

Abstract
Glutamine (Q) expansion diseases are a family of degenerative disorders caused by the lengthening of CAG triplet repeats present in the coding sequences of seemingly unrelated genes whose mutant proteins drive pathogenesis. Despite all the molecular evidence for the genetic basis of these diseases, how mutant poly-Q proteins promote cell death and drive pathogenesis remains controversial. In this report, we show a specific interaction between the mutant androgen receptor (AR), a protein associated with spinal and bulbar muscular atrophy (SBMA), and the nuclear protein PTIP (Pax Transactivation-domain Interacting Protein), a protein with an unusually long Q-rich domain that functions in DNA repair. Upon exposure to ionizing radiation, PTIP localizes to nuclear foci that are sites of DNA damage and repair. However, the expression of poly-Q AR sequesters PTIP away from radiation-induced nuclear foci. This results in sensitivity to DNA-damaging agents and chromosomal instabilities. In a mouse model of SBMA, evidence for DNA damage is detected in muscle cell nuclei and muscular atrophy is accelerated when one copy of the gene encoding PTIP is removed. These data provide a new paradigm for understanding the mechanisms of cellular degeneration observed in poly-Q expansion diseases.
INTRODUCTION
Expanded polyglutamine (poly-Q) tracts cause nine chronic and progressive degenerative diseases, including Huntington's disease, spinal and bulbar muscular atrophy (SBMA), dentatorubral-pallidoluysian atrophy and several dominantly inherited spinocerebellar ataxias. The age of onset and severity correlates with the length of the poly-Q expansion associated with each disease gene. The expanded poly-Q tracts promote protein polymerization, aggregation and insolubility, yet the precise cause of neuromuscular dysfunction and cell death is still controversial (1,2). Nuclear localization of poly-Q androgen receptor (AR) (3,4) and Ataxin1 (5) is essential for disease pathogenesis, suggesting that the nucleus is a common site of toxicity in the poly-Q disorders. Consistent with this notion is the occurrence of nuclear inclusions, one pathologic hallmark of these diseases. Yet, recent evidence suggests that nuclear aggregates do not correlate with disease (6–8) and that smaller, soluble protein species are the drivers of neuronal death (9,10). Nuclear poly-Q proteins are known to interact with a variety of transcriptional regulators and are thought to affect neuronal gene expression patterns (11–17). However, it remains unclear whether changes in gene expression are the cause or the result of disease progression (18).
SBMA is an X-linked, adult onset neuromuscular disorder caused by poly-Q expansion in the AR (19). In affected adult SBMA males, ligand-dependent poly-Q AR unfolding triggers a progressive neuromuscular disorder with clinical onset in adolescence to adulthood that is often characterized initially by muscle cramps and elevated serum creatine kinase (20,21). These features commonly precede overt muscle weakness, which inevitably develops as the disease progresses and is most severe in the proximal limb and bulbar muscles. The clinical features of SBMA, also called Kennedy's disease, correlate with a loss of motor neurons in the brainstem and spinal cord and with marked myopathic and neurogenic changes in skeletal muscle (22,23).
A connection between genomic stability, poly-Q proteins and cell death was recently suggested through the association of poly-Q huntingtin with DNA damage and repair defects (24). Furthermore, the activation of the DNA damage response appears to precede the formation of nuclear protein aggregates (25). Yet, the mechanism linking poly-Q proteins to genomic instability is unknown. While the wild-type (WT) proteins associated with poly-Q diseases can have variable Q tracts ranging into the upper 30s, the mutant proteins associated with disease have poly-Q tracts that can exceed 100 amino acid residues, suggesting that abnormal protein–protein interactions mediated by this domain may contribute to pathology.
The BRCT (Brcal C-terminal)-domain protein PTIP (Pax Transactivation-domain Interacting Protein) functions in both DNA damage and repair pathways and in the histone H3K4 (histone H3, lysine 4) methylation pathways (26,27). In addition to six BRCT domains, PTIP is highly conserved from flies to mice and contains a Q-rich domain, positioned between BRCT domains 2 and 3, that is more than 54% Q in mice and men. Upon DNA damage, PTIP localizes to nuclear foci and interacts with 53BP1 (p53-binding protein 1) and Rad50 (28,29), two proteins involved in sensing and repairing DNA damage. In cell culture, PTIP knock-downs exhibit fewer 53BP1-positive radiation-induced foci, suggesting that PTIP localization to sites of double-stranded breaks (DSBs) is an early event after DNA damage (29). The dual role for PTIP in gene regulation and DSB repair is best illustrated in B cells which require PTIP for efficient immunoglobulin isotype switching (30,31), a process dependent on both transcription and DSB repair. Given that the loss of PTIP function is embryonic lethal in mice (32) and flies (33) and that PTIP function is necessary for differentiation in a variety of cell types, altering the normal function of PTIP is likely to be detrimental and may underlie the pathology seen in poly-Q expansion disease.
In this report, we postulated that PTIP may interact with poly-Q expansion disease proteins through its naturally occurring Q domain and that this interaction may inhibit essential functions like DNA repair and gene regulation. Here, we show a specific interaction between the poly-Q mutant AR and PTIP. Poly-Q AR activates the DNA damage response, but prevents PTIP from localizing to ionizing radiation (IR)-induced nuclear foci and thus attenuates the DNA repair response. Expression of poly-Q AR is sufficient to induce DNA damage and chromosomal instability. Moreover, in a mouse model of SBMA, which carries a poly-Q AR allele and exhibits evidence of the DNA damage response, reducing the PTIP gene dosage accelerates muscular atrophy. These data demonstrate that toxicity in poly-Q expansion disease can be caused by sequestration of PTIP and suppression of normal DNA repair.
RESULTS
Unusually long Q-rich domains in PTIPs
Human and mouse PTIPs have two N-terminal BRCT domains, a Q-rich domain and four C-terminal BRCT domains. Such BRCT domains are found in many proteins that are involved in the DNA damage response, including BRCA1, MDC1, XRCC1 and TopBP1. Some BRCT domain pairs are known to bind P-serine, especially those phosphorylated by the ATM (Ataxia Telangiectasia Mutated) kinase (34,35). The highly conserved Q-domain is unusual in its length and spans ∼190 amino acids of which 54% are Q. The Q-domain in mice and humans contains multiple stretches of 5–6 Qs, yet the conserved Drosophila PTIP contains an even larger Q-domain, predicted to be nearly 1500 amino acid long, with a subdomain of 150 amino acids that is 86% Q (Table 1). The functions of such a large Q-rich domain are unknown. However, these naturally occurring Q-domains may facilitate interaction with other proteins.
Table 1.
Q-rich domains
| Species | Protein | Sequence (% Q) |
|---|---|---|
| Mouse | PTIP | Amino acids 397–577 (52.5%) |
| Amino acids 477–541 (63%) | ||
| Human | PTIP | Amino acids 402–590 (54.5%) |
| Chicken | PTIP | Amino acids 408–596 (57.7%) |
| Xenopus | PTIP | Amino acids 370–779 (49.5%) |
| Drosophila | PTIP | Amino acids 196–1673 (43.9%) |
| Amino acids 205–265 (86.9%) | ||
| Amino acids 864–911 (77.1%) |
The PTIP localizes to γ-H2AX- and 53BP1-positive nuclear foci induced by IR. We examined whether the Q-domain was sufficient to localize PTIP to these radiation-induced nuclear foci (Fig. 1). Transient transfection of just the Q-domain, linked to a nuclear targeting sequence, did not result in protein localization to nuclear foci after radiation (Fig. 1A). However, deletion of the Q-domain from a full-length PTIP-EGFP fusion protein did alter the pattern of PTIP localization after radiation (Fig. 1B). These data demonstrate that the Q-domain is necessary but not sufficient for targeting PTIP to nuclear foci after exposure to IR.
Figure 1.

Effects of the Q domain on PTIP localization. (A) Full-length PTIP or the Q domain only, linked to a nuclear localization signal, was expressed with flag-epitope tags in HEK293 cells and subject to 5 Gy IR (+IR). Cells were stained 1 h post IR with anti-flag and anti-53BP1 as indicated. (B) HEK293 cells expressing either a full-length EGFP-PTIP fusion protein or a Q domain deletion construct (EGFP-PTIP-ΔQ) were exposed to 5 Gy IR and imaged fixed 1 h post irradiation. EGFP was visualized by fluorescent microscopy and nuclei stained with Dapi.
Poly-Q AR inhibits PTIP localization to radiation-induced nuclear foci
The PTIP is associated with the DNA damage response and with histone H3K4 methylation. Thus, we hypothesized that a poly-Q expansion protein might interfere with an essential PTIP function, perhaps by interacting with and sequestering PTIP or other proteins via the expanded poly Q-domain. To test whether a poly-Q disease protein can interact with PTIP in the DNA damage response, we expressed WT (AR24Q) or poly-Q AR (AR113Q) in HEK293 cells and assayed for PTIP localization after IR. By adding the synthetic AR ligand R1881 for a short time only, we could focus our analysis on cells at early stages before the appearance of large intranuclear aggregates, as evidence suggests that the soluble species mediate poly-Q toxicity (9,10).
Transfected PTIP localizes to nuclear foci upon the exposure of cells to IR. However, the transient expression of AR113Q but not AR24Q resulted in a ligand-dependent decrease in the localization of full-length PTIP to nuclear foci in HEK293 cells expressing flag-PTIP (Fig. 2A and C). After exposure to 5 Gy IR, this affect was best observed in neighboring cells in which an AR113Q-expressing cell was juxtaposed to a non-expressing cell. In the absence of ligand, cytoplasmic AR113Q did not affect nuclear PTIP localization to foci; neither did the WT AR24Q protein in the presence of ligand. However, nuclear AR113Q significantly (P< 0.01) reduced the number of PTIP-positive nuclear foci (Fig. 2C).
Figure 2.
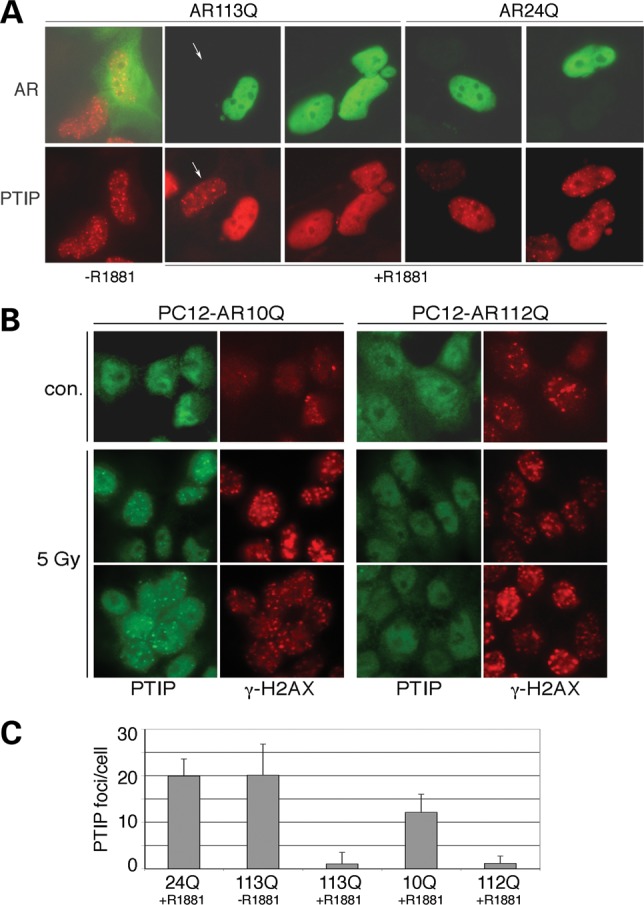
Poly-Q AR inhibits PTIP from localization to radiation-induced nuclear foci. (A) Cells stably expressing flag-tagged PTIP HEK293 cells were transfected with AR113Q or AR24Q and stained with antibodies against AR (green) or flag-PTIP (red). All cells were subject to 5 Gy IR and the same field is shown in the top and bottom panels. Without ligand, AR113Q has no effect on PTIP foci; however, PTIP foci are reduced in AR113Q-expressing cells in the presence of R1881. PTIP foci are unaffected in cells not expressing AR113Q (arrow). AR24Q has little effect on PTIP foci formation in response to IR, as PTIP is in foci regardless of whether AR24Q is coexpressed. (B) Endogenous PTIP was examined in PC12 Cells stably expressing either AR10Q or AR112Q before and 1 h after 5 Gy IR. PTIP (green) is observed in nuclear foci after irradiation in AR10Q-expressing rat PC12 cells but not in AR112Q-expressing PC12 cells. The γ-H2AX (red) staining marks IR-induced foci 1 h after 5 Gy exposure and is similar in both cell types. However, more γ-H2AX are observed in AR112Q cells in the absence of IR. (C) Quantitation of IR-induced PTIP-positive foci in transfected HEK293 cells (24Q, 113Q) and in PC12 cells (10Q, 112Q). All cells were subject to 5 Gy IR and stained for PTIP. The numbers of foci were counted in cells expressing AR24Q with ligand, AR113Q without ligand, AR113Q with ligand, AR10Q with ligand and AR112Q with ligand. The mean number of foci/cell is shown with error bars being 1SEM. Note that the SEM is greater than the mean for 113Q + R1881, because many cells had zero PTIP-positive foci. The Student's t-test for two independent variables gave P-values of <0.01 for 113Q + R1881 and 112Q + R1881 compared with espective controls.
We next utilized a model PC12 cell system that stably expresses either AR10Q or AR112Q under the control of a DOX-inducible promoter (36) (Fig. 2B and C). While endogenous PTIP is more difficult to detect, we confirmed that in the PC12 cells, AR112Q expression significantly (P< 0.01) reduced the numbers of endogenous PTIP containing foci after exposure to IR. Yet, AR112Q did not affect the numbers of γ-H2AX-positive foci, which mark the DSB induced by IR. These data show that nuclear AR112Q specifically inhibits PTIP localization to radiation-induced nuclear foci. Also of note, the AR112Q-expressing cells always had more γ-H2AX foci in the absence of IR. These data suggest that AR112Q cells have a consistent low level of DNA damage that may not repair efficiently because of this defect in PTIP recruitment to nuclear foci.
PTIP interacts with expanded poly-Q AR
We next examined interactions between PTIP and AR by immunoprecipitation in PC12 and HEK293 cells. In the absence of irradiation, PTIP was co-precipitated from PC12 lysates with antibodies against AR but only in the cells expressing AR112Q (Fig. 3A). Furthermore, the co-precipitated PTIP migrated slightly higher than the normal 130-kD PTIP. This higher PTIP form was already evident in the input samples but only in cells expressing AR112Q and was induced by treatment with the ligand R1881.
Figure 3.

Interactions between PTIP and poly-Q AR. (A) PC12 cells stably expressing AR10Q or AR112Q were induced with synthetic ligand R1881 and protein lysates immunoprecipitated with anti-AR antibodies as indicated. Addition of ligand induced a slower migrating PTIP isoform (input) in AR112Q cells. The AR112Q protein binds to this slower migrating form of PTIP, whereas AR10Q shows no interaction. (B) The slower migrating form of PTIP is specific for AR112Q PC12 cells after the addition of ligand (arrows). The presence of the slower PTIP isoform is inhibited by the DNA-PKcs inhibitor Nu7026 but not by the ATM inhibitor Ku55933. (C) Lambda phosphatase reduces the slower migrating form of PTIP in AR112Q-expressing cell lysates. (D) Western blot of lysates from transient expression of AR24Q and AR113Q in HEK293 cells. Note slower migrating PTIP isoform is not radiation inducible and only appears when poly-Q AR is expressed. (E) AR112Q induces ATM phosphorylation in PC12 cells and higher levels of γ-H2AX even in the absence of IR. 5 Gy IR activates P-ATM and increases γ-H2AX in both cell types. (F) Western blot of lysates from AR10Q and AR112Q PC12 cells showing p53 phosphorylation in response to IR. Note, P-p53 levels in AR112Q cells are elevated prior to radiation.
The higher migrating form of PTIP could be the result of phosphorylation in the AR112Q-expressing PC12 cells. Thus, we examined the effect of protein kinase inhibitors specific for ATM or DNA-PKcs, two kinases induced by DNA damage. The DNA-PKcs, but not the ATM, inhibitor prevented the formation of the slower migrating PTIP that was observed exclusively in AR112Q cells (Fig. 3B). The slower migrating PTIP isoform is likely due to phosphorylation as the treatment of lysates with lambda phosphatase converts the slower migrating PTIP isoform to a single species at ∼130 kD (Fig. 3C).
A similar slow migrating PTIP isoform was also observed upon the transient transfection of AR113Q into HEK293 cells (Fig. 3D). Interestingly, the slower migrating PTIP isoform was not induced by IR alone in cells expressing the WT AR. Only the presence of poly-Q AR induced the larger PTIP isoform and this was independent of irradiation, suggesting that the P-PTIP seen in AR112Q cells was not just an effect of specifically activating the DNA damage response but the result of nuclear poly-Q AR. Most significantly, AR112Q activated the DNA damage response in the absence of IR, as seen by higher levels of γ-H2AX and P-ATM compared with AR10Q-expressing cells (Fig. 3E). We also examined the levels of P-p53 in both AR10Q and AR112Q cells, before and after IR. As expected, 2 or 5 Gy induced high levels of P-p53. However, non-irradiated AR112Q cells treated with R1881 already had higher levels of P-p53 compared with the AR10Q cells, again suggesting that they are subject to some type of DNA stress (Fig. 3F). These data clearly show the activation of the DNA damage response upon the expression of nuclear poly-Q AR and the induction of a unique PTIP isoform.
Poly-Q AR activates the DNA damage response and promotes genomic instability
To confirm the activation of the DNA damage response and to examine the resolution of radiation-induced nuclear foci over time, we stained AR10Q and AR112Q PC12 cells for γ-H2AX and 53BP1 over time (Fig. 4). AR112Q cells already had increased numbers of γ-H2AX-positive foci prior to irradiation, consistent with western blotting data for γ-H2AX. By 1 h post irradiation, nearly 100% of AR10Q and AR112Q cells had 10 or more γ-H2AX-positive foci. However, the number of 53BP1 foci and the percentage of cells with 10 or more foci were less in the AR112Q-expressing cells. By 4 h post IR, more AR112Q cells still had more than 10 γ-H2AX foci and also had more 53BP1 foci at this time. By 24 h post irradiation, the AR10Q cells appeared similar to non-irradiated cells, whereas the AR112Q cells still had significantly more γ-H2AX and 53BP1 foci. These data suggest that by sequestering PTIP to prevent localization to nuclear foci, AR112Q expression attenuates the process of DNA repair.
Figure 4.

Resolution of IR-induced nuclear foci. (A) PC12 cells expressing AR10Q or AR112Q were cultured with R1881 and stained with antibodies against γ-H2AX or 53BP1 before (zero) or 1, 4 or 24 h post irradiation. (B) The percentages of cells with 10 or more γ-H2AX-positive nuclear foci were counted in eight independent images for each time point. (C) The percentages of cells with 10 or more 53BP1 nuclear foci were counted from eight fields at each time point. Statistically differences were calculated by the Student's t-test; *P< 0.01 for all comparisons except the 1-h γ-H2AX samples.
The attenuation of DNA repair could result in sensitivity to damaging agents and genetic instability. To examine this more directly, we assessed genomic instability in HEK293 cells expressing either AR24Q or AR113Q and in PC12 cells expressing either AR10Q or AR112Q and treated with ligand. Proliferation, survival and karyotypes were analyzed for all cell types. In HEK293 cells, karyotypes from untransfected cells or cells expressing AR24Q did not exhibit any chromatid breaks or triradials, whereas 12% of karyotypes from AR113Q-expressing cells had either a break or a triradial structure (Table 2, Fig. 5A). The chromosomal abnormalities seen in transiently transfected 293 cells were in the absence of irradiation. For the PC12 cells, poly-Q AR also increased the rate of chromosomal abnormalities significantly (Table 3), both prior to and after 5 Gy IR. Expression of poly-Q AR in HEK293 cells reduced cell proliferation after a low dose (2 Gy) of IR (Fig. 5B). However, overexpression of flag-PTIP rescued the IR sensitivity conferred by AR113Q alone in these cells, suggesting that endogenous PTIP was limiting. Similarly, poly-Q AR-expressing cells were more sensitive to camptothecin (CPT), a reagent that generates DSBs during S-phase (Fig. 5C). The AR112Q PC12 cells were also more sensitive to mitomycin C (MMC), a DNA cross-linking agent (Fig. 5D).
Table 2.
Chromosomal abnormalities in HEK293 cells
| Cells | n | Triradials | Gaps/breaks | Total |
|---|---|---|---|---|
| HEK293 | 50 | 0 | 0 | 0 |
| AR24Q | 31 | 0 | 0 | 0 |
| AR113Q | 50 | 1 | 5 | 6 (12%) |
Figure 5.

Genomic instabilities in poly-Q AR-expressing cells. (A) Representative karyotypes are shown with chromosomal abnormalities in AR113Q-expressing HEK293 cells (top panels) or in AR112Q-expressing PC12 cells (bottom panels); triradials, gaps and breaks are indicated by the arrow. (B) Growth curves of HEK293 cells and cells transiently expressing AR24Q, AR113Q or AR113Q + flagPTIP after exposure to 2 Gy IR. Note that overexpression of PTIP rescues AR113Q-dependent effects. (C) Growth curves of HEK293 cells, AR24Q- or AR113Q-expressing cells after treatment with 40 nm CPT. (D) MMC sensitivity of PC12 cells expressing either AR10Q or AR112Q. Survival after 8 days in culture is indicated.
Table 3.
Chromosomal abnormalities in PC12 cells
| Cells | IR (Gy) | n | Triradials | Gaps/breaks | Total |
|---|---|---|---|---|---|
| AR10Q | 0 | 50 | 0 | 1 | 1 (2%) |
| AR112Q | 0 | 48 | 0 | 6 | 6 (12.5%) |
| AR10Q | 5 | 50 | 2 | 10 | 12 (24%) |
| AR112Q | 5 | 50 | 8 | 22 | 30 (60%) |
The cumulative data suggest that AR113Q interrupts an essential function of PTIP in DNA damage sensing or repair. However, sequestration could impact gene expression patterns because PTIP is also part of an MLL3/4 (KMT2C/D) histone methyltransferase complex. Thus, we tested for the effects of WT AR and poly-Q AR on transcription using two different assays. First, we co-transfected AR24Q or AR113Q with Pax2 into a Pax2/5/8 transcription reporter cell system that depends on PTIP (27,31). The ability of Pax2 to activate reporter gene expression was not influenced by the presence of AR113Q (Fig. 6A). Secondly, we examined the gene expression patterns by Affymetrix microarrays in response to ligand addition in AR10Q- and AR112Q-expressing PC12 cells (Fig. 6B). Cells expressing AR10Q showed more than 1200 genes with significant changes in mRNA levels 24 h after ligand addition. These genes were also changed upon ligand addition in AR112Q cells, although the absolute level of activation was reduced for the genes showing the greatest change. Nevertheless, the pattern of activation and repression was very similar. These data are consistent with prior studies demonstrating that the expanded Q tract mediates a partial loss of AR function (37,38) and argue that there are not gross, qualitative deficits in the transcriptional response between AR10Q- and AR112Q-expressing cells.
Figure 6.

Effects of AR112Q on gene expression. (A) HEK293 cells containing an integrated PTIP/Pax reporter gene were transiently transfected with expression plasmids as indicated and lysates western blotted for the EGFP reporter gene. Note, Pax2 activates the EGFP reporter but is unaffected by either AR24Q or AR113Q. (B) Affymetrix microarray analysis of R1881-dependent gene expression in PC12 cells expressing either AR10Q or AR112Q. For AR10Q, 1261 genes were either activated or repressed more than 0.6-fold (log2 scale) in response to ligand addition. These genes are plotted on the x-axis from highest activated to the highest repressed (1–1261). The level of activation or repression in AR112Q cells is then overlaid on this graph, with each gene represented by a single point. Note that the overall pattern of activation and repression is similar between AR10Q and AR112Q, although the absolute level of activation or repression is generally lower for AR112Q.
PTIP gene dosage accelerates SBMA in a mouse model
In male mice carrying a poly-Q expansion AR gene, muscular atrophy and degeneration are observed over time. Thus, we examined age-matched WT and AR113Q-expressing mice for evidence of activation of the DNA damage response. At 21 weeks of age, AR113Q-expressing mice had significantly higher numbers of γ-H2AX-positive foci in muscle cell nuclei (Fig. 7). AR113Q muscle cell nuclei were generally lager and misshapen and also contained AR-positive aggregates; however, these did not co-localize with γ-H2AX (Fig. 7A). Coincident with the greater number of γ-H2AX-positive foci in AR113Q mouse muscle was increased the expression of Ku70 (Fig. 7B) and 53BP1 (Fig. 7C), two proteins that mediate the DNA damage response. Also, antibodies against P-ATM (Fig. 7D) and P-DNA-Pkcs (Fig. 7E) showed more intense staining in AR113Q nuclei, consistent with the activation of the DNA damage response. These data suggest enhanced DNA damage and activation of the DNA damage response is a hallmark of AR113Q expression in mouse muscle nuclei.
Figure 7.

Evidence for the activation of the DNA damage response in AR113Q mice. Immunohistochemistry from muscle sections of AR113Q (113Q) and wt mice are shown in (A)–(E). Genotypes are as indicated. Cell nuclei are stained blue with Dapi. (A) Co-staining of muscle cell nuclei for γ-H2AX (green) and AR (red). Note green nuclear foci are distinct from the AR aggregates seen in AR113Q muscle cells compared to age matched WT controls. (B) Co-staining of γ-H2AX (green) and Ku70 (red) shows increased Ku70 and γ-H2AX foci in AR113Q muscle nuclei. (C) Co-staining for γ-H2AX (green) and 53BP1 (red) shows increased levels of 53BP1 in AR113Q muscle nuclei. (D) Co-staining of P-ATM (green) and 53BP1 (red) shows increased levels P-ATM and 53BP1 in AR113Q muscle cell nuclei. (E) Staining for P-DNA-PKcs (red) shows increased levels in AR113Q nuclei compared with WTs. (F) Quantitation of γ-H2AX foci in sections of muscle from 21-week-old wt or 21- or 28-week-old AR113Q mice. Ten images were taken from each muscle sample, with each image containing more than 50 nuclei, and the number of γ-H2AX foci counted. Data are represented as the average number of foci per nucleus. The P< 0.01 (*) when compared with wt samples. Error bars are 1SEM.
We also examined the spinal cord for evidence of DNA damage and activation of the damage response. Unlike the muscle tissue of AR113Q mice at 21 weeks of age, we did not detect significant differences in staining for 53BP1, γ-H2AX or P-ATM in sections of ventral spinal cord where motor neuron cell bodies are located (Fig. 8). This is consistent with the gross phenotypes reported in AR113Q mice, which include defects in muscle fibers and motor neuron axonal transport but no motor neuron cell death (39,40).
Figure 8.

Immunostaining of WT and AR113Q spinal cord. Representative sections of the ventral spinal cord are shown stained with antibodies for the indicated proteins. Comparing the WT to AR113Q spinal cord, we did not see any increased evidence of DNA damage or activation of the DNA damage response in nuclei from regions of ventral motor neurons.
If sequestration of PTIP reduces the efficiency of the DNA damage response and thus underlies the phenotype observed in AR113Q mice, then a reduction in PTIP levels should enhance the severity and progression of the disease in mice. Thus, we examined the phenotype in AR113Q mice that carried either one or two copies of the Paxip1 gene, which encodes the PTIP. Paxip1 homozygous null embryos are post-gastrulation lethal (32), although Paxip1+/− mice are viable and fertile. We crossed AR113Q/+ heterozygous females to Paxip1+/− heterozygous males and genotyped offspring at weaning. Of 78 weanlings with the genotype AR113Q;Paxip1+/+, 51% were males and 49% females. However, of the weanlings with the genotype AR113Q;Paxip1+/−, we recorded 33% males and 67% females (n= 77). This was a significant deviation from the expected sex ratios, as judged by the Fisher's two-sided exact test (P= 0.035), suggesting that a reduction in PTIP was detrimental in males.
The surviving male AR113Q;Paxip1+/− mice were allowed to age and were compared with their AR113Q;Paxip1+/+ littermates. In order to test whether reduced Paxip1 gene dosage impacted the SBMA phenotype, we examined the proximal hind limb muscles of the affected male mice (40) (Fig. 9). We measured both the cross-sectional area of individual muscle fibers and total muscle mass in surviving mice. The average cross-sectional area of muscle fibers was reduced by 25% in AR113Q mice carrying one copy of Paxip1 compared with AR113Q mice with a WT genetic background. Similarly, the muscle mass was also reduced by ∼25% in the Paxip1+/− background. In the absence of the AR113Q allele, reduced Paxip1 gene dosage slightly increased the muscle fiber size, although muscle mass was unchanged in the Paxip1 heterozygotes. Thus, the muscular atrophy in AR113Q mice was accelerated by the loss of one Paxip1 allele. These genetic interaction data are consistent with our interpretation that AR113Q sequesters the PTIP in muscle cells to attenuate a DNA damage sensing or repair pathway.
Figure 9.

Enhanced muscle atrophy through reduced Paxip1 gene dosage. (A) Distribution of the cross-sectional area of muscle fibers from histological sections of proximal hind limb muscle taken from AR113Q male mice carrying either 1 or 2 copies of the Paxip1 gene which encodes PTIP (P< 0.001). (B) The cross-sectional area of muscle fibers of all examined genotypes (mean ± SEM) (***P< 0.001). (C) Hind limb muscle mass (mean ± SEM) (***P< 0.001, *P< 0.05, n.s., not significant). (D) Representative histological sections of muscle fibers taken from AR113Q mice with indicated genetic background. Scale bar = 50 μm.
We also considered that PTIP haploinsufficiency might accentuate motor neuron pathology in SBMA mice. Since our prior characterization of this model indicated that a sensitive measure of motor neuron dysfunction is the induction of genes in skeletal muscle that are responsive to denervation (MyoD, acetylcholine receptor α-subunit, myogenin), we checked their expression by qPCR. We found that PTIP haploinsufficiency did not alter the expression of these genes (data not shown). These findings are consistent with our observations of DNA damage in skeletal muscle and show an effect of PTIP gene dosage on muscle fiber size but not neuronal dysfunction.
DISCUSSION
The underlying causes of cell death in the family of poly-Q expansion diseases are still controversial even though the genes and associated proteins have been studied extensively. We present data that link the poly-Q AR protein to the ubiquitous nuclear protein PTIP, a multifunctional protein associated with DNA repair and gene expression. In cell culture, poly-Q but not WT AR binds to PTIP and prevents PTIP localization to nuclear foci upon DNA damage. Expression of AR112Q also induces low levels of P-p53, increases sensitivity to DNA-damaging agents and generates chromosomal instabilities. Furthermore, in an animal model of SBMA that exhibits a significant DNA damage response, a reduction in PTIP dosage increases muscle atrophy, indicating that PTIP levels, and by inference PTIP function, are important in mediating the cellular pathology in response to the poly-Q AR. The data suggest that SBMA skeletal muscle is particularly sensitive to impairments of PTIP-dependent pathways.
The poly-Q AR-expressing cells in culture are hypersensitive to a broad array of DNA-damaging agents, including IR, CPT and MMC. Chromosomal abnormalities are observed prior to the irradiation of cells expressing poly-Q AR and these abnormalities are more prevalent after exposure to radiation. In cell culture, the attenuation of the DNA repair process is likely due to a partial loss of PTIP function. This may phenocopy a hypomorphic PTIP mutation. Such mutations have been characterized previously, as a single point mutation can impact DSB repair, without affecting the ability of PTIP to mediate histone H3K4 methylation (30). The Q-domain, which is necessary for targeting PTIP to radiation-induced nuclear foci, may interact with other components of the DNA repair machinery with which the poly-Q AR now competes. Binding of poly-Q AR may prevent PTIP from localizing to radiation-induced foci but not to sites of transcription initiation that require MLL3/4-mediated H3K4 methylation.
One of the most striking features of poly-Q AR expression in cells was the generation of a slower migrating, modified PTIP isoform. This PTIP isoform had never been observed in any other cell system and was not induced by IR alone in cells expressing the WT AR. Our data suggest that the larger isoform is due to phosphorylation, as its presence is inhibited by DNA-PKcs inhibitors but not by ATM kinase inhibitors. Furthermore, the larger PTIP isoform can be reduced by the pretreatment of lysates with phosphatases. Yet, if DNA-PKcs activation were enough to phosphorylate PTIP, we would expect the large isoform to be inducible by radiation in WT AR-expressing cells. Instead, this larger isoform appears to be the result of a specific interaction with the poly-Q AR protein. The data suggest that unique post-translational modifications of endogenous proteins can be mediated by nuclear poly-Q AR and that such modifications can impact normal PTIP function. Similarly, an expanded Q-tract in SCA1 has been shown to alter the native protein complexes formed by ataxin 1 (41). Here, poly-Q expansion in the AR not only leads to aberrant association with PTIP but also triggers its post-translational modification. Whether DNA-Pkcs is the kinase responsible and which serine or threonine residues on PTIP are subject to phosphorylation will need to be determined in order to fully understand this phenomenon.
The interaction of poly-Q proteins with transcriptional co-regulators has attracted considerable attention as a pathogenic mechanism (11–17). Like PTIP, some of these proteins contain Q-rich domains and are present in limiting quantity within the nucleus. It has been suggested recently that the toxicity of the poly-Q AR is mediated by aberrant, ligand-dependent interactions with components of the transcriptional regulatory apparatus (42). As PTIP functions in both DNA repair and gene regulatory pathways, we considered the possibility that disruption of the PTIP function as a regulator of histone H3K4 methylation contributes to toxicity. The DNA-binding proteins Pax2 and Pax5 recruit PTIP to chromatin, a necessary step for the assembly of the MLL3/4 histone methyltransferase complex (27,43). Expression of AR113Q has no effect on the ability of Pax2 to activate a chromatin reporter gene that was previously used to define the PTIP function. Furthermore, we show that more than 1200 genes are activated or repressed by the addition of ligand to PC12 cells expressing AR10Q. The overwhelming majority of these genes are still activated or repressed in cells expressing AR112Q upon ligand addition, although the absolute levels of activation and repression are changed. Thus, gene expression analysis showed that expansion of the AR's Q tract leads to a partial loss of function rather than to gross alterations in the transcriptional response. It is possible that this partial loss of transactivation function could be due to reduced histone H3K4 methylation, which may be attributable to the PTIP/Mll3/4 complex. However, this would not explain the reduced levels of AR112Q-mediated gene repression observed. Nevertheless, the data strongly suggest that the poly-Q AR primarily disrupts PTIP function in DNA repair pathways and not in the histone H3K4 methylation pathway associated with transcription activation.
Cellular degeneration in poly-Q expansion diseases is a relatively slow process compared with the life of the individual and likely reflects the end result of chronic, low-level cellular injury in mitotically inactive cell types. Our findings suggest that DNA repair pathways are important in the pathology associated with poly-Q expansion. In normal dividing cells, DNA damage is an ongoing phenomenon that is continuously repaired in, but perhaps less so in poly-Q-expressing cells. IR and DNA-damaging agents are tools to enhance this damage and allow for experimental manipulation and quantitation. However, in vivo the affected cells are non-dividing and are not generally subject to radiation. In the mouse, conditional inactivation of proteins known to function in the repair of DNA DSBs, such as Rad50 (44) or NBS1 (45), indicates that mitotically active cells are highly susceptible to mutations in the DNA repair pathways but that non-dividing, terminally differentiated cells are less susceptible. Given the shorter life span of mice, perhaps it is not surprising that the effects of reduced DNA repair are not that apparent when compared with the slowly progressing pathologies found in humans with poly-Q expansion diseases which affect primarily non-dividing cells.
In the case of AR, the poly-Q tract in the knock-in mouse models must be significantly longer than those found in human patients with Kennedy's disease (40). Thus, it seems necessary to stress the system further so that the relevant pathologies are observed sooner in the mouse models. Another difference between mouse and humans is the lack of motor neuron death in the mouse models, although a distal axonopathy and myopathy are observed (19,39). Again, these differences may reflect the shorter life-span of mice and the need for neuromuscular feedback to drive the motor neuron pathology. Interestingly, the AR113Q mice also show significant defects in spermatogenesis (46), as AR is highly expressed in the testis. These testis defects are very similar to what is observed upon a loss of PTIP (K. Schwab and G.R.Dressler, submitted) and other DNA repair proteins (47,48) in the testis of adult male mice.
Rather than induce DNA damage directly, the data indicate that poly-Q AR proteins attenuate the DNA damage response, which may thus lead to an increase in the accumulation of mutations, cellular dysfunction and ultimately cell death. That this is mediated, at least in part, by interactions with PTIP is further substantiated by our genetic gene dosage experiments in AR113Q-expressing mice. The DNA repair proteins also are implicated in the inherent genetic instabilities of CAG triplet repeats, which are subject to generational expansion. Any attenuation of DNA repair is likely to increase the frequency of CAG expansion (49,50). Thus, our model would also help explain the genetic anticipation observed in many poly-Q-mediated diseases, as the propensity for CAG expansions to increase with decreasing DNA repair efficiency in a feed-forward manner.
MATERIALS AND METHODS
Antibodies
Antibodies to the AR (N20), Ku70, AR and 53BP1 (H-300) were from Santa Cruz Biotechnology. Mouse anti-γ-H2AX (Ser139) was from UpstateBiotech. Antibodies to the Mre11, P-ATM (Ser198) and P-p53 (Ser15) were from Cell Signaling Technology. Rabbit anti-PTIP antibodies against residues 316–591 have been described (27).
Plasmid construction
Plasmids encoding AR24Q and AR113Q were as described (38). The plasmid p3xFLAG-PTIP was generated previously (27). Plasmid p3xFLAG-PTIP-Q (amino acids 181–590) was generated by inserting a cDNA coding for the Q-rich domain with HindIII and XbaI sites into 3xFLAG-CMV10. A sequence encoding the nuclear localization signal (DPKKKRKV) has been fused in frame at the N terminus of each PTIP fragment to direct the protein into the nucleus.
Cell culture and transfection
Stable PC12 Tet-on AR10Q or AR112Q cells were generated previously (36) and cultured in phenol red-free DMEM with 10% charcoal-stripped heat inactivated horse serum (Invitrogen) and 5% charcoal-stripped heat inactivated bovine serum (Clontech), 100 U/ml penicillin/streptomycin (Gibco®), 200 µg/ml hygromycin (Invitrogen) and 100 µg/ml G418 (Gibco®) 5% CO2 at 37°C. Expression of AR was induced with 500 ng/ml of doxycycline (Clontech) for 48 h. HEK293 cells (ATCC) were cultured in a DMEM with 10% heat inactivated bovine serum (Gibco), 100 U/ml penicillin/streptomycin (Gibco) in 5% CO2 at 37°C. HEK293 cells with the PRS4-EGFP reporter gene were generated previously (27). Stable HEK293 cells expressing full-length 3xFLAG-PTIP were generated by transfecting HEK293 cells with plasmid p3xFLAG-PTIP using Fugene 6 (Roche Molecular Biochemical). Stable clones were selected with 600 µg/ml of G418 for 10 days and then screened for PTIP expression through western blotting using an anti-flag antibody. Cells expressing AR24Q or AR113Q were generated by transient transfection in HEK293 cells using Fugene 6. 24 h after transfection, cells were treated with 10 nm of the synthetic androgen R1881 (New England Nuclear) for 48 h and then harvested for further analysis. Similarly, cells expressing the PTIP-Q domain were generated by transient transfection into HEK293 cells using Fugene 6.
Western blot analysis and immunoprecipitation
For western blot analysis, the expression of AR in PC12 cells was induced with 500 ng of Dox for 24 h, and then treated with 10 nm R1881 for an additional 48 h. An ATM inhibitor Ku55933 (5 µm) or a DNA-PKcs inhibitor Nu7026 (50 µm) was added together with R1881. Transiently transfected HEK293 cells were treated with R1881 24 h after transfection for an additional 48 h. Cells were resuspended in lysis buffer (50 mm Tris–HCl, pH 7.4, with 150 mm NaCl, 1 mm EDTA and 1% Triton X-100) supplemented with protease inhibitors (Roche) and phosphatase inhibitors cocktail (Roche). Proteins were separated on a 6% sodium dodecyl sulfate (SDS)–polyacrylamide gel. Proteins were transferred to an Immobilon-FL PVDF membrane (Millipore) and detected with an indicated antibody.
For immunoprecipitations, cells were lysed in lysis buffer supplemented with protease inhibitors and phosphatase inhibitors. Five hundred micrograms of protein extract was incubated with 2 μg of indicated antibody overnight and then 30 µl of protein-A beads for 2 h (Invitrogen), followed by five washes in TBS buffer. The immunoprecipitates were eluted in 2× Tris–glycine SDS sample buffer and analyzed with western blot.
Immunofluorescence staining
Cells grown on chamber slide (Thermo Scientific) were irradiated with indicated dose and then recovered for the indicated time. The cells were fixed with 4% paraformaldehyde (PFA) for 10 min and permeabilized in 0.5% Triton phosphate-buffered saline (PBS) for 10 min. The cells were incubated with indicated primary antibodies diluted in 2% goat serum for 60 min. After washing with PBST (PBS with 0.1% Tween 20) three times, the cells were incubated with fluorescence-conjugated secondary antibodies for 60 min. Nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI) and then mounted with cover slides. The experiment was carried out at room temperature.
Muscle tissues were fixed overnight in 4% PFA and embedded in paraffin. Sections were dewaxed, rehydrated and microwaved for 10 min in a citric acid-based antigen unmasking solution (Vector Laboratories, Burlingame, CA, USA). Sections were permeabilized with 0.3% Triton X-100 in PBS and blocked with 10% goat serum in PBS. Primary antibodies were incubated for 2 h at room temperature in PBS, 0.1% Triton and 2% goat serum. Sections were washed twice and incubated with the secondary fluorescent antibodies and DAPI in PBS, 0.1% Triton and 2% goat serum for 1 h at room temperature. The sections were washed again and mounted in Mowiol. Stained and fluorescent-labeled sections were analyzed under a Nikon ES800 microscope. Micrographs were taken with a digital spot camera, using equivalent exposure times among sections.
In vitro PTIP dephosphorylation assay
Cell lysate from AR112Q PC12 cells enriched for phosphorylated PTIP was incubated in dephosphorylation buffer (50 mm HEPES, 100 mm NaCl, 2 mm DTT, 0.01% Brij 35, pH 7.5 @ 25°, 1 mm MnCl2) with 200 or 1000 U of Lambda Protein Phosphatase (New England BioLabs) at 30°C for 60 min.
Cellular sensitivities to DNA-damage agents
For IR treatment, cells were exposed at indicated dose. For CPT treatment, HEK293 cells were transiently transfected AR24Q or AR113Q and incubated with 40 nm CPT for 3 days. Cells were harvested at indicated days and counted with Invitrogen automated cell counter. For MMC treatment, PC12 cells were incubated with MMC at concentration range 0–320 nm. After 8 days of incubation, cell numbers were counted.
Chromosomal breakage analysis
HEK293 cells were transiently transfected with AR24Q or AR113Q using Fugene 6. 24 h after transfection, cells were treated with 10 nm R1881 for additional 48 h. Colcemid (GIBCO/BRL; KaryoMAX Colcemid solution) was added to the cultures (100 ng/ml), and the cultures were incubated for 3 h to arrest proliferating cells at metaphases. Metaphases were generated using standard procedures. Slides were then stained with DAPI. All imaging was performed on an Olympus BX-61 microscope equipped with an interferometer driven by a desktop computer and specialized software (ASI, Vista, CA, USA).
Mouse model of SBMA
The AR knock-in mice with targeted AR113Q CAG repeat expansion in exon 1 (40) and Paxip1 null alleles (32) have been described. The AR113Q/+ female mice were crossed to AR+:Paxip1+/− male mice to generate male littermates carrying either AR113Q;Paxip1+/− or AR113Q;Paxip1+/+ alleles.
To measure muscle weight and fiber size, mice were euthanized by isoflurane, and the tibialis anterior (TA) and soleus muscles were harvested. The TA muscle was weighed with an electronic analytical balance (Mettler Toledo AG104). The Soleus muscle was frozen in isopentane pre-chilled by liquid nitrogen. The muscle was cut in a cross-section orientation at a thickness of 5 µm with Leica Cryocut 1800 and H&E staining was performed. Digital images were captured using a Zeiss Axioplan 2 imaging system. The area of each muscle fiber was defined using Adobe Photoshop CS4, and 100 adjacent fibers from each section were measured. The pixel number was converted to µm2 according to scale.
Microarrays
Expression of AR10Q and AR112Q in PC12 cells was induced by the addition of 500 μg/ml of doxycycline for 24 h, followed by treatment with R1881 for an additional 24 h prior to RNA extraction. Total RNA was isolated from the PC12 cell lines using Trizol (Invitrogen). Microarray experiments were carried out using Affymetrix GeneChip Rat Genome 230 2.0 arrays that processed in the UMCCC Affymetrix and cDNA Microarray Core Facility at University of Michigan, Ann Arbor. Statistical analysis was performed according to the published methods (51,52).
FUNDING
This work was supported in part by NIH (DK054740 and DK073722 to G.R.D. and NS055746 to A.P.L.).
ACKNOWLEDGEMENTS
We thank Joe Washburn and Craig Johnson of the University of Michigan microarray core facility for data generation and statistical analysis.
Conflict of Interest statement. None declared.
References
- 1.La Spada A.R., Taylor J.P. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat. Rev. Genet. 2010;11:247–258. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takahashi T., Katada S., Onodera O. Polyglutamine diseases: where does toxicity come from? what is toxicity? where are we going? J. Mol. Cell. Biol. 2010;2:180–191. doi: 10.1093/jmcb/mjq005. [DOI] [PubMed] [Google Scholar]
- 3.Takeyama K., Ito S., Yamamoto A., Tanimoto H., Furutani T., Kanuka H., Miura M., Tabata T., Kato S. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron. 2002;35:855–864. doi: 10.1016/s0896-6273(02)00875-9. [DOI] [PubMed] [Google Scholar]
- 4.Montie H.L., Cho M.S., Holder L., Liu Y., Tsvetkov A.S., Finkbeiner S., Merry D.E. Cytoplasmic retention of polyglutamine-expanded androgen receptor ameliorates disease via autophagy in a mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2009;18:1937–1950. doi: 10.1093/hmg/ddp115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klement I.A., Skinner P.J., Kaytor M.D., Yi H., Hersch S.M., Clark H.B., Zoghbi H.Y., Orr H.T. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 6.Huynh D.P., Figueroa K., Hoang N., Pulst S.M. Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat. Genet. 2000;26:44–50. doi: 10.1038/79162. [DOI] [PubMed] [Google Scholar]
- 7.Bowman A.B., Yoo S.Y., Dantuma N.P., Zoghbi H.Y. Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum. Mol. Genet. 2005;14:679–691. doi: 10.1093/hmg/ddi064. [DOI] [PubMed] [Google Scholar]
- 8.Slow E.J., Graham R.K., Osmand A.P., Devon R.S., Lu G., Deng Y., Pearson J., Vaid K., Bissada N., Wetzel R., et al. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc. Natl Acad. Sci. USA. 2005;102:11402–11407. doi: 10.1073/pnas.0503634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arrasate M., Mitra S., Schweitzer E.S., Segal M.R., Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 10.Saudou F., Finkbeiner S., Devys D., Greenberg M.E. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 11.Dunah A.W., Jeong H., Griffin A., Kim Y.M., Standaert D.G., Hersch S.M., Mouradian M.M., Young A.B., Tanese N., Krainc D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science. 2002;296:2238–2243. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- 12.McCampbell A., Taylor J.P., Taye A.A., Robitschek J., Li M., Walcott J., Merry D., Chai Y., Paulson H., Sobue G., et al. CREB-binding protein sequestration by expanded polyglutamine. Hum. Mol. Genet. 2000;9:2197–2202. doi: 10.1093/hmg/9.14.2197. [DOI] [PubMed] [Google Scholar]
- 13.Nucifora F.C., Jr, Sasaki M., Peters M.F., Huang H., Cooper J.K., Yamada M., Takahashi H., Tsuji S., Troncoso J., Dawson V.L., et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 14.Steffan J.S., Kazantsev A., Spasic-Boskovic O., Greenwald M., Zhu Y.Z., Gohler H., Wanker E.E., Bates G.P., Housman D.E., Thompson L.M. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl Acad. Sci. USA. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsuda H., Jafar-Nejad H., Patel A.J., Sun Y., Chen H.K., Rose M.F., Venken K.J., Botas J., Orr H.T., Bellen H.J., et al. The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/Senseless proteins. Cell. 2005;122:633–644. doi: 10.1016/j.cell.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 16.Cui L., Jeong H., Borovecki F., Parkhurst C.N., Tanese N., Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 17.Zhai W., Jeong H., Cui L., Krainc D., Tjian R. In vitro analysis of huntingtin-mediated transcriptional repression reveals multiple transcription factor targets. Cell. 2005;123:1241–1253. doi: 10.1016/j.cell.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 18.Helmlinger D., Tora L., Devys D. Transcriptional alterations and chromatin remodeling in polyglutamine diseases. Trends Genet. 2006;22:562–570. doi: 10.1016/j.tig.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 19.Jordan C.L., Lieberman A.P. Spinal and bulbar muscular atrophy: a motoneuron or muscle disease? Curr. Opin. Pharmacol. 2008;8:752–758. doi: 10.1016/j.coph.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katsuno M., Adachi H., Waza M., Banno H., Suzuki K., Tanaka F., Doyu M., Sobue G. Pathogenesis, animal models and therapeutics in spinal and bulbar muscular atrophy (SBMA) Exp. Neurol. 2006;200:8–18. doi: 10.1016/j.expneurol.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 21.Sperfeld A.D., Karitzky J., Brummer D., Schreiber H., Haussler J., Ludolph A.C., Hanemann C.O. X-linked bulbospinal neuronopathy: Kennedy disease. Arch. Neurol. 2002;59:1921–1926. doi: 10.1001/archneur.59.12.1921. [DOI] [PubMed] [Google Scholar]
- 22.Kennedy W.R., Alter M., Sung J.H. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology. 1968;18:671–680. doi: 10.1212/wnl.18.7.671. [DOI] [PubMed] [Google Scholar]
- 23.Sobue G., Hashizume Y., Mukai E., Hirayama M., Mitsuma T., Takahashi A. X-linked recessive bulbospinal neuronopathy. A clinicopathological study. Brain. 1989;112(Pt 1):209–232. doi: 10.1093/brain/112.1.209. [DOI] [PubMed] [Google Scholar]
- 24.Enokido Y., Tamura T., Ito H., Arumughan A., Komuro A., Shiwaku H., Sone M., Foulle R., Sawada H., Ishiguro H., et al. Mutant huntingtin impairs Ku70-mediated DNA repair. J. Cell Biol. 2010;189:425–443. doi: 10.1083/jcb.200905138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Illuzzi J., Yerkes S., Parekh-Olmedo H., Kmiec E.B. DNA breakage and induction of DNA damage response proteins precede the appearance of visible mutant huntingtin aggregates. J. Neurosci. Res. 2009;87:733–747. doi: 10.1002/jnr.21881. [DOI] [PubMed] [Google Scholar]
- 26.Cho Y.W., Hong T., Hong S., Guo H., Yu H., Kim D., Guszczynski T., Dressler G.R., Copeland T.D., Kalkum M., et al. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 2007;282:20395–20406. doi: 10.1074/jbc.M701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel S.R., Kim D., Levitan I., Dressler G.R. The BRCT-domain containing protein PTIP links PAX2 to a histone H3, lysine 4 methyltransferase complex. Dev. Cell. 2007;13:580–592. doi: 10.1016/j.devcel.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gong Z., Cho Y.W., Kim J.E., Ge K., Chen J. Accumulation of Pax2 transactivation domain interaction protein (PTIP) at sites of DNA breaks via RNF8-dependent pathway is required for cell survival after DNA damage. J. Biol. Chem. 2009;284:7284–7293. doi: 10.1074/jbc.M809158200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu J., Prindle M.J., Dressler G.R., Yu X. PTIP regulates 53BP1 and SMC1 at the DNA damage sites. J. Biol. Chem. 2009;284:18078–18084. doi: 10.1074/jbc.M109.002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daniel J.A., Santos M.A., Wang Z., Zang C., Schwab K.R., Jankovic M., Filsuf D., Chen H.T., Gazumyan A., Yamane A., et al. PTIP promotes chromatin changes critical for immunoglobulin class switch recombination. Science. 2010;329:917–923. doi: 10.1126/science.1187942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwab K.R., Patel S.R., Dressler G.R. Role of PTIP in class switch recombination and long-range chromatin interactions at the immunoglobulin heavy chain locus. Mol. Cell. Biol. 2011;31:1503–1511. doi: 10.1128/MCB.00990-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho E.A., Prindle M.J., Dressler G.R. BRCT domain-containing protein PTIP is essential for progression through mitosis. Mol. Cell. Biol. 2003;23:1666–1673. doi: 10.1128/MCB.23.5.1666-1673.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fang M., Ren H., Liu J., Cadigan K.M., Patel S.R., Dressler G.R. Drosophila ptip is essential for anterior/posterior patterning in development and interacts with the PcG and trxG pathways. Development. 2009;136:1929–1938. doi: 10.1242/dev.026559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manke I.A., Lowery D.M., Nguyen A., Yaffe M.B. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003;302:636–639. doi: 10.1126/science.1088877. [DOI] [PubMed] [Google Scholar]
- 35.Yu X., Chini C.C., He M., Mer G., Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 36.Walcott J.L., Merry D.E. Ligand promotes intranuclear inclusions in a novel cell model of spinal and bulbar muscular atrophy. J. Biol. Chem. 2002;277:50855–50859. doi: 10.1074/jbc.M209466200. [DOI] [PubMed] [Google Scholar]
- 37.Grierson A.J., Mootoosamy R.C., Miller C.C. Polyglutamine repeat length influences human androgen receptor/c-Jun mediated transcription. Neurosci. Lett. 1999;277:9–12. doi: 10.1016/s0304-3940(99)00844-7. [DOI] [PubMed] [Google Scholar]
- 38.Lieberman A.P., Harmison G., Strand A.D., Olson J.M., Fischbeck K.H. Altered transcriptional regulation in cells expressing the expanded polyglutamine androgen receptor. Hum. Mol. Genet. 2002;11:1967–1976. doi: 10.1093/hmg/11.17.1967. [DOI] [PubMed] [Google Scholar]
- 39.Kemp M.Q., Poort J.L., Baqri R.M., Lieberman A.P., Breedlove S.M., Miller K.E., Jordan C.L. Impaired motoneuronal retrograde transport in two models of SBMA implicates two sites of androgen action. Hum. Mol. Genet. 2011;20:4475–4490. doi: 10.1093/hmg/ddr380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu Z., Dadgar N., Albertelli M., Gruis K., Jordan C., Robins D.M., Lieberman A.P. Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J. Clin. Invest. 2006;116:2663–2672. doi: 10.1172/JCI28773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lim J., Crespo-Barreto J., Jafar-Nejad P., Bowman A.B., Richman R., Hill D.E., Orr H.T., Zoghbi H.Y. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature. 2008;452:713–718. doi: 10.1038/nature06731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nedelsky N.B., Pennuto M., Smith R.B., Palazzolo I., Moore J., Nie Z., Neale G., Taylor J.P. Native functions of the androgen receptor are essential to pathogenesis in a Drosophila model of spinobulbar muscular atrophy. Neuron. 2010;67:936–952. doi: 10.1016/j.neuron.2010.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McManus S., Ebert A., Salvagiotto G., Medvedovic J., Sun Q., Tamir I., Jaritz M., Tagoh H., Busslinger M. The transcription factor Pax5 regulates its target genes by recruiting chromatin-modifying proteins in committed B cells. EMBO J. 2011;30:2388–2404. doi: 10.1038/emboj.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adelman C.A., De S., Petrini J.H. Rad50 is dispensable for the maintenance and viability of postmitotic tissues. Mol. Cell. Biol. 2009;29:483–492. doi: 10.1128/MCB.01525-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Demuth I., Frappart P.O., Hildebrand G., Melchers A., Lobitz S., Stockl L., Varon R., Herceg Z., Sperling K., Wang Z.Q., et al. An inducible null mutant murine model of Nijmegen breakage syndrome proves the essential function of NBS1 in chromosomal stability and cell viability. Hum. Mol. Genet. 2004;13:2385–2397. doi: 10.1093/hmg/ddh278. [DOI] [PubMed] [Google Scholar]
- 46.Yu Z., Dadgar N., Albertelli M., Scheller A., Albin R.L., Robins D.M., Lieberman A.P. Abnormalities of germ cell maturation and sertoli cell cytoskeleton in androgen receptor 113 CAG knock-in mice reveal toxic effects of the mutant protein. Am. J. Pathol. 2006;168:195–204. doi: 10.2353/ajpath.2006.050619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fernandez-Capetillo O., Mahadevaiah S.K., Celeste A., Romanienko P.J., Camerini-Otero R.D., Bonner W.M., Manova K., Burgoyne P., Nussenzweig A. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev. Cell. 2003;4:497–508. doi: 10.1016/s1534-5807(03)00093-5. [DOI] [PubMed] [Google Scholar]
- 48.Xu X., Aprelikova O., Moens P., Deng C.X., Furth P.A. Impaired meiotic DNA-damage repair and lack of crossing-over during spermatogenesis in BRCA1 full-length isoform deficient mice. Development. 2003;130:2001–2012. doi: 10.1242/dev.00410. [DOI] [PubMed] [Google Scholar]
- 49.Richard G.F., Goellner G.M., McMurray C.T., Haber J.E. Recombination-induced CAG trinucleotide repeat expansions in yeast involve the MRE11-RAD50-XRS2 complex. EMBO J. 2000;19:2381–2390. doi: 10.1093/emboj/19.10.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sundararajan R., Freudenreich C.H. Expanded CAG/CTG repeat DNA induces a checkpoint response that impacts cell proliferation in Saccharomyces cerevisiae. PLoS Genet. 2011;7:e1001339. doi: 10.1371/journal.pgen.1001339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ritchie M.E., Diyagama D., Neilson J., van Laar R., Dobrovic A., Holloway A., Smyth G.K. Empirical array quality weights in the analysis of microarray data. BMC Bioinformatics. 2006;7:261. doi: 10.1186/1471-2105-7-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Irizarry R.A., Wu Z., Jaffee H.A. Comparison of Affymetrix GeneChip expression measures. Bioinformatics. 2006;22:789–794. doi: 10.1093/bioinformatics/btk046. [DOI] [PubMed] [Google Scholar]