDB 3QZS).
Through discovery of potent small molecule inhibitors (Figure 2),6 BET family members have been demonstrated to be druggable as defined by Hopkins et al.,7 a definition that will be used throughout the paper: proteins able (or predicted to be able) to bind drug-like molecules (not necessarily a drug). Bromodomain inhibitors have been investigated as potential therapeutics in multiple disease areas.8 A short hairpin RNA screen suggested that inhibition of the BET family may be a therapeutic strategy for AML.9 Through discovery of pan-BET family inhibitor GSK1210151A from the isoxazole class, it has been suggested that inhibition of the BET family may be a therapeutic strategy for MLL-fusion leukemia, and pan-BET family inhibitor GSK525762A, from the benzodiazepine class, has demonstrated anti-inflammatory potential in mouse models of inflammatory disease and sepsis.6,10 Inhibitors of other bromodomains (CREBBP and PCAF) have been found (Figure 2),11 but none show the submicromolar inhibition reported for BET family inhibitors so far. Bromodomains are currently an underexplored protein family in both basic biology and drug discovery, however, therapeutic potential is becoming increasingly recognized. With many bromodomain structures publicly available, this led us to investigate the structure-based druggability across the protein family.
Figure 2.

Selected published bromodomain inhibitors.
From an initial inspection of various bromodomain binding sites, we hypothesized that not all bromodomains would be as druggable as the BET family and a wide range of druggabilities would be observed. Further, we wanted to identify variations in the amino acids within the binding site that correlated with predicted druggability.
Prediction of the druggability of a novel protein target allows realistic expectations of hit rates before any screening effort is undertaken. For a less druggable target, the acceptable potencies and associated ligand efficiencies are likely to be lower than for a more druggable one and there is an associated risk of not finding tractable hit matter. In this scenario, alternative strategies may be sought such as higher screening concentrations, the use of larger and more diverse libraries, or the choice of screening technique employed.
One analysis of the druggability across a protein family was performed by Campagna-Slater et al. on another epigenetic target family, the histone methyltransferases.12 In this study, SiteMap was used alongside the degree of buried surface area of the bound cofactor to assess the druggability. All of the histone methyltransferases were predicted to be druggable with Dscores from SiteMap ranging from 0.96 to 1.13 but with the degree of buried surface area of the cofactor showing some variability.
Another study has also recently been published, performed by Santiago et al. primarily on methyl-lysine binding proteins, but bromodomain members were used for comparison.13 SiteMap was also used in this work, and the authors suggest the methyl-lysine binding proteins to be less druggable than bromodomains. However, they only consider the eight members of the BET family that may not be representative of the family as a whole.
Druggability Methods
Many structure-based druggability prediction methods have been published in recent years, these include DLID,14 DoGSiteScorer,15 the EBI’s DrugEBIlity,16 DrugPred,17 fPocket,18 MAPPOD,19 SCREEN,20 and SiteMap.21 Reviews by Hajduk et al.22 and Fauman et al.23 cover a number of these methods and some of the challenges in computational druggability assessment.
To assess the druggability of bromodomain proteins, we required a tool that is readily available but more importantly allows water molecules to be included in the analysis. This is necessary as we have identified five water molecules that appear to be conserved for most bromodomains and reduce the overall volume of the pocket. To our knowledge, the only tool that fulfils both of these criteria is SiteMap.
The use of SiteMap is consistent with the analyses of the histone methyltransferases and methyl lysine binding proteins highlighted above. A detailed validation of this method has been published, with SiteMap accurately identifying 86% of the ligand binding sites from a set of 538 complexes of the PDBBind database as the top scoring site.21 Further validation has been performed on the druggability assessment by Schmidke et al., demonstrating comparable performance of SiteMap to fPocket on their nonredundant data set (NRDD).18 SiteMap uses the same definition of druggable as we are using here and uses contributions from the volume of the pocket, the enclosure, and the degree of hydrophobicity to assess druggability. The main output from SiteMap is two druggability assessment scores: SiteScore (eq 1) and Dscore (eq 2) where n is the number of site points, e is the enclosure score, and p is the hydrophilic score.
| 1 |
| 2 |
Both scores take contributions from the same properties but with different coefficients. Both scores use a cap of 100 for the number of site points (for our analysis only two structures reach this cap), and SiteScore uses a cap of 1.0 for the hydrophilic score, whereas Dscore is not capped. For our data set, the two scores have high correlation, with R2 equal to 0.92. Because of the high correlation with SiteScore and the suggestion that it is more discriminatory of druggable and undruggable sites,21 Dscore was selected to be used alone in our analyses.
SiteMap was applied to a filtered set of the published bromodomain structures extracted from the PDB,24 and a wide range of predicted druggabilities was observed, from difficult to druggable. From this initial druggability assessment, the Dscores were compared with the clustering generated from whole sequence similarity of structure-based alignments by Filippakopoulos et al.4 This analysis showed that whole sequence similarity alone did not sufficiently explain the trends in the druggability that were observed, therefore we inspected the binding sites and identified unique amino acid signatures that showed better correlation with observed druggabilities. We propose that this new classification is more relevant to small molecule binding than whole sequence similarity due to its focus on the binding site residues. It allows druggability prediction of bromodomains without structural characterization and will aid the selection of templates for homology models by comparison to members within the same classification. Crucially, it also enables the medicinal chemist to identify family members that are likely to bind the same inhibitors as the targeted bromodomain, which can be explored either for lead hopping or selectivity screening.
Materials and Methods
Nomenclature
Many bromodomain-containing proteins possess multiple bromodomains but also exist as different sequence isoforms. When referring to a single bromodomain of one isoform, we have used this format: bromodomain-containing protein name, followed by isoform if present (A/B/C if isoforms are identical), followed by the bromodomain number. For example, the second bromodomain of the B isoform of BRD8 would be shortened to BRD8B(2), and the single bromodomain of BAZ1A, which is identical between isoforms A and B, would be shortened to BAZ1A(A/B).
When referring to different chains within a PDB file, we have used this format: PDB code followed by a letter corresponding to the number of the protein chain within the file. For example, the second chain of the protein BRD1 in PDB 3RCW would be shortened to 3RCW_B.
Protein Preparation
Protein chains within each PDB file were separated, ligands and nonconserved water molecules removed, and protonation states assigned using Protonate3D in MOE.25 Forty-six chains from 14 PDB files with unresolved binding site residues were filtered out (Supporting Information, Table S1). Bound state, resolution, presence of unresolved side chains, and presence of conserved water molecules were recorded for each chain. For TAF1(A/B), whereby both bromodomains have been crystallized within one peptide chain, these were separated and treated individually. Individual chains were then preprocessed using the Protein Preparation Wizard26 in Maestro27 with “Assign bond orders”, “Create disulfide bonds”, and “Convert selenomethionines to methionines” options selected.
Druggability Assessment Using SiteMap
The preprocessed chains were submitted to SiteMap using default parameters and with “Identify top-ranked potential receptor binding sites” to avoid any bias from using ligands/peptides to define pockets. The minimum number of site points per pocket identified needed to be reduced to 14 from 15 for PB1(A/B/C)(1). KAc binding sites were then selected from all identified sites and all outputted values recorded.
Structure Overlays and Sequence Alignment
Structure overlays were performed in MOE using the “align” module and the blosum62 matrix with default settings.25 As used in the full sequence alignment by Filippakopoulos et al.,4 we have also used BRD4(A/B)(1) as a reference sequence for numbering of the residues.
Generation of Figures of Structural Models
Figures were generated using MOE. Surfaces are color-coded using the pocket coloring from MOE with green indicating enclosed surface of the protein and white indicating exposed.
Graph Generation
Figure 4 and Figure 5 were generated using Aabel.28
Figure 4.

(A) Plot of Dscores obtained when conserved water molecules were included in analysis against the same structures with all water molecules removed. Linear line of best fit added to plot. (B) Histogram of same data showing distribution of scores with normal distribution fitted to this data. Colors indicate druggability classification: red, druggable; yellow, intermediate; white, difficult.
Figure 5.

Box-plots showing range and distribution of druggability for each bromodomain across available structures passing imposed filters (including presence of binding site water molecules). Ranked by median Dscore. Colors indicate druggability classification: red, druggable; yellow, intermediate; white, difficult. (a) Four outliers removed from druggability assessment (see group 4 text).
Classification Tree Generation
The classification tree (Figure 7) was created using iMindMap.29
Figure 7.

Bromodomain classification tree generated on the basis of eight binding site amino acid signatures showing bromodomain druggability. Numbering and branch colors consistent with groupings from Table 2. Druggability classification colors consistent with Figure 4 and Figure 5. Druggabilities of ASH1L, ATAD2B, BRPF1B, PB1(A/B/C)(3), PB1(A/B/C)(4), and SMARCA2B assessed using structures failing imposed filters included (see group text).
Results and Discussion
Having selected SiteMap to assess druggability, the next step was to collect the available crystal structures from the PDB. This yielded 105 different PDB entries covering 33 of the 61 unique human bromodomains. These PDB entries were then separated into the separate protein chains, as each protein chain within a crystal structure can be of a different conformation, and any chains with unresolved residues in the binding site were removed.
Significance of Water Molecules
Through inspection of available bromodomain structures, it was apparent that five water molecules are conserved across most bromodomain KAc binding sites. No publicly available structures demonstrate the displacement of any of these by a ligand (Figures 1A, 3, and Supporting Information, Figure S1), suggesting that the water molecules are an important feature of the binding pocket. Frequently, water molecules are removed prior to druggability assessment and we decided to determine druggability in the presence and absence of these conserved water molecules to assess their effect on the Dscore. All five water molecules could be identified in structures of 23 of the 28 unique bromodomains passing the requirement of a structure without unresolved binding site residues, although not all of the water molecules were always present in the same structure due to limitations of protein crystallography (most frequently in low resolution structures). To maximize our coverage of the observed protein conformations while ensuring that all assessed structures contained the same number of water molecules, for structures with missing water molecules, structures of the same protein with the missing water molecules were aligned and the missing water molecules were included from the other structure(s). For SMARCA4, a high resolution (1.50 Å) structure and one bound with NMP were available and both of these structures demonstrate only four of the five water molecules present, so the druggability assessment has been assessed as such, raising the number of bromodomains initially considered to 24. Details of all the water molecules included can be found in Supporting Information, Table S3.
Figure 3.
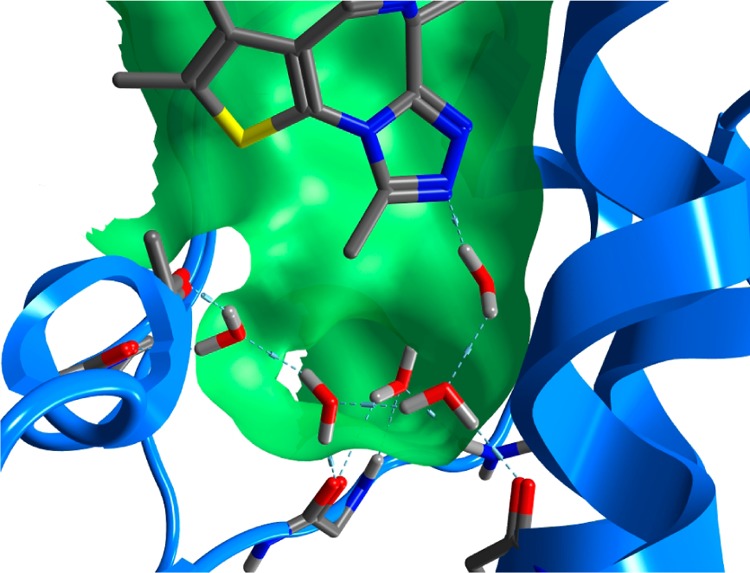
Conserved water molecules in the binding site of BRD4(A/B)(1) (PDB 3MXF).
The absence of these water molecules led to a larger identified pocket and consequently a higher druggability score, with most of the bromodomains classified in the druggable range (Dscore >0.85). Crucially, without the water molecules, a smaller range of scores was observed, making the assessment less discriminative between sites (Figure 4). Given that these water molecules enclose the pocket and have a significant effect on the druggability, all subsequent analysis was performed with the water molecules present. Inclusion of all five water molecules also allows direct comparison between bromodomains.
Druggability Assessment by SiteMap
The 178 qualifying protein chains (24 of the 61 unique human bromodomains) were prepared and submitted to SiteMap druggability assessment. A wide range of druggabilities was observed for the bromodomains from difficult (Dscore <0.75) (e.g., BAZ2B PDB 3Q2F, Dscore = 0.52) to druggable (Dscore >0.85) (e.g., PCAF PDB 3GG3_B, Dscore = 1.08). Scores in between these two have been classified as intermediate (e.g., CREBBP PDB 3P1E_B, Dscore = 0.82) (Figure 5). Details of all outputted scores from this assessment can be found in Supporting Information, Table S3.
Most bromodomains contain a small and tight binding site to recognize KAc of the protein substrate. This conveys a basic level of druggability as demonstrated by BAZ2B (PDB 3Q2F, Dscore 0.52) and the potential to bind a small fragment with acceptable ligand efficiency30 (NMP, BAZ2B LE 0.29).31 The differences between the sites stem from the environments around that small and tight pocket.
For the bromodomain family as a whole, even the lowest scoring bromodomain (PB1(A/B/C)(1)) would be classed as more druggable than a protein–protein interaction with a large (>1000 Å2) and fairly featureless interface. SiteMap would fail to identify binding sites such as this (e.g., SIAH1 PDB 2A25). At the other end of the druggability scale, some bromodomains have demonstrated comparable predicted druggability to what are currently considered druggable targets such as protein kinases (e.g., Aurora A, PDB 1MQ4, Dscore = 0.96) or Hsp90 (PDB 1AM1, Dscore = 0.99).
Effect of Bound State on Predicted Druggability
Apo crystal structures give only one conformational snapshot of a protein. When a ligand or peptide binds, the observed conformation of the protein may change to potentially induce a more druggable pocket. This has been seen for the Bcl-2 family of proteins whereby small molecule inhibitors bind to pockets not observed in the apo or peptide bound structures and show improved potency.32 This is in line with an increase in the SiteMap predicted druggabilities of the Bcl-xL binding sites from apo (PDB 1R2D_A, Dscore = 0.76) to ligand bound (ABT-737, PDB 2YXJ, Dscore = 0.95). To assess whether this could be the case for bromodomains, those with both apo and ligand or peptide bound (holo) structures available were collated and the Dscores from SiteMap compared (Table 1).
Table 1. Analysis of Effect of Ligand Binding on the Predicted Druggability of Bromodomains with Both Apo and Holo Structures Available That Passed the Imposed Filtersb.
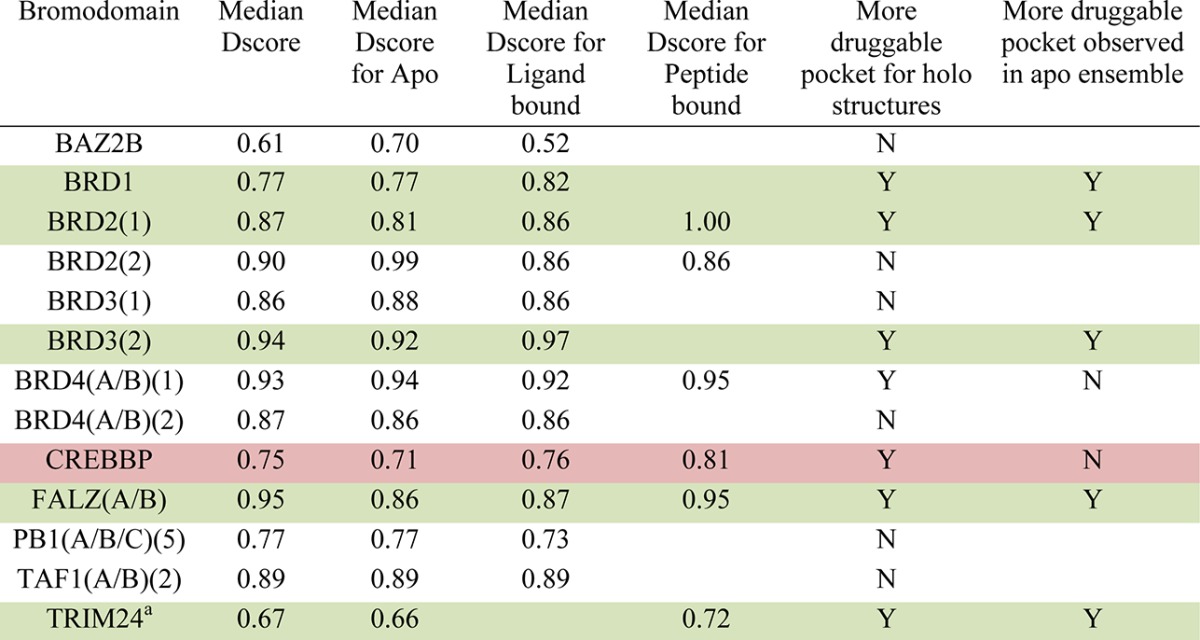
Dscores after four outliers were removed (see group 4 text for details).
Colors: white, bromodomains for which a more druggable pocket was not observed for the holo structures; green, bromodomains for which a more druggable pocket was observed for the holo structures but a comparably druggable pocket was observed in the ensemble of apo structures; red, bromodomains for which a more druggable pocket was observed for the holo structures and a comparably druggable pocket was not observed in the ensemble of apo structures.
From Table 1, it can be seen that only CREBBP of the 12 bromodomains with apo and holo structures available show evidence of a more druggable pocket (0.05–0.1 increase in median Dscore) in the presence of a ligand or peptide that is not observed in the ensemble of apo structures. The median Dscore for the entire ensemble of structures was classed as intermediate at 0.75, but the highest scoring structure was classed as druggable at 0.89 (PDB 3P1C_B) when bound with KAc.
Three bromodomains (BRD2(2), BAZ2B, and PB1(A/B/C)(5)) unexpectedly show reduced median druggability for the ligand or peptide bound structures when compared to the apo. Comparably druggable conformations of the holo structures for BRD2(2) and PB1(A/B/C)(5) within the range of the apo are observed in the full ensemble, but this is not the case for BAZ2B. These effects will be discussed later, in the context of the similarity of each bromodomain with other members of the family. Other than for CREBBP, in general it does not appear that ligand binding is able to induce a significantly more druggable structure than is observed in the apo ensemble for the bromodomain family.
Grouping of Bromodomains by Common Binding Site Features
Having completed the initial computational druggability assessment, the next step was to identify trends within the data set. When compared with the clustering generated from whole sequence similarity performed by Filippakopoulos et al.,4 a lack of correlation was observed such as the first and second bromodomains of TAF1 being placed in the same cluster despite Dscores at either end of the scale. It is not surprising to see a lack of correlation between druggability and whole sequence similarity, as when dealing with a druggability assessment it is the nature of the binding site that affects the score, not the rest of the domain. For this reason, we decided to inspect the structures for binding site for features that vary across the bromodomain family and can be used to order the members of the family into groups.
This led to the identification of eight groups characterizing 49 of the 61 unique bromodomains. Each of these groups is defined by the presence of a unique signature of up to three amino acid residues that is shared by all members of that particular group and gives a characteristic shape to the KAc binding site (Table 2, Figure 9, and Supporting Information, Figure S1). Taken together, the amino acid residues of all group signatures span seven residues that enclose the KAc binding site. These were position 81, which is a tryptophan in the BET family and forms what has been termed the ZA-channel,6 the two residues facing the binding pocket on the ZA-loop, both leucine at position 92 and 94 in the BET family, the residue at position 140, which is most commonly asparagine and forms the key hydrogen bond donor interaction with the KAc carbonyl, residues 144 and 146 on the BC-loop and C-helix, which enclose the hydrophobic shelf in the BET family, and residue 149, which although not enclosing the pocket does influence the position of residue 81, which can have a large effect on both the ZA-channel and hydrophobic shelf. Residue 145 on the C-helix has also been included in the analysis, which although not part of any of the signatures has been used in further differentiating some of the bromodomains within each of the groups. Thus, in total, eight residues have been used to characterize the bromodomain binding sites (Figure 6).
Table 2. Summary of the Signature Residues Used to Define the Nine Bromodomain Groupings and the Range of Median Dscore for the Group.

Number of bromodomains with available structures passing the filters out of the number of members in the group.
Median Dscores.
Median Dscore with four outliers removed (see Group 4 text).
Figure 9.
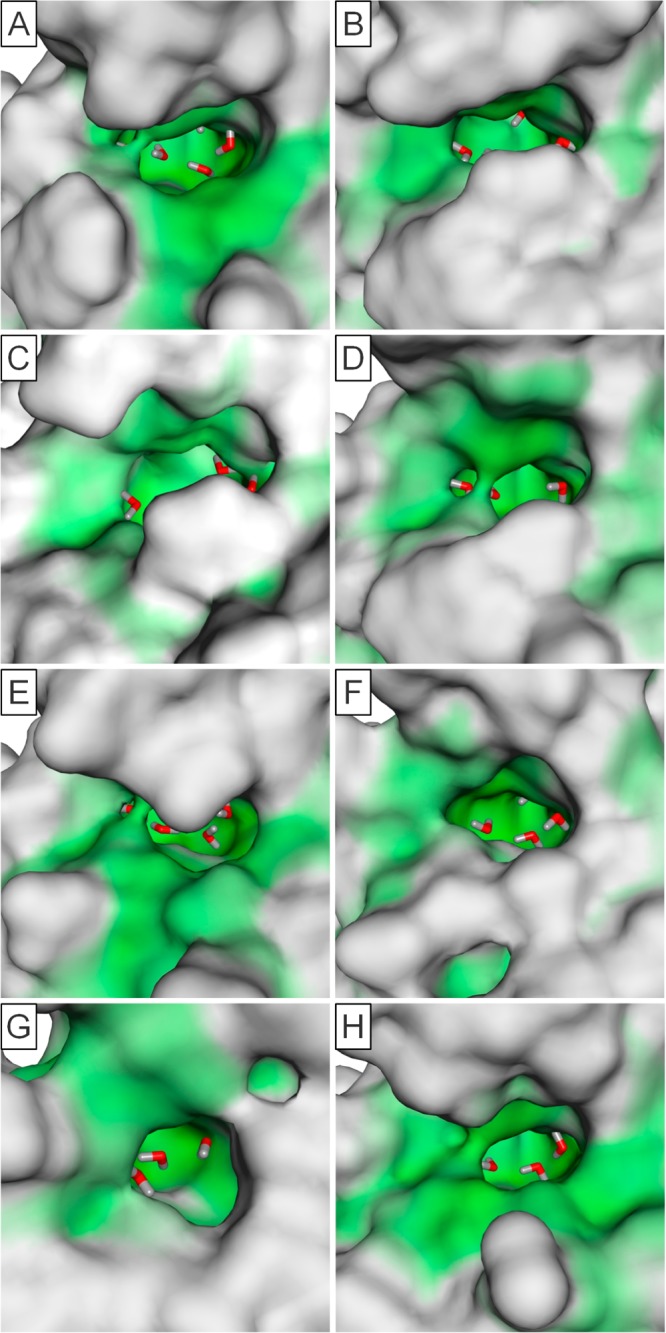
Bromodomain binding site similarity groups exemplified by the surfaces of representative example. Bromodomains aligned with BRD4(A/B)(1) PDB 3MXF and colors generated using MOE Pocket coloring: green = enclosed and white = exposed. Pocket colors are used to highlight binding sites and does not represent pockets identified by SiteMap used for druggability assessment. Images captured from the same viewpoint except image G at the same orientation as Figure 6. (A) BRD4(A/B)(1) PDB 3MXF structure representative of group 1. (B) PCAF PDB 3GG3_B structure representative of group 2. (C) BRD1 PDB 3RCW_A structure representative of Group 3. (D) BAZ2B PDB 3G0L structure representative of group 4. (E) PHIP(2) PDB 3MB3 structure representative of group 5. (F) SMARCA4 PDB 2GRC structure representative of group 7. (G) PB1(A/B/C)(1) PDB 3IU5 structure representative of group 8. (H) CREBBP PDB 3P1C_B structure.
Figure 6.

Eight residues around the binding site used in analysis (BRD4(A/B)(1) used as reference, PDB 3MXF). (A) Binding site residues shown as ribbon representation. (B) Binding site residues shown with transparent surface representation.
To determine which residues were present at each position, available bromodomain structures were overlaid with the reference structure, BRD4(A/B)(1) (PDB 3MXF), and the eight binding site residues were recorded that best aligned with the BRD4(A/B)(1) residues (Supporting Information, Table S5 and Figure S1). For five of the eight identified residues (140, 144, 145, 146, and 149), the spacial alignment corresponded with the sequence alignment, making the identification of the residues for the bromodomains without a structure straightforward. For the other three residues, due to the variation in length and position of the ZA-loop, spatial alignment did not always correspond with the sequence alignment. Here we have used the residue that aligned best in space with the BRD4(A/B)(1) structure, as this should be more relevant to the nature of the binding pocket. For bromodomains without a structure, the matching residue from a protein within the same grouping from the alignment of Filippakopoulos et al.4 was used (e.g., between BRPF1B and BRPF3). In a few cases, this was not always possible due to the lack of a sufficiently homologous bromodomain with a structure being available (e.g., MLL1 or TRIM28). These bromodomains have been excluded from groups that are characterized by the ZA-loop residues (81, 92, and 94).
Generation and Analysis of Binding-Site Classification Tree
Using the binding site groupings obtained, a qualitative classification tree was generated (Figure 7). This allowed plotting of the predicted druggabilities to visualize where the most druggable groups are as well relationships between the groups. These included the relationship between CREBBP and EP300 with the BET family as they share the extended length of the ZA-loop, the relationship between groups 2 and 3 (Y or F at position 146), and between groups 5 and 6 (Y or F at position 94) (see group texts for more details).
Furthermore, we further divided several groups into subclassifications such as the separation of the BAZ family within group 4 by exploring changes in whole sequence similarity that may have an effect on the overall fold of the bromodomains such as the ZA loop position or more subtle changes in the binding site that have smaller effects on the pockets than the signature residues. These subclassifications will be described in the context of each group.
We believe that this grouping better explains the trend in druggability assessment than whole sequence similarity, but also that this grouping will predict small molecule selectivity patterns more accurately due to the focus on the binding site. This should prove useful when determining selectivity of inhibitors and the potential to identify possibilities to transfer hit matter from one bromodomain to another. Another potential use of this grouping is for the building of a homology model. If the use of the model is to predict the binding mode of a small molecule inhibitor or to select compounds in a virtual screening approach, then the choice of template is very important. The bromodomains grouped together here share binding site features, and thus members of the same group should represent the best templates for building homology models for binding mode prediction.
Whole Sequence Clustering versus Binding Site Grouping
When comparing the clustering based on whole sequence similarity to the grouping performed here, differences can be observed (Figure 8). The two classifications are not dramatically different (42/61 placed in the same group), which is not surprising, but there is sufficient difference to suggest that when dealing with small molecule inhibitors the binding site classification may be more informative.
Figure 8.
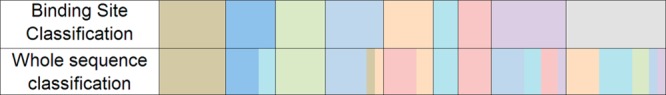
Comparison between composition of groups from binding site and whole sequence classifications. Groups from left to right in same number order as Table 2 with the same coloring. Whole sequence classification colors generated from binding site classification group which shares highest percentage similarity.
An example of the differences is BAZ1A(A/B) that shares whole sequence similarity with the BET family but does not share binding site similarity with this family. It is therefore unlikely to bind similar ligands and likely possesses druggability similar to that of the group it has been placed in by binding site classification (group 4). Another example is that ATAD2A and ATAD2B are placed in the same cluster as group 3 by whole sequence similarity but do not share binding site similarity with this group or any other bromodomain.
Each of the binding site classification groups will now be discussed individually, commenting on their druggability, but also any trends or inconsistencies within the groups.
Features of Group 1 (BET Family) Conveying Druggability
The BET family of bromodomains was classified as druggable, which correlates with the fact that a number of potent small molecule inhibitors have been found.6 The comparatively high druggability for this family can be explained by a more enclosed upper part of the pocket and thus additional surface area for interaction with small molecules not present in other less druggable bromodomains. This is predominantly due to the presence of a tryptophan residue at position 81 and a methionine residue at position 149 that influences the position of the tryptophan residue, forming the ZA channel. On the other side of the pocket, the ZA loop is longer than most other bromodomains, providing additional surface that can be utilized for interaction with small molecules. These features result in above-average druggability of the sites (Figure 9A).
Given the high similarity of the pockets, it is not surprising that nonselective BET family inhibitors have been found, although there are subtle differences between the first and second bromodomains of the BET family that could be exploited for selectivity. The entire BET family has the same ZA loop residues facing the pocket, but the first bromodomains possess an aspartate at position 144 whereas the second bromodomains possess a histidine. At position 145, the entire BET family has an acidic residue but this changes between aspartate and glutamate. We have separated this group in the classification tree into the first and second bromodomains to reflect these small changes.
An outlier in the druggability assessment was seen for a peptide bound structure of BRD2(2) (PDB 2E3K_B, Dscore = 0.64). The reason for this low score was the position of the tryptophan at position 81. For the rest of the BET family (and other BRD2(2) structures), this residue is directed toward the binding site creating the ZA channel (Figure 10A). In this outlier, the tryptophan residue is directed away from the pocket, opening the pocket significantly (Figure 10B) and inducing a pocket more like CREBBP or EP300 (leucine at position 81) (Figure 10C), greatly reducing the druggability. For the 56 BET family structures passing the initial filters, this is the only one for which the tryptophan is oriented away from the binding site, suggesting that this is an unusual conformation and does not appear relevant to small molecule inhibitor binding.
Figure 10.

Comparison of BRD2(2) structures. Surface colors consistent with Figure 9. (A) BRD2(2) PDB 3E3K_C structure showing typical BET family conformation. (B) BRD2(2) PDB 2E3K_B structure showing atypical conformation. (C) CREBBP PDB 3DWY_A structure showing similarity to BRD2(2) atypical conformation.
Features of Groups 2 and 3
Group 2 consists of six members. Four of these (CECR2, FALZ(A/B), GCN5L2, and PCAF) were classified at the high end of the druggability scale by SiteMap and are within the same cluster by whole sequence similarity. The key features of these pockets are the aromatic residue at position 146 compared with a small hydrophobic residue in many other bromodomains and a tryptophan at position 81. Together, these signature residues provide a significant amount of hydrophobic surface on this side of the pocket. The ZA loop is two amino acids shorter than the BET family, but this part of the pocket is still sufficiently enclosed to provide high druggability (Figure 9B). These bromodomains represent a family that demonstrate high predicted druggability but to date have not been exploited with high affinity compounds.
Two outliers were seen for the bromodomain FALZ(A/B) (PDB 2F6N_A and 2FSA_C), which both scored significantly less than the other nine structures passing the imposed filters of this bromodomain. When inspecting the structures and comparing them with more druggable conformations of FALZ(A/B), no obvious changes could be seen, as was the case with BRD2(2). However, when examined more closely, it could be seen that both the ZA loop and the BC loop are moved slightly away from the pocket, inducing a more open conformation and thus reducing the druggability. The four structures that are peptide bound do not demonstrate this more open conformation and may be stabilized in the closed conformation by the presence of the peptide.
Interestingly, the remaining two members of group 2 (TAF1(A/B)(2) and TAF1L(2)) possess the same signature residues but are not present in the same whole sequence classification as the other members of group 2. TAF1(A/B)(2) also scored in the druggable range (Dscore = 0.89), however, TAF1L(2) scored in the difficult range (Dscore = 0.73), possibly due to the ZA loop and tryptophan 81 positions opening the binding site. With only one structure available, this could be an example of a false negative, with an effect similar to the outliers of FALZ(A/B) (PDB 2F6N_A and 2FSA_C), and with further conformational sampling of the ZA loop and tryptophan 81, a more druggable conformation could be observed. TAF1(A/B)(2) and TAF1L(2) differ to the other members of group 2 by the residues at position 94, 145, and 149 within the binding site as well as having reduced sequence similarity; for these reasons they have been given their own subclassification in the classification tree.
Group 3 contains six bromodomains, which all fall into the same classification from the whole sequence similarity and are related to the group 2 by the presence of the aromatic residue at position 146, enclosing this part of the pocket. They differ by the lack of the tryptophan at position 81 opening the ZA channel, so the druggability scores are somewhat lower, placing them in the intermediate category (Figure 9C). Within the group, BRD7 and BRD9 have been given their own subclassification due to the changes in ZA loop residues and having tyrosine rather than phenylalanine at position 146.
Although a crystal structure for BRPF1B was not available, an NMR structure (PDB 2D9E) was and passed the filters other than the presence of the conserved water molecules. When SiteMap druggability assessment was applied to the ensemble of structures, a median Dscore of 1.04 was obtained (Supporting Information, Table S4) and is slightly higher but in a similar range to the Dscore values obtained from the other members of the group without water present. Using the lines of best fit from Figure 4A and a subset of the values from this group, estimates of the Dscore with water molecules were achieved of 0.91 and 0.97 respectively. These values are higher than other members of the group and places this bromodomain in the druggable category.
Features of Group 4
The four members of the BAZ family cluster together by binding site similarity within group 4, unlike the whole sequence similarity classification. The group is characterized by a shorter ZA loop than the BET family, with no residue overlapping with leucine 92 from BRD4(A/B)(1) in space, making the pocket fairly open and reducing druggability. The BAZ family share the tryptophan at position 81 with the BET family, but this does not form the same ZA channel due to the change in residue at position 149 (Figure 9D). For the BET family, this is a methionine, which restricts the movement of the tryptophan forming the ZA channel, but in the BAZ family, this residue is small (alanine or cysteine), which results in movement of the tryptophan toward residue 149, removing the ZA channel and hydrophobic shelf present in the BET family and heavily reducing the druggability into the difficult category.
The BAZ family is joined by TRIM24, TRIM33A, and TRIM66 in this group, and although these do not possess a tryptophan at position 81, they share a very similar ZA loop, with no residue overlapping with the Leu92 from the BET family. This open part of the pocket, and the lack of a ZA channel, give these bromodomains similar pockets to the BAZ family. They have been given their own subclassification due to this change in position 81 from tryptophan to a leucine or valine.
Four structures of TRIM24 score highly in the druggability assessment and appear to be outliers (Supporting Information, Table S4). When inspecting the sites identified by SiteMap, it was apparent that the favorable score is not solely due to the KAc binding site but also an extended site ranging from the KAc binding site to the interface between the bromodomain and the adjacent PHD. For this reason, the analysis performed here has excluded these data points. The KAc binding site is better assessed by the other generated scores, placing it in the difficult range, but there may be small pockets close to the KAc binding site which could be exploited by using fragments followed by a linking effort.
BAZ2B surprisingly indicated reduced druggability of the ligand bound structure when compared to the apo (0.18 reduction in Dscore). From the initial definition of druggability, it would be expected that in general, holo structures should be as druggable if not more so than their apo counterparts. When comparing the two BAZ2B structures (PDB 3G0L and 3Q2F), there are only subtle differences between the two conformations of the binding site, namely that for the ligand bound structure the pocket is slightly narrower due to movements of the ZA loop and BC loop, increasing the enclosure score (0.61–0.69). This narrowing most likely occurs to maximize contact with the flat heteroaromatic part of the ligand, but in doing this reduces the volume of the most enclosed part of the pocket (105 Å3 to 92 Å3). For SiteMap druggability assessment (particularly for low druggability sites), the reduction in volume counts more toward the Dscore than the increase in enclosure, so the overall effect is to reduce the predicted druggability of the ligand bound structure relative to the apo.
Features of Groups 5 and 6
Group 5 is characterized by the presence of an aromatic residue at position 94, the effect of this is to provide a “lid” to the pocket, thus increasing the enclosure and therefore the druggability (Figure 9E).
Structures of PB1(A/B/C)(3) and PB1(A/B/C)(4) were available (PDB 3K2J and 3TLP), but these structures were excluded from the initial analysis due to them missing some of the conserved water molecules. To include them in the analysis and to allow direct comparison of the Dscores, water molecules from the highly similar PB1(A/B/C)(2) were included through alignment of the structures. This yielded median druggabilities for the two bromodomains of 0.57 and 0.70, respectively, placing them in the difficult category (Supporting Information, Table S4).
PHIP(2) scored highest and was placed in the druggable range. The second, third, fourth, and sixth bromodomains of PB1 also fall into this grouping but none show as high a druggability as PHIP(2) and also show less whole-sequence similarity, and this has been indicated with a different subclassification with PB1A(6) given its own subclassification due to a four-residue shorter ZA loop. The PHIP(2) structure does have a ligand bound, so this reduced druggability of the PB1 members could either be due to lack of protein conformational sampling (ZA loop position) and be a false negative or could be due to more subtle effects influencing the overall conformation of the protein and therefore the druggability.
The members of group 6 also share this aromatic residue at position 94, but without any available structures it is difficult to say whether these would be druggable like PHIP(2) or more challenging like many of the PB1 bromodomains. Unlike group 5, group 6 members cluster together by whole sequence similarity, however, there are six other bromodomains that share whole sequence similarity with group 6 but do not appear to share binding site similarity.
Features of Group 7
The four proteins in this group all fall into the same classification by whole sequence similarity. By whole sequence similarity, group 7 is joined by four other bromodomains (PB1(A/B/C)(2–4) and PB1A(6)) but do not share the signature residues and do not fall into this group. PB1(A/B/C)(5) was classified in the intermediate druggability range but SMARCA4 as difficult. All of these proteins possess a shorter ZA loop than the BET family, reducing the surface available for interaction with small molecules. The shape of the KAc binding pockets are also different to those of the BET family in the available structures, with the location of the aromatic residue at position 139 moved toward the pocket and leucine at position 87 rather than the valine present in most other bromodomains. This induces a wider entrance, opening the tight binding pocket at the base of the binding site (Figures 9F and 11A). For this reason, it is expected that this group could bind ligands differently to the other groups, as the tightest, most conserved part of the binding site is significantly different.
Figure 11.

(A) PB1(A/B/C)(5) structure representative of Group 7. (B) PB1(A/B/C)(5) structure demonstrating unusual conformation. (C) Overlaid backbones of usual conformation (PDB 3G0J_A) in green and unusual (PDB 3G0J_B) in blue. Orientation and coloring consistent with Figure 9.
One structure of PB1(A/B/C)(5) that failed the original filters on the presence of the conserved water molecules (PDB 3G0J_B) showed a particularly unusual conformation that is unlike any conformation of this bromodomain or any other bromodomain (Figure 11B). The ZA loop is moved toward the Z′ helix, which is not possible in many other bromodomains due to the presence of alanine at position 81 (Figure 11C) but may also be allowed due to the change in shape of the binding site discussed previously. The effect of this is to close this part of the pocket, which for many of the other bromodomains is the location of the ZA channel. This results in an increase in hydrophobicity of the remaining pocket and reduced preference for the conserved water molecules. When SiteMap druggability assessment was applied to this structure (with a single water molecule at the base of the pocket) a Dscore of 0.87 was achieved, which is significantly higher than any other conformation of this protein and places it in the druggable range. This unusual conformation of the protein may represent an opportunity for inhibiting this bromodomain selectively over any other, as this unusual conformation is not expected to be common and may be unique to PB1(A/B/C)(5). This conformation may also allow for substitution of the water molecules, which appear to be highly conserved for most other bromodomains.
As was the case for BRPF1B, no crystal structure was available for SMARCA2B, but an NMR structure was available that passed the filters imposed apart from the presence of the conserved water molecules (PDB 2DAT). When SiteMap druggability assessment was applied to the ensemble of structures, a median Dscore of 0.81 was achieved (Supporting Information, Table S4), which is higher than the Dscore for SMARCA4 without water present but lower than the Dscore for PB1(A/B/C)(5). When converted, as with BRPF1B, using the Dscore values with and without water molecules of the other bromodomains and the other members of the group from Figure 4A, estimates of the Dscore with water molecules of 0.53 and 0.64 were obtained. This places SMARCA2B in the difficult range with comparable predicted druggability to SMARCA4, which shares high whole sequence similarity.
Features of Group 8
For the binding of KAc to bromodomains, a key hydrogen bond is formed between the carbonyl of the acetyl group and a donor from either an asparagine or threonine residue at position 140.3 For group 8, this key residue is replaced with tyrosine (eight bromodomains) or aspartic acid (MLL1: although protein construct has been engineered and may not be true for full length protein). This changes the nature of the pocket significantly, and it has been suggested that these domains may not bind KAc, or if they do, the manner in which they do would be unlike most other bromodomains.4 MLL1 has been given its own subclassification due to it possessing an aspartate at position 140 as have the SP family due to their high sequence similarity to each other over the other members.
A structure for PB1(A/B/C)(1) is available which shows how tyrosine 140 reduces the size of the pocket (Figure 9G), and SiteMap assessed this site as the least druggable of the bromodomains with only a very small pocket being identified. With such a low assessed druggability, the only opportunity to target this site with a small molecule inhibitor would be to displace the conserved water molecules. But, even with the water molecules removed, the site only achieves a Dscore in the low end of the intermediate range suggesting that PB1(1) would be very challenging to bind small molecules to the equivalent of the KAc binding site of other bromodomains.
A structure for ASH1L (PDB 3MQM), another member of this group, is also available, but the conserved water molecules are not present as is the case for PB1(A/B/C)(1), so it was removed by the initial filter. The site scored comparably to PB1(A/B/C)(1) without water molecules with a median Dscore of 0.74, suggesting that this bromodomain would be similarly difficult to target with a small molecule inhibitor.
Features of Other Bromodomains
The remaining bromodomains failed to be placed into any groups larger than two, with little similarity to any of the other groupings described here. Of those with available structures, none showed any particular druggability as assessed by SiteMap, except CREBBP, which was classified in the intermediate range. CREBBP is interesting as it possesses the same longer ZA loop as the BET family, with similar residues facing the binding site that provide similar interaction potential. However, the tryptophan at position 81 in the BET family, which forms the ZA channel and hydrophobic shelf, is a considerably smaller leucine, resulting in a loss of these features and a decrease in predicted druggability (Figure 9H). Another unusual feature is the presence of an arginine at position 145, which provides the potential to form charged interactions with this strongly basic center. Flexibility of both the ZA loop and the unusual arginine could explain the large changes in predicted druggability of CREBBP, with the highest scoring protein conformation being placed in the druggable category and the lowest in the difficult. With high sequence similarity and binding site similarity, the bromodomain of EP300 would be expected to bind similar ligands to CREBBP and have similar potential for a more druggable pocket to be induced.
Similarly to PB1(A/B/C)(3) and PB1(A/B/C)(4), a structure of ATAD2B (PDB 3LXJ_D) was available that was filtered out due to missing two of the conserved water molecules. All five water molecules were present in a structure of the similar ATAD2A (PDB 3DAI), and through aligning the two structures, the missing water molecules of ATAD2B were included. SiteMap druggability assessment yielded a score of 0.64, placing this bromodomain in the difficult category.
Implications for Selectivity of Bromodomain Inhibitors
When targeting any protein with small molecule inhibitors, selectivity is often desired. For the bromodomains, the highly conserved small and tight binding site (binding acetyl part of KAc) at the base of the pocket makes prediction of selectivity for a low molecular weight (<200 Da) fragment challenging as it is the environment around this site which will determine the selectivity for larger molecules. From this analysis, the first proteins that should be tested for selectivity issues would be those within the same group (Figure 7). There are, however, some similarities between groups that may give rise to comparable binding of small molecules. The groups that are related to each other that have been previously discussed (groups 2 and 3 and groups 5 and 6) may bind similar ligands, but there are differences that may be exploited for selectivity. Also as discussed, CREBBP and EP300 show some similarity through the ZA loop to the BET family, but also PHIP(2) and WDR9(A/B)(2) show some similarity in the shape of the binding site with the same hydrophobic shelf and above average druggability. Other than these, selectivity would be expected between groups for molecules larger than small fragments.
Translation of Druggability to Full Protein Conformational Ensemble
Adequately assessing the full ensemble of protein conformations is an issue that affects any prediction that uses crystal structures, as by their nature a static image is observed. To address this issue, we have used as many experimentally observed conformations of the bromodomains as possible, and scores generated by the druggability assessment do vary between conformations of the same protein, including different protein chains within the same crystal structure. For this reason, we cannot rule out that some proteins that were classified as difficult or intermediate may be false negatives and may have the potential to be druggable with additional conformational sampling. However, the range of scores observed for different conformations of the same bromodomain appear to be less than those between different bromodomains due to changes in the eight selected residues identified here (Figure 5). For this reason, it is possible that new bromodomain conformers may show slightly increased druggability than predicted from the currently available structures (e.g., intermediate instead of difficult or druggable instead of intermediate). However, it is unlikely that large leaps will occur (e.g., for a protein classified as difficult to move into the druggable category), and other than a few special cases discussed in the text (BRD2(2), FALZ(A/B) and CREBBP), these have not so far been observed.
Conclusion
Predicted druggabilities of available bromodomain structures were assessed and a range of scores observed. The BET family members were predicted to be druggable, consistent with literature evidence. One group (group 2) showed comparable or increased predicted druggability relative to the BET family and represents a currently unexplored group of proteins that may have relevance in drug discovery as their biology is revealed. Many of the other bromodomains showed lower predicted druggability and some of these were classed as difficult based on their Dscore. The comparatively low score suggests that these will show lower hit rates in screening efforts and that it will be more challenging to identify and optimize hit matter. However, it should be noted that even these bromodomains are far more druggable than featureless protein–protein interactions.
Trends within the data set were then sought and rationalized by unique signatures characterizing the binding pockets, leading to a new classification of the bromodomains into groups with similar amino acids in key positions and similar predicted druggabilities. This classification showed significant differences to the whole sequence classification, suggesting that it may prove more useful to drug discovery directed toward the acetyl-lysine binding site.
Our proposed classification also allows medicinal chemists who work on a particular bromodomain to identify other family members that are likely to bind similar inhibitors. This information can be explored to select proteins for counterscreening or to identify bromodomain inhibitors that can be explored in a target hopping approach.
Furthermore, our results highlight the significance of water molecules in the computational analysis of bromodomain binding sites. A number of conserved water molecules occupy the base of the pocket and so far no example has been reported in which these have been replaced by small molecules. For this reason, all bromodomains have been treated equally with all of these water molecules kept as part of the binding site and the druggability assessment performed as such. The corresponding assessment without the water molecules present has also been performed, which places more of the bromodomains in the druggable category and, crucially, appears to increase the observed score more for the less druggable sites, making it less discriminatory between druggable and difficult sites.
This work represents the first analysis of this type for the bromodomain family and will prove useful for drug discovery projects aiming to identify inhibitors of the acetyl-lysine binding site of bromodomains.
Acknowledgments
Lewis Vidler is funded by Cancer Research UK Grant No. C309/A11369. We acknowledge NHS funding to the NIHR Biomedical Research Centre and funding from Cancer Research UK Grant No. C309/A8274. Stefan Knapp receives funding from the Structural Genomics Consortium, a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada, GlaxoSmithKline, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Eli Lilly, Pfizer, Abbott, Takeda, the Novartis Research Foundation and the Wellcome Trust. We thank Prof. Julian Blagg, Dr. Berry Matijssen, Sarah Langdon, Nicholas Firth and Sally McGrath for helpful discussions. We thank the reviewers for their insightful and helpful comments.
Glossary
Abbreviations Used
- ASH1L
absent, small, or homeotic 1-like
- ATAD
AAA domain-containing protein
- BAZ
bromodomain adjacent to zinc finger domain protein
- BET
bromodomain and extra terminal
- BRD
bromodomain-containing protein
- BRPF
bromodomain and PHD finger containing protein
- BRWD
bromodomain and WD repeat-containing protein
- CBP
CREB binding protein
- CECR2
cat eye syndrome critical region protein 2
- CREB
cAMP response element modulator
- CREBBP
CREB binding protein
- EBI
European Bioinformatics Institute
- EMBL
European Molecular Biology Laboratory
- Da
daltons
- DEAF-1
deformed, epidermal autoregulatory factor-1
- DLID
drug-like density
- Dscore
druggability score
- EP300
E1A binding protein 300
- FALZ
fetal Alzheimer antigen
- GCN5L2
general control of amino acid synthesis protein 5-like 2
- KAc
acetyl-lysine
- med
median
- MLL
mixed lineage leukemia
- MOE
molecular operating environment
- MYND
myeloid, nervy and DEAF-1
- no.
number
- P300
E1A binding protein 300
- PB
polybromo
- PCAF
P300/CBP-associated factor
- PH
Pleckstrin homology
- PHD
plant homeodomain
- PHIP
PH-interacting protein
- PRKCBP1
protein kinase C binding protein 1
- SCREEN
surface cavity recognition and evaluation
- SIAH
seven in absentia homologue
- SMARCA
SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A
- SP
speckled
- SWI/SNF
switch/sucrose nonfermentable
- TAF
TBP-associated factor
- TBP
TATA-box-binding protein
- TRIM
tripartite motif-containing protein
- WDR
WD repeat containing protein
- ZMYND
zinc finger MYND domain-containing protein
Supporting Information Available
Details of structures filtered out with unresolved residues; details of structures filtered out for missing water molecules; output from SiteMap for structures passing filters; output from SiteMap for structures that have been excluded from Figures 4 and 5 and Table 1 but have been subsequently used in analysis; eight identified binding site residues, representative PDB code, and median druggability for each bromodomain; pockets identified and selected binding site residues for eight representative bromodomains from Figure 9. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Kouzarides T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Jenuwein T.; Allis C. D. Translating the Histone Code. Science 2001, 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- Zeng L.; Zhou M. M. Bromodomain: An acetyl-lysine binding domain. FEBS Lett. 2002, 513, 124–128. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J.-P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Müller S.; Gingras A.-C.; Pawson T.; Arrowsmith C.; Knapp S. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutjaba S.; Zeng L.; Zhou M. M. Structure and acetyl-lysine recognition of the bromodomain. Oncogene 2007, 26, 5521–5527. [DOI] [PubMed] [Google Scholar]
- a Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chung C.-w.; Coste H.; White J. H.; Mirguet O.; Wilde J.; Gosmini R. L.; Delves C.; Magny S. M.; Woodward R.; Hughes S. A.; Boursier E. V.; Flynn H.; Bouillot A. M.; Bamborough P.; Brusq J.-M. G.; Gellibert F. J.; Jones E. J.; Riou A. M.; Homes P.; Martin S. L.; Uings I. J.; Toum J.; Clement C. A.; Boullay A.-B.; Grimley R. L.; Blandel F. M.; Prinjha R. K.; Lee K.; Kirilovsky J.; Nicodeme E. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J. Med. Chem. 2011, 54, 3827–3838. [DOI] [PubMed] [Google Scholar]; c Dawson M. A.; Prinjha R. K.; Dittman A.; Giotopoulos G.; Bantscheff M.; Chan W.-i.; Robson S. C.; Chung C.-w.; Hopf C.; Savitski M. M.; Huthmacher C.; Gudgin E.; Lugo D.; Beinke S.; Chapman T. D.; Roberts E. J.; Soden P. E.; Auger K. R.; Mirguet O.; Doehner K.; Delwel R.; Burnett A.; Jeffrey P.; Drewes G.; Lee K.; Huntly B. J. P.; Kouzarides T. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R. The druggable genome. Nature Rev. Drug Discovery 2002, 1, 727–730. [DOI] [PubMed] [Google Scholar]
- Muller S.; Filippakopoulos P.; Knapp S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13, e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J.; Shi J.; Wang E.; Rappaport A. R.; Herrmann H.; Sison E. A.; Magoon D.; Qi J.; Blatt K.; Wunderlich M.; Taylor M. J.; Johns C.; Chicas A.; Mulloy J. C.; Kogan S. C.; Brown P.; Valent P.; Bradner J. E.; Lowe S. W.; Vakoc C. R. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E.; Jeffrey K. L.; Schaefer U.; Beinke S.; Dewell S.; Chung C.-w.; Chandwani R.; Marazzi I.; Wilson P.; Coste H.; White J.; Kirilovsky J.; Rice C. M.; Lora J. M.; Prinjha R. K.; Lee K.; Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hewings D. S.; Wang M.; Philpott M.; Fedorov O.; Uttarkar S.; Filippakopoulos P.; Picaud S.; Vuppusetty C.; Marsden B.; Knapp S.; Conway S. J.; Heightman T. D. 3,5-Dimethylisoxazoles Act as Acetyl-lysine-mimetic Bromodomain Ligands. J. Med. Chem. 2011, 54, 6761–6770. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sachchidanand; Resnick-Silverman L.; Yan S.; Mutjaba S.; Liu W. J.; Zeng L.; Manfredi J. J.; Zhou M. M. Target structure-based discovery of small molecules that block human p53 and CREB binding protein association. Chem. Biol. 2006, 13, 81–90. [DOI] [PubMed] [Google Scholar]; c Borah J. C.; Mujtaba S.; Karakikes I.; Zeng L.; Muller M.; Patel J.; Moshkina N.; Morohashi K.; Zhang W.; Gerona-Navarro G.; Hajjar R. J.; Zhou M. M. A small molecule binding to the coactivator CREB-binding protein blocks apoptosis in cardiomyocytes. Chem. Biol. 2011, 4, 531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zeng L.; Li J.; Muller M.; Yan S.; Mujtaba S.; Pan C.; Wang Z.; Zhou M.-M. Selective Small Molecules Blocking HIV-1 Tat and Coactivator PCAF Association. J. Am. Chem. Soc. 2005, 127, 2376–2377. [DOI] [PubMed] [Google Scholar]
- Campagna-Slater V.; Mok M. W.; Nguyen K. T.; Feher M.; Najmanovich R.; Schapira M. Structural Chemistry of the Histone Methyltransferases Cofactor Binding Site. J. Chem Inf. Model. 2011, 51, 612–623. [DOI] [PubMed] [Google Scholar]
- Santiago C.; Nguyen K.; Schapira M. Druggability of methyl-lysine binding sites. J Comput.-Aided. Mol. Des. 2011, 25, 1171–1178. [DOI] [PubMed] [Google Scholar]
- Sheridan R. P.; Maiorov V. N.; Holloway M. K.; Cornell W. D.; Gao Y. D. Drug-like density: a method of quantifying the “bindability” of a protein target based on a very large set of pockets and drug-like ligands from the Protein Data Bank. J. Chem. Inf. Model. 2010, 50, 2029–2040. [DOI] [PubMed] [Google Scholar]
- a Volkamer A.; Griewel A.; Grombacher T.; Rarey M. Analyzing the Topology of Active Sites: On the Prediction of Pockets and Subpockets. J. Chem. Inf. Model. 2010, 50, 2041–2052. [DOI] [PubMed] [Google Scholar]; b Volkamer A.; Kuhn D.; Grombacher T.; Rippmann F.; Rarey M. Combining Global and Local Measures for Structure-Based Druggability Predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [DOI] [PubMed] [Google Scholar]
- DrugEBIlity; EMBL-EBI: Wellcome Trust Genome Campus: Hinxton, Cambridgeshire, CB10 1SD, UK; https://www.ebi.ac.uk/chembl/drugebility/.
- Krasowski A.; Muthas D.; Sarkar A.; Schmitt S.; Brenk R. DrugPred: A Structure-Based Approach To Predict Protein Druggability Developed Using an Extensive Nonredundant Data Set. J. Chem. Inf. Model. 2011, 51, 2829–2842. [DOI] [PubMed] [Google Scholar]
- a Schmidke P.; Le Guilloux V.; Maupetit J.; Tufféry P. fpocket: online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010, 38, W582–W589. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schmidke P.; Barril X. Understanding and Predicting Druggability. A High-Throughput Method for Detection of Drug Binding Sites. J. Med. Chem. 2010, 53, 5858–5867. [DOI] [PubMed] [Google Scholar]
- Cheng A.; Coleman R. G.; Smyth K. T.; Cao Q.; Soulard P.; Caffrey D. R.; Salzberg A. C.; Huang E. S. Structure-based maximal affinity model predicts small-molecules druggability. Nature Biotechnol. 2007, 25, 71–75. [DOI] [PubMed] [Google Scholar]
- Nayal M.; Honig B. On the nature of cavities on protein surfaces: application to the identification of drug-binding sites. Proteins 2006, 63, 892–906. [DOI] [PubMed] [Google Scholar]
- a SiteMap 2.5; Schrödinger, LLC: 101 SW Main Street, Suite 1300, Portland, OR 97204, U.S.A.; http://www.schrodinger.com/.; b Halgren T. A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [DOI] [PubMed] [Google Scholar]
- Hajduk P. J.; Huth J. R.; Tse C. Predicting protein druggability. Drug Discovery Today 2005, 10, 1675–1682. [DOI] [PubMed] [Google Scholar]
- Fauman E. B.; Rai B. K.; Huang E. S. Structure-based druggability assessment—identifying suitable targets for small molecule therapeutics. Curr. Opin. Chem. Biol. 2011, 15, 463–468. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOE 2010.10; Chemical Computing Group: Montreal, Quebec, Canada; http://www.chemcomp.com/.
- Protein Preparation Wizard; Schrödinger, LLC: 101 SW Main Street, Suite 1300, Portland, OR 97204, U.S.A.; http://www.schrodinger.com/.
- Maestro 9.2; Schrödinger, LLC: 101 SW Main Street, Suite 1300, Portland, OR 97204, U.S.A.; http://www.schrodinger.com/.
- Aabel 3; Gigawiz Ltd. Co.: P.O. Box 33109, Tulisa, OK 74153, U.S.A.; http://www.gigawiz.com/.
- iMindMap 5.43 Basic; ThinkBuzan: Regus House, Falcon Drive, Cardiff, CF10 4RU, U.K.; http://thinkbuzan.com/.
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Philpott M.; Yang J.; Tumber T.; Fedorov O.; Uttarkar S.; Filippakopoulos P.; Picaud S.; Keates T.; Felletar I.; Ciulli A.; Knapp S.; Heightman T. Bromodomain-peptide displacement assays for interactome mapping and inhibitor discovery. Mol. BioSyst. 2011, 7, 2899–2908. [DOI] [PubMed] [Google Scholar]
- a Oltersdorf T.; Elmore S. W.; Shoemaker A. R.; Armstrong R. C.; Augeri D. J.; Belli B. A.; Bruncko M.; Deckwerth T. L.; Dinges J.; Hajduk P. J.; Joseph M. K.; Kitada S.; Korsmeyer S. J.; Kunzer A. R.; Letai A.; Li C.; Mitten M. J.; Nettesheim D. G.; Ng S.; Nimmer P. M.; O’Connor J. M.; Oleksijew A.; Petros A. M.; Reed J. C.; Shen W.; Tahir S. K.; Thompson C. B.; Tomaselli K. J.; Wang B.; Wendt M. D.; Zhang H.; Fesik S. W.; Rosenberg S. H. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [DOI] [PubMed] [Google Scholar]; b Bruncko M.; Oost T. K.; Belli B. A.; Ding H.; Joseph M. K.; Kunzer A.; Martineau D.; McClellan W. J.; Mitten M.; Ng S. C.; Nimmer P. M.; Oltersdorf T.; Park C. M.; Petros A. M.; Shoemaker A. R.; Song X.; Wang X.; Wendt M. D.; Zhang H.; Fesik S. W.; Rosenberg S. H.; Elmore S. W. Studies leading to potent, dual inhibitors of Bcl-2 and Bcl-xL. J. Med. Chem. 2007, 50, 641–662. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.