

Abstract
Background
Retinoids have shown antiproliferative and chemopreventive activity. We analyzed data from a randomized, placebo-controlled chemoprevention trial to determine whether a 3-month treatment with either 9-cis-retinoic acid (RA) or 13-cis-RA and α-tocopherol reduced Ki-67, a proliferation biomarker, in the bronchial epithelium.
Methods
Former smokers (n = 225) were randomly assigned to receive 3 months of daily oral 9-cis-RA (100 mg), 13-cis-RA (1 mg/kg) and α-tocopherol (1200 IU), or placebo. Bronchoscopic biopsy specimens obtained before and after treatment were immunohistochemically assessed for changes in the Ki-67 proliferative index (i.e., percentage of cells with Ki-67–positive nuclear staining) in the basal and parabasal layers of the bronchial epithelium. Per-subject and per–biopsy site analyses were conducted. Multicovariable analyses, including a mixed-effects model and a generalized estimating equations model, were used to investigate the treatment effect (Ki-67 labeling index and percentage of bronchial epithelial biopsy sites with a Ki-67 index ≥ 5%) with adjustment for multiple covariates, such as smoking history and metaplasia. Coefficient estimates and 95% confidence intervals (CIs) were obtained from the models. All statistical tests were two-sided.
Results
In per-subject analyses, Ki-67 labeling in the basal layer was not changed by any treatment; the percentage of subjects with a high Ki-67 labeling in the parabasal layer dropped statistically significantly after treatment with 13-cis-RA and α-tocopherol treatment (P = .04) compared with placebo, but the drop was not statistically significant after 9-cis-RA treatment (P = .17). A similar effect was observed in the parabasal layer in a per-site analysis; the percentage of sites with high Ki-67 labeling dropped statistically significantly after 9-cis-RA treatment (coefficient estimate = −0.72, 95% CI = −1.24 to −0.20; P = .007) compared with placebo, and after 13-cis-RA and α-tocopherol treatment (coefficient estimate = −0.66, 95% CI = −1.15 to −0.17; P = .008).
Conclusions
In per-subject analyses, treatment with 13-cis-RA and α-tocopherol, compared with placebo, was statistically significantly associated with reduced bronchial epithelial cell proliferation; treatment with 9-cis-RA was not. In per-site analyses, statistically significant associations were obtained with both treatments.
Lung cancer remains the leading cause of cancer death for both men and women in the United States, accounting for more deaths than breast, prostate, and colon cancers combined (1). Because lung cancer is frequently diagnosed at a late stage, improvements in long-term cure have been difficult to achieve, despite a variety of new therapeutic approaches (2). Tobacco smoking accounts for 90% of the attributable risk for lung cancer (3), but lung cancer risk remains elevated for many years after smoking cessation and never decreases to the level in never smokers (4,5). Nearly 50% of newly diagnosed lung cancers occur in former smokers (6,7), a result that supports the hypothesis that, during the prior years of smoking exposure, a tumorigenesis process is initiated that continues to evolve after smoking cessation. Consequently, there is continued interest in developing strategies to slow the rate of progression to lung cancer, especially in individuals at high risk of lung cancer (8). However, such high-risk individuals are difficult to identify. One means to identify such individuals is by their smoking history. A smoking history of 20 pack-years is associated with a 10%−15% lifetime risk for developing lung cancer (9). Chemoprevention trials studying such high-risk individuals with cancer incidence as the primary endpoint may thus require decades of follow-up and tens of thousands of participants (10). More recent chemoprevention trials with hundreds of participants have used intermediate biomarker endpoints with shorter follow-up times to identify potentially useful strategies to delay the onset of lung cancer in current and former smokers (11,12).
Many potentially useful biomarkers for lung cancer chemoprevention trials have been identified by their differential expression in lung cancer tissue and normal lung tissue. These biomarkers must be detectable in bronchial epithelial biopsy specimens from individuals thought to be at increased lung cancer risk, and they should be evaluable for modulation during chemopreventive intervention (13-16). Histopathologic markers such as bronchial metaplasia and dysplasia have been used in chemoprevention trials because they can be detected in bronchial biopsy samples from approximately 80% of current smokers, if there are sufficient numbers of biopsy specimens (14) or if regions with abnormal bronchoscopic fluorescence emission patterns are selected for biopsy examination (15). However, squamous metaplasia becomes undetectable with smoking cessation in approximately 50% of the individuals who have stopped smoking (14) and so has limited utility in chemoprevention studies that are evaluating former smokers.
Loss of the retinoic acid receptor β (RAR-β) expression is also a frequent fi nding in the bronchial epithelium of individuals with a smoking history of 20 pack-years (17). In vitro studies have demonstrated that retinoid treatment can restore RAR-β expression and slow proliferation of premalignant and malignant cell lines (18). At high doses, retinoids, such as 13-cis-retinoic acid (13-cis-RA), have shown activity in preventing second primary head and neck cancers (19) and in reducing the extent of oral leukoplakia in association with increased RAR-β expression (20).
In large lung cancer prevention trials that evaluated combinations of agents, including vitamins A or E, retinoids, β-carotene, and/or N-acetylcysteine, retinoid treatment was associated with increased lung cancer incidence among current smokers but with a non–statistically signifi cant decreased incidence among former and never smokers (21-24). High-dose 13-cis-RA could not reverse bronchial metaplasia in current smokers, but a trend toward reversion was observed in a small subset of former smokers (14).
To build on observations indicating that retinoids might have chemopreventive efficacy in former smokers, we explored the use of retinoids in a randomized, placebo-controlled chemoprevention trial, involving a group of former smokers with a smoking history of at least 20 pack-years, that used modulation of RAR-β expression as the primary intermediate endpoint biomarker (25,26). One group of subjects received a combination treatment of 13-cis-RA and α-tocopherol because α-tocopherol has been shown to reduce the side effects of 13-cis-RA, including mucocutaneous toxicity, increased liver function abnormalities, and hypertriglyceridemia (25). The second group of former smokers received 9-cis-RA alone because 9-cis-RA has been shown to bind to both RARs and retinoid X receptors and thus might have effects beyond that of 13-cis-RA (27). The third group received placebo treatment. As reported previously (26), treatment with 9-cis-RA, but not 13-cis-RA and α-tocopherol, was associated with a statistically significant increase in RAR-β expression in the bronchial epithelium (P = .03). The strength of this fi nding, however, was limited because only 30% of the pretreatment biopsy sites lacked RAR-β expression. Treatment with 9-cis-RA or 13-cis-RA and α-tocopherol was also associated with a reduction in the fraction of biopsy specimens with metaplasia after a 3-month treatment, whereas the fraction of bronchial sites with metaplasia in the placebo arm increased slightly during the trial period. However, the strength of this finding was limited because only 6.9% of the samples showed metaplasia at the outset of the treatment regimen. Thus, the impact of the intervention could be assessed in only a small subset of the participants presenting with modulable endpoints.
The Ki-67 index may be a useful intermediate endpoint biomarker because one hallmark of lung tumorigenesis is dysregulated proliferation. Studies from our group and those of others have demonstrated that Ki-67 labeling indices (i.e., percentage of cells with Ki-67–positive nuclear staining) are higher throughout the bronchial epithelium in current smokers than in nonsmokers 28). With tobacco smoking cessation, Ki-67 labeling indices, especially in the parabasal layers of the epithelium, dropped statistically significantly within 1 year (28). However, increased Ki-67 labeling indices remained detectable and measurable as a continuous variable for more than 20 years, even in the absence of squamous metaplasia (28). Moreover, a fraction of subjects had statistically significantly increased Ki-67 levels for several years after smoking cessation, possibly indicating a continuing and evolving tumorigenesis process in these individuals (28,29).
To investigate whether 9-cis-RA and 13-cis-RA have antiproliferative effects in vivo, we compared Ki-67 labeling indices obtained before and after the intervention in subjects who participated in the trial. We asked whether these retinoids showed antiproliferative effects in the subjects who participated in the randomized, placebo-controlled chemoprevention trial by comparing the Ki-67 labeling indices in the bronchoscopic biopsy specimens obtained before and after a 3-month intervention period. Because distinct biologic regulatory pathways may determine the proliferative status of the basal, parabasal, and superficial layers of the bronchial epithelium, we characterized the effects of retinoid treatment on all three epithelial layers separately.
Subjects and Methods
Subjects
The entry requirements for the chemoprevention trial have been previously described in detail (26). Briefly, the trial was designed to include former smokers, defined as individuals with a smoking history of at least 20 pack-years who had stopped smoking at least 1 year before study entry. Subjects were allowed to have a prior smoking-associated cancer but were required to be tumor-free for 6 months before enrollment on this study. Potential participants were not to have taken more than 25 000 IU of vitamin A or other retinoids within 3 months of study entry and were required to abstain from consuming vitamin A dietary supplements while on treatment in this study. The study was approved by the Institutional Review Board at the University of Texas M. D. Anderson Cancer Center and by the U.S. Department of Health and Human Services. All eligible participants provided written informed consent before treatment randomization. Male and female subjects were not analyzed separately.
Trial Design and Treatment
The clinical study design was a three-arm, double-blinded, placebo-controlled, randomized chemoprevention trial to evaluate 13-cis-RA (1 mg/kg) and α-tocopherol (1200 IU), 9-cis-RA (100 mg), and placebo in former smokers. Because prior toxicity data from phase I trials of 9-cis-RA did not extend beyond a 3-month treatment period (30,31), the duration of retinoid treatment in this trial was 3 months. The primary treatment endpoint was the restoration of RAR-β expression after a 3-month treatment period. The trial was also designed to examine the impact of a 3-month treatment on other markers in the bronchial epithelium, including Ki-67 labeling indices, loss of heterozygosity, p53 expression, and chromosome polysomy. A total of 240 subjects were registered onto the trial, and 225 subjects were randomly assigned to treatment. Forty-six subjects withdrew from study before completing 3 months of treatment (11 for toxicity and 35 for job relocation). Dose reductions were required in 87 subjects, including 49 in the 9-cis-RA group, 30 in the 13-cis-RA and α-tocopherol group, and eight in the placebo group.
Participants were to have stopped smoking at least 12 months before study entry. However, assessments of serum cotinine levels drawn at registration and at 3 months indicated that seven subjects may have experienced substantial smoke exposure (i.e., they may have resumed smoking) during their participation in the study (because of the presence of a serum cotinine level of ≥20 ng/mL), including three in the 9-cis-RA group, one in the 13-cis-RA and α-tocopherol group, and three in the placebo group. Because we have previously found that current smoking status is associated with statistically signifi cantly higher Ki-67 labeling indices (28), data analyses were carried out that either included or excluded those with elevated serum cotinine levels.
Biopsy Specimens
Before the initiation of treatment and after 3 months of treatment, bronchial biopsy specimens were obtained from the following six predefined sites in the bronchial tree: the main carina, the carina between the right upper lobe and the bronchus intermedius, the carina between the right middle lobe and the right lower lobe, the carina of the posterior basal segment of the right lower lobe, the carina between the left upper lobe and the left lower lobe, and the carina of the posterior basal segment of the left lower lobe. The specimens were fixed in buffered formalin, embedded in paraffin, and sectioned (4-μm thick). Ten sections per biopsy site were stained with hematoxylin–eosin and evaluated for metaplasia index or the presence of dysplasia by a pathologist (J. Y. Ro) who was blinded to the timing of the biopsy and the associated treatment group.
Immunohistochemical Analysis of Ki-67
One 4-μm section from each biopsy site was immunocytochemically stained for Ki-67 expression, as previously described (28). Briefly, a section of HeLa cells was placed on each sample slide to serve as a positive internal control for the immunostaining procedure. The tissue sections were then deparaffinized in xylene, rehydrated through an alcohol series, and immersed in a solution of 3% hydrogen peroxide in methanol for 15 minutes to block endogenous peroxidase activity. Antigen retrieval was accomplished by treatment with a 0.01 M citrate buffer (pH = 6.0, Zymed Laboratories, Inc, South San Francisco, CA) and microwave heating (800-W microwave at 100% power for two 4-minute periods). The slides were then immunostained with MIB-1 mouse anti–Ki-67 monoclonal antibody (Zymed Laboratories, Inc) and a biotinylated anti-mouse immunoglobulin G second antibody (Vector Laboratories, Inc, Burlingame, CA). The antibody reactions were visualized by use of the ABC peroxidase kit (Vector Laboratories, Inc) with diaminobenzidine (0.5 mg/mL) and 0.6% hydrogen peroxide. The slides were lightly counterstained with hematoxylin and mounted in Eukit (Calibrated Instruments, Hawthorne, NY).
The fraction of Ki-67–positive cells was determined separately in the basal layer, the parabasal layers, and, when present, the superficial layers of the bronchial biopsy specimens. Ki-67 labeling indices were expressed as the percentage of cells with positive nuclear staining. Slides with inadequate control HeLa cell labeling or those that lacked evaluable epithelium were excluded from the analyses. Ki-67 labeling indices were analyzed on a per–biopsy site basis and on a per-subject basis (the average of all biopsy sites that could be evaluated from a participant at a particular time point). The Ki-67 labeling index for the parabasal layer was dichotomized to high and low groups by use of 5% as a cut point (the 75th percentile of the Ki-67 labeling index of the parabasal layer for subjects who had quit smoking for at least 10 years and were without squamous metaplasia) (28). Similar conclusions were found with Ki-67 labeling index cut points between 4% and 5.5%, representing 46.6% and 33.5% of patients with high Ki-67 labeling indices, respectively.
Of 240 subjects registered in the clinical trial, baseline biopsy specimens for Ki-67 analysis were obtained from 231 subjects (1076 evaluable biopsy sites). Of these 231 subjects, 225 were randomly assigned to the treatment arms, including 74 in the 9-cis-RA group, 77 in the 13-cis-RA and α-tocopherol group, and 74 in the placebo group. Of the 225 subjects who were randomly assigned, 177 completed both baseline and 3-month bronchoscopy examinations, with 165 of them (48 subjects in the 9-cis-RA group, 62 in the 13-cis-RA and α-tocopherol group, and 55 in the placebo group) having both baseline and 3-month Ki-67 evaluations, for a total of 1495 Ki-67 observations. Sixty-six participants had baseline Ki-67 evaluations only (of these 66 participants, 56 belonged to a treatment group), and four participants had 3-month Ki-67 evaluations only.
Statistical Analysis
Summary statistics, including frequency, tabulation, mean (and 95% confidence intervals [CIs]), and median (and range), characterized the distributions of Ki-67 labeling indices in the basal and parabasal layers between subject groups. The mean Ki-67 index over the six potential biopsy sites was computed when the subject was used as the analysis unit. The Kruskal–Wallis test was used to compare continuous variables among the three treatment groups. The chi-square test or the Fisher’s exact test was used to test the statistical significance of the association between two categorical variables. The Wilcoxon signed rank test (32) was used to test changes in Ki-67 labeling indices by subject before and after treatment within each treatment group. To increase the efficiency of the statistical analysis, we also used the biopsy site as the unit of analysis under the assumption that the site was nested within the subject. Appropriate statistical methods were used to account for the correlation of sites within the same subject. For these multicovariable analyses, we used a mixed-effects model and a generalized estimating equations model (33), to test the treatment effect on Ki-67 labeling indices and dichotomized Ki-67 index for the basal layer and parabasal layer, adjusting for multiple covariates that affect Ki-67 levels, such as number of years since smoking cessation (1−2, 2−5, or ≥5 versus 0 years), squamous metaplasia (presence or absence), RAR-β expression (positive or negative), treatment arm, and time point (0 or 3 months). Generalized estimating equation analysis was carried out by assuming an unstructured working correlation matrix. When the mixed-effects model was used, a logarithmic transformation was applied to Ki-67 labeling indices to satisfy the Gaussian distribution assumption in the model. In the multicovariable analysis, the primary interest is to compare the modulation of Ki-67 over time (from baseline to 3 months after treatment) between the two treated groups and the placebo group. The model-based coefficient estimates and their 95% confidence intervals were reported. All statistical tests were two-sided, with a 5% type I error rate. Statistical analysis was performed by use of standard statistical software, including SAS release 9.1 (SAS Institute, Cary, NC) and S-Plus version 7 (Insightful, Inc, Seattle, WA). A power calculation was not done.
Results
Subject and Treatment-Associated Characteristics
The details of the study population and the compliance, response, and toxicity in this randomized, placebo-controlled trial have been reported previously (26). However, for completeness, the characteristics and relevant treatment-associated characteristics of the 225 randomly assigned subjects are summarized briefly (Table 1). The treatment groups were well balanced with regard to age, sex, race, history of smoking-related cancer, numbers of smoking pack-years and quit-years (the number of years since smoking cessation), and metaplasia index (the frequency of biopsy sites with metaplasia). However, when interpreting Ki-67 assessments, it is important to note that participants in the placebo group were slightly older and had a longer smoking history than those in either treatment group (P = .24 and .07, respectively).
Table 1.
Characteristics of the 225 randomly assigned subjects according to treatment group*
| Characteristic | Placebo (n = 74) | 13-cis-RA + AT (n = 77) | 9-cis-RA (n = 74) | P value† |
|---|---|---|---|---|
| Sex, No. (%) | .65 | |||
| Male | 45 (60.8) | 42 (54.6) | 40 (54.1) | |
| Female | 29 (39.2) | 35 (45.5 | 34 (45.9) | |
| Race, No. (%) | .82 | |||
| White | 65 (87.8) | 67 (87.0) | 69 (93.2) | |
| African American | 6 (8.1) | 6 (7.8) | 4 (5.4) | |
| Hispanic | 3 (4.1) | 3 (3.9) | 1 (1.4) | |
| Asian | 0 (0.0) | 1 (1.3) | 0 (0.0) | |
| Smoking-related cancer‡, No. (%) | .40 | |||
| No | 64 (86.5) | 69 (89.6) | 69 (93.2) | |
| Yes | 10 (13.5) | 8 (10.4) | 5 (6.8) | |
| Age, y | .24 | |||
| Mean ± SD | 58.4 ± 9.1 | 56.8 ± 10.0 | 56.0 ± 10.0 | |
| Median (range) | 58.9 (34.9–75.6) | 56.5 (34.0–78.2) | 55.1 (35.9–78.1) | |
| No. of years of smoking | .07 | |||
| Mean ± SD | 30.2 ± 9.9 | 26.6 ± 8.4 | 26.7 ± 9.4 | |
| Median (range) | 30 (15–50) | 25 (11–49) | 25 (10–50) | |
| No. of packs smoked per day | .56 | |||
| Mean ± SD | 1.8 ± 0.7 | 1.8 ± 0.8 | 1.9 ± 0.8 | |
| Median (range) | 1.8 (1–4) | 1.5 (0.8–4) | 2 (0.8–4) | |
| No. of pack-years | .28 | |||
| Mean ± SD | 53.9 ± 31.1 | 46.1 ± 23.7 | 50.1 ± 28.8 | |
| Median (range) | 44 (20–152) | 39 (20–136) | 40 (20–136) | |
| No. of smoking quit-years§ | .79 | |||
| Mean ± SD | 10.2 ± 8.1 | 10.9 ± 9.4 | 11.2 ± 8.9 | |
| Median (range) | 9.7 (1.0–35.2) | 8.2 (1.0–46.0) | 8.4 (1.0–48.1) |
RA = retinoic acid; AT = α-tocopherol; SD = standard deviation.
The Kruskal–Wallis test was performed to test the equal median of continuous variables among the three treatment groups. The chi-square test (for sex and smoking-related cancer) or Fisher’s exact test (for race) was performed to test the association between two categorical variables. All statistical tests were two-sided.
All smoking-related cancers were lung cancers, except for two head-and-neck cancers and one bladder cancer in the 13-cis-RA and AT group and one head-and-neck cancer in the 9-cis-RA group.
No. of smoking quit-years was calculated from data from 218 participants whose smoking status was confirmed by serum cotinine level. Seven participants (three in the placebo group, one in the 13-cis-RA group, and three in the 9-cis-RA group) with serum cotinine levels of more than 20 ng/mL at baseline were considered to be active smokers and so were excluded from this calculation.
9-cis-Retinoic Acid or 13-cis-Retinoic Acid Treatment and Proliferation in the Bronchial Epithelium
Among all 225 randomly assigned subjects with Ki-67 measurements, the baseline Ki-67 labeling indices were higher in the parabasal layers (5.79%, 95% CI = 5.01% to 6.56%) than in the basal layers (2.32%, 95% CI = 2.02% to 2.63%), consistent with what has previously been described in the bronchial epithelium of former smokers (28). No statistically significant differences were observed in the baseline proliferative status of the bronchial epithelium of subjects randomly assigned to any of the three treatment groups (Table 2). However, subjects assigned to the placebo group had slightly higher mean baseline Ki-67 labeling indices in the basal and parabasal layers than subjects assigned to the active treatment groups. Among all 225 randomly assigned subjects, little or no change after the 3-month treatment period was observed in Ki-67 labeling indices in the basal layer of subjects receiving 9-cis-RA (from 2.34% to 2.39%) or 13-cis-RA and α-tocopherol (from 2.22% to 2.19%), but Ki-67 labeling indices of the basal layer in the placebo group increased slightly between pretreatment and 3-month treatment specimens (from 2.41% to 2.93%). However, none of these within-group differences reached statistical significance (P = .94, .66, and .24, respectively).
Table 2.
Ki-67 index: Characteristics and modulation by treatment group*
| Placebo (n = 74)
|
13-cis-RA + AT (n = 77)
|
9-cis-RA (n = 74)
|
||||
|---|---|---|---|---|---|---|
| Epithelial layer | Mean (95% CI) | Median (range) | Mean (95% CI) | Median (range) | Mean (95% CI) | Median (range) |
| Basal layer | ||||||
| Baseline† | 2.41 (1.85 to 2.97) | 1.70 (0 to 15.03) | 2.22 (1.79 to 2.65) | 1.78 (0 to 11.16) | 2.34 (1.71 to 2.97) | 1.58 (0 to 18.17) |
| 3 months† | 2.93 (2.22 to 3.65) | 2.32 (0 to 14.53) | 2.19 (1.64 to 2.74) | 1.75 (0 to 11.62) | 2.39 (1.41 to 3.37) | 1.54 (0.23 to 23.16) |
| Modulation‡ | 0.39 (−0.41 to 1.18) | 0.10 (−12.12 to 11.14) | 0.07 (−0.67 to 0.81) | −0.23 (−9.87 to 9.7) | 0.11 (−1.27 to 1.48) | −0.01 (−17.88 to 22.45) |
| P value§ | .24 | .66 | .94 | |||
| Parabasal layer | ||||||
| Baseline† | 6.10 (4.64 to 7.57) | 3.96 (0.76 to 33.62) | 5.46 (4.31 to 6.62) | 3.57 (0 to 27.70) | 5.81 (4.33 to 7.28) | 3.41 (0 to 31.58) |
| 3 months† | 7.16 (5.12 to 9.20) | 4.59 (0 to 38.10) | 4.24 (3.28 to 5.21) | 3.44 (0 to 20.23) | 4.03 (3.01 to 5.05) | 2.77 (0.28 to 15.40) |
| Modulation‡ | 0.67 (−1.55 to 2.89) | 0.08 (−29.77 to 22.78) | −1.08 (−2.65 to 0.48) | −0.81 (−23.11 to 17.34) | −1.65 (−3.48 to 0.19) | −0.34 (−31.31 to 8.26) |
| P value§ | .59 | .13 | .24 | |||
RA = retinoic acid; AT = α-tocopherol; CI = confidence interval. Ki-67 index is expressed as the mean (95% CI) and the median (range).
The statistics for baseline (n = 73 in the placebo group, n = 75 in the 13-cis-RA group, and n = 73 in the 9-cis-RA group) and 3-month (n = 56 in the placebo group, n = 64 in the 13-cis-RA group, and n = 49 in the 9-cis-RA group) measurements were based on all available samples.
Modulation of Ki-67 index (n = 55 in placebo group, n = 62 in 13-cis-RA group, and n = 48 in 9-cis-RA group) was calculated only on participants who had both baseline and 3-month measurements (Ki-67 at 3 months – Ki-67 at baseline); shown as mean (95% CI) and median (range).
Wilcoxon signed rank test was performed to test the statistical significance of the change in the median Ki-67 modulation in each treatment group. All statistical tests were two-sided.
A different pattern was observed in the parabasal bronchial epithelial layers. Although the Ki-67 labeling index in the parabasal layer of subjects in the placebo group had increased after 3 months of treatment (from 6.10% to 7.16%), the labeling index in the parabasal layer of subjects in the 9-cis-RA (5.81% to 4.03%) or 13-cis-RA and α-tocopherol (from 5.46% to 4.24%) groups appeared to decrease after 3 months of treatment. By subtracting the Ki-67 values at 3 months from those at baseline in the subset of 165 subjects with both baseline and 3-month evaluable Ki-67 results, we determined the degree of Ki-67 modulation on individual patients. Treatment with either 9-cis-RA or 13-cis-RA and α-tocopherol was associated with a decreased Ki-67 labeling index in the parabasal layers (−1.65% and −1.08%, respectively), whereas treatment with placebo was associated with a slight increase in the Ki-67 labeling index (0.67%) after treatment (Table 2). However, no within-group difference reached statistical significance (P = .24, .13, and .59, respectively).
One potentially confounding aspect of the study was that seven subjects (one in the 13-cis-RA and α-tocopherol group, three in the 9-cis-RA group, and three in the placebo group) appeared to have been substantially smoke exposed either before or during the course of the trial, as judged by serum cotinine measurements (i.e., they had likely resumed smoking). We had previously observed (28) that Ki-67 labeling indices are profoundly higher in current smokers than in former smokers and that it takes nearly a year after smoking cessation for Ki-67 labeling indices in the bronchial epithelium to drop substantially. The association between retinoid treatment and the Ki-67 labeling indices was therefore reanalyzed after excluding the seven subjects with high serum cotinine levels. In the basal layer analyses, exclusion of the suspected smoke-exposed subjects did not alter the Ki-67 labeling index for any treatment group. In the parabasal layer analyses, the exclusion made the increased labeling index in the placebo group less apparent but did not alter the decreased Ki-67 labeling index for the groups receiving 9-cis-RA or 13-cis-RA and α-tocopherol (Fig. 1).
Fig. 1.
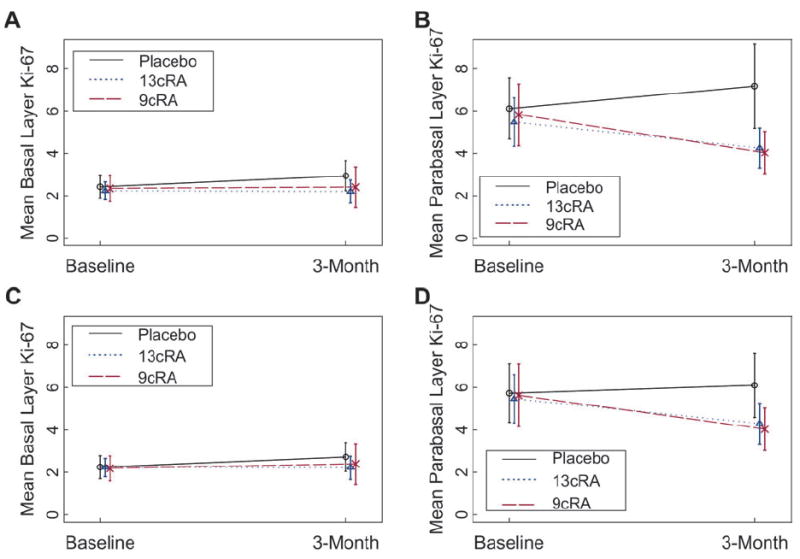
Comparisons of a 3-month treatment with 13-cis-retinoic acid (13cRA) and α-tocopherol (AT), 9cRA, or placebo on Ki-67 proliferation indices in the basal (A and C) and parabasal (B and D) bronchial epithelial cell layers of former smokers. A and B) Data from all 225 subjects. C and D) Data from 218 subjects. In these analyses, data were excluded from seven subjects with suspected ongoing tobacco smoke exposure because of increased serum cotinine levels. The subject was used as the analysis unit. Results are expressed as the mean Ki-67 index over time; error bars = 95% confidence intervals.
To better determine the relative impact of either 9-cis-RA or 13-cis-RA and α-tocopherol treatment on the Ki-67 labeling index of the bronchial epithelium compared with placebo treatment and to increase the efficiency of the statistical analysis, we carried out a multicovariable analysis by use of a mixed model, with bronchial site as a unit. In this analysis, we assumed that the site was nested within the subject, and we adjusted for features previously found to be associated with increased Ki-67 labeling indices (with logarithmic transformation), including the presence of squamous metaplasia, the expression of RAR-β, and the time since smoking cessation. This analysis showed again that these variables were associated with the Ki-67 labeling indices in the basal and parabasal layers (Table 3). Treatment with 13-cis-RA and α-tocopherol, compared with placebo, was statistically significantly associated with decreased Ki-67 labeling indices in the basal (coefficient estimate = −0.14, 95% CI = −0.28 to −0.003; P = .045) and parabasal (coefficient estimate = −0.15, 95% CI = −0.28 to −0.01; P = .04) layers. There was also a trend for the association between 9-cis-RA treatment and decreased Ki-67 labeling indices, especially in the parabasal layers (coefficient estimate = −0.12, 95% CI = −0.26 to 0.02; P = .097). Similar conclusions were obtained from an analysis with the less model-dependent generalized estimating equation approach (data not shown).
Table 3.
Multicovariable analysis by the mixed-effects model for testing the effect of covariates on log10(Ki-67 index), using the biopsy site as the analysis unit with the assumption that sites were nested in subjects and were correlated (1768 sites in 225 patients)*
| Comparison | Basal layer
|
Parabasal layer
|
||
|---|---|---|---|---|
| Coefficient estimate (95% CI) | P value† | Coefficient estimate (95% CI) | P value† | |
| No. of smoking quit-years | ||||
| 1–2 vs 0 | −0.28 (−0.47 to −0.09) | .003 | −0.21 (−0.40 to −0.015) | .03 |
| 2–5 vs 0 | −0.39 (−0.58 to −0.20) | <.001 | −0.27 (0.46 to −0.08) | .005 |
| ≥5 vs 0 | −0.31 (−0.49 to −0.14) | <.001 | −0.25 (−0.42 to −0.07) | .007 |
| Squamous metaplasia (1 vs 0)‡ | 0.54 (0.43 to 0.65) | <.001 | 0.74 (0.63 to 0.85) | <.001 |
| RAR-β | 0.14 (0.07 to 0.20) | <.001 | 0.15 (0.08 to 0.22) | <.001 |
| Treatment group | ||||
| 13-cis-RA + AT vs placebo | 0.07 (−0.02 to 0.16) | .11 | −0.02 (−0.11 to 0.09) | .64 |
| 9-cis-RA vs placebo | −0.06 (−0.15 to 0.03) | .21 | −0.12 (−0.22 to −0.03) | .008 |
| Time (3 mo vs baseline) | 0.15 (0.05 to 0.24) | .004 | 0.11 (0.01 to 0.21) | .03 |
| Group × time interaction | ||||
| 13-cis-RA + AT at 3 mo | −0.14 (−0.28 to −0.003) | .045 | −0.15 (−0.28 to −0.01) | .04 |
| 9-cis-RA at 3 mo | −0.10 (−0.24 to 0.04) | .17 | −0.12 (−0.26 to 0.02) | .097 |
CI = confidence interval; RAR-β = retinoic acid receptor β; RA = retinoic acid; AT = α-tocopherol.
The t test from a mixed-effects model was used. All statistical tests were two-sided.
0 = no squamous metaplasia; 1 = squamous metaplasia.
We dichotomized the parabasal layer data into low- or high-proliferative biopsy site groups by use of a Ki-67 labeling index cutoff of 5 to better define the impact of the treatments on subjects entering the trial who had relatively high Ki-67 labeling indices and who might best benefit from an antiproliferative strategy. By use of this cut point on data for the parabasal layer, 39.7%, 37.0%, and 40.0% of subjects fell into the high Ki-67 labeling index category before treatment in the groups randomly assigned to placebo, 9-cis-RA, or 13-cis-RA and α-tocopherol, respectively. The fraction of patients with high Ki-67 labeling indices increased to 48.2% after treatment in the placebo group but decreased to 28.6% and 26.6% after treatment with 9-cis-RA or 13-cis-RA and α-tocopherol, respectively. In an analysis that used the biopsy site as the analysis unit and the assumption that the site is nested within a subject, a generalized estimating equation model that was adjusted for years of smoking, packs per day, and metaplasia demonstrated that a 3-month treatment with either 9-cis-RA or 13-cis-RA and α-tocopherol was associated with a statistically significant decreased fraction of sites in the bronchial epithelium with a high Ki-67 index (coefficient estimate = −0.72, 95% CI = −1.24 to −0.20, P = .007, and coefficient estimate = −0.66, 95% CI = −1.15 to −0.17, P = .008, respectively) (Table 4). A similar trend was also observed when the analysis was carried out with the subject as the analysis unit (P = .17 and P = .04, respectively).
Table 4.
Multicovariable analysis by the generalized estimating equations model for testing the effect of covariates on parabasal layer Ki-67 index with values of 5% or more, using the biopsy site as the analysis unit with the assumption that sites were nested in subjects and were correlated (1768 sites in 225 patients)*
| Covariates | Coefficient estimate (95% CI) | P value† |
|---|---|---|
| No. of smoking quit-years | ||
| 1–2 vs 0 | −1.35 (−2.09 to −0.61) | <.001 |
| 2–5 vs 0 | −1.50 (−2.24 to −0.76) | <.001 |
| ≥5 vs 0 | −1.54 (−2.23 to −0.85) | <.001 |
| Squamous metaplasia (1 vs 0)‡ | 2.35 (1.90 to 2.80) | <.001 |
| Treatment group | ||
| 13-cis-RA + AT vs placebo | 0.07 (−0.26 to 0.40) | .68 |
| 9-cis-RA vs placebo | −0.08 (−0.43 to 0.27) | .67 |
| Time (3 mo vs baseline) | 0.55 (0.22 to 0.87) | .001 |
| Group × time interaction | ||
| 13-cis-RA + AT at 3 mo | −0.66 (−1.15 to −0.17) | .008 |
| 9-cis-RA at 3 mo | −0.72 (−1.24 to −0.20) | .007 |
CI = confidence interval; RA = retinoic acid; AT = α-tocopherol.
The Z test from the generalized estimating equations model was used. All statistical tests were two-sided.
0 = no squamous metaplasia; 1 = squamous metaplasia.
Discussion
In per-subject analyses, treatment with 13-cis-RA and α-tocopherol, compared with placebo, was statistically significantly associated with reduced bronchial epithelial cell proliferation in the parabasal layer; treatment with 9-cis-RA was not. In per-site analyses, a 3-month treatment with either 9-cis-RA or 13-cis-RA and α-tocopherol was statistically significantly associated with decreased proliferation (as assessed by the Ki-67 labeling index) in the parabasal layer of the bronchial epithelium of former chronic smokers, compared with placebo treatment. The antiproliferative activity of the retinoids was most pronounced in the parabasal layers of the bronchial epithelium.
The results of this study support the contention that smoking status can influence and limit the efficacy of chemopreventive intervention. In a previous biomarker-associated chemoprevention trial (14), we examined the impact of a 6-month treatment of 13-cis-RA on proliferation in the bronchial epithelium in current smokers by use of proliferating-cell nuclear antigen (PCNA) labeling indices, RAR-β expression, and squamous metaplasia indices as study endpoints. Although there was an apparent effect on RAR-β expression in that study, there was little measurable impact of 13-cis-RA on proliferation and metaplasia in individuals who continued to smoke. Nevertheless, we did observe a non–statistically significant trend toward decreasing PCNA labeling indices in individuals who stopped smoking during the 6-month treatment period. However, because we also found (28) that proliferation levels decreased in the bronchial epithelium of individuals in the first 6–12 months after smoking cessation in association with reversal of squamous metaplasia, it was difficult to ascribe decreased bronchial epithelial proliferation to 13-cis-RA treatment in that study.
The current study explored the impact of 9-cis-RA or 13-cis-RA and α-tocopherol in former smokers, as defined by individuals with a smoking history of at least 20 pack-years who had not smoked for at least 12 months before entry onto the trial. After this time of smoking cessation, squamous metaplasia and the levels of proliferative activity should have already decreased to a reduced, albeit still abnormal, steady-state level (28). Indeed, the finding that the degree of proliferative activity in the bronchial epithelium remained relatively stable in the placebo group (except in those individuals who might have resumed smoking tobacco) supports the notion that a baseline level had been achieved and that the variation in baseline values represents differences between individuals rather than variation in Ki-67 assessment. Moreover, the finding that proliferation indices decreased after treatment in the subjects in both the 9-cis-RA and 13-cis-RA and α-tocopherol groups indicates that retinoids may have a chemopreventive role in former smokers. However, because 9-cis-RA dose reductions and toxicity-based withdrawals were frequently required, in future trials, it will be important to incorporate these retinoids into combination regimens in which lower doses can be used.
A number of markers of proliferation have been explored in the chemoprevention setting as intermediate markers of response, including PCNA, minichromosome maintenance protein 2, and phosphorylated histone H3 (34-36). Each marker is useful for characterizing different aspects of proliferation, and each has its limitations. For example, early studies demonstrated increased PCNA labeling in the bronchial epithelium of chronic smokers; the levels were strongly associated with histologic changes in the aerodigestive tract (37) and could be modulated with chemopreventive intervention (34). Minichromosome maintenance protein 2 is a part of the DNA replication complex and is found in cells preparing for and undergoing DNA synthesis, but its expression is reduced later in the cell cycle. Phosphorylated histone H3 is a biomarker for cells in mitosis, but it has a limited dynamic range and is thus less useful as an intermediate biomarker in this setting. Ki-67 is useful for identifying the fraction of cells that have exited quiescent state, but it does not necessarily indicate that the cells are committed to movement through the cell cycle. The expression of each of these markers has been previously shown to increase as lung tissues exhibit more advanced premalignant histologies (28,34-36), and such proliferation markers have been shown to have prognostic implications in non–small-cell lung carcinoma (38-42). None of these proliferation markers, however, has been validated as a risk biomarker in a prospective chemoprevention study. Thus, the relationship between decreased expression of proliferation markers in the bronchial epithelium and delayed lung cancer onset will need to be validated in prospective studies in high-risk individuals.
We chose Ki-67 as the intermediate endpoint biomarker for this study for several reasons. First, we previously showed (28) that Ki-67 labeling indices are generally higher in lesions with metaplasia and dysplasia than in lesions without them. Second, Ki-67 labeling indices were strongly associated with increases in the frequency of bronchial sites with metaplasia and the number of packs smoked per day, indicating that Ki-67 assessments reflect a field effect and are minimally dependent on the particular biopsy site evaluated (28,43). Third, although Ki-67 labeling has been previously demonstrated (28) to decrease substantially in the first year after smoking cessation, a lifestyle change that is associated with decreased lung cancer risk (4,5), the Ki-67 labeling index still remains abnormally high even in the absence of squamous metaplasia and can be subject to measurable modulation (28). Fourth, Ki-67 labeling indices could be assessed as a continuous variable, and so modulation could be measured in all subjects. As a result, we were able to demonstrate that both 9-cis-RA and 13-cis-RA and α-tocopherol treatments could lower the levels of proliferation in the bronchial epithelium of former smokers.
We previously reported (26) that treatment with 9-cis-RA, but not with 13-cis-RA and α-tocopherol, reversed the loss of RAR-β expression in the bronchial epithelium of this same group of subjects. One limitation of our study is that, for three reasons, we could not carry out case-by-case comparisons between changes in Ki-67 labeling indices and RAR-β expression status to better understand the molecular basis for proliferative changes with treatment. First, only 30% of the subjects showed loss of RAR-β mRNA expression before treatment in at least one of six bronchial biopsy sites, whereas Ki-67 labeling indices were assessed in all bronchial sites. Second, because of limitations in availability of validated antibodies to RAR-β, the protein levels of RAR-β could not be assessed in these small biopsy sections, and so it was difficult to address the question of why proliferation, as measured by the Ki-67 labeling index, was repressed when the critical retinoid receptor was undetectable at baseline. Third, the in situ hybridization technique used in the previous report (26) did not distinguish between different isoforms and variants of RAR-β. It is now known that several isoforms and variants of RAR-β exist that are controlled by distinct regulatory pathways and have distinct functions and biologic consequences in normal and malignant cells. For example, although RAR-β2 is the most frequently expressed form that is inducible by retinoic acid and is suppressed in many premalignant and malignant cells and tissues (44), RAR-β4 is increased in certain cancers and may act as an oncogene [e.g., see (45,46)]. Therefore, when we assessed these tissues for RAR-β mRNA expression, it was not clear whether the mRNA detected would encode a tumor suppressor or a tumor promoter. It may be for these reasons that we found that the Ki-67 results provide strong independent information in a multicovariable analysis (Table 3) that included RAR-β expression as one of the covariates.
Although the use of Ki-67 labeling indices as an intermediate marker of response allowed us to examine retinoid impact in all treated subjects, the analysis was still somewhat limited by the generally low proliferation levels in former smokers at baseline, which made it difficult to detect a statistically significant reduction in proliferation in subjects with low pretreatment proliferation indices. For this reason, we dichotomized the subject population into those with low and those with high pretreatment proliferation levels (by use of a parabasal layer Ki-67 proliferation index cutoff of 5, which represented the 75th percentile value among former smokers). In this analysis, we observed a strong association between both 9-cis-RA and 13-cis-RA and α-tocopherol treatments and proliferation activity in the bronchial epithelium of former smokers (P = .008 and P = .007, respectively). Although the use of this particular cutoff point might be somewhat arbitrary, we hypothesize that former smokers with the highest Ki-67 values are at the highest risk of lung cancer and the use of such a cutoff value would permit us to examine the treatment effect on the high-risk group. Although similar conclusions were obtained by slightly varying the cut-point value, this cutoff point should be validated in future studies.
This finding of decreased proliferation in the bronchial epithelium after retinoid treatment is important when changes in the bronchial epithelium of placebo-treated former smokers are taken into account. That is, some subjects in the placebo group exhibited an increase in bronchial epithelial proliferation during a 3-month observation period, whereas no increased proliferation was observed in either retinoid treatment group. This finding may also be important if individuals with the highest pretreatment proliferation levels in the bronchial epithelium are at the highest risk of lung cancer and are likely to be the subjects who may gain the greatest benefit from chemopreventive intervention (34,43).
We distinguished between proliferation in the basal and parabasal epithelial layers in the three treatment groups for two reasons. First, proliferation in the parabasal layers appeared to be higher than that in the basal layer of the bronchial epithelium; thus, a treatment-related change could be more easily observed. Second, the factors that regulate proliferation in the basal layers and in the parabasal layers might be different. For example, in regenerating epithelial tissues, the initiation of proliferation might occur in the basal layer and then expand into the parabasal layers, in which negative regulatory factors might control the degree of tissue expansion. Indeed, we found that retinoid treatment was more strongly related to modulation of proliferation in the parabasal layers than to modulation of proliferation in the basal layer of the bronchial epithelium. These results support those from a small non–placebo-controlled trial of celecoxib in active smokers that also demonstrated decreased Ki-67 labeling indices in the parabasal layers of the bronchial epithelium (47). Because lung tumorigenesis is likely to be a multistep process of clonal outgrowth and clonal evolution that is driven by genetic instability (48), we expect that decreased proliferation would slow lung tumor development by slowing clonal expansion, decreasing the target population for subsequent genetic change, and decreasing ongoing genetic instability associated with dysregulated growth control.
The molecular underpinnings of the potential differences in growth regulatory properties between the basal and parabasal layers of the bronchial epithelium are still poorly understood and deserve greater examination. Although early histopathologic changes (e.g., metaplasia and dysplasia) in the bronchial epithelium give the appearance of an expanding basal layer, it is not known whether all epithelial layers are equally capable of participating in multistep tumorigenesis. It is also still not known whether it is important to target the growth properties of the basal cells, the parabasal cells, or a subset of these cells for long-term chemopreventive effect. It will therefore be important to distinguish the effects of molecularly targeted agents on the basal and parabasal bronchial epithelial layers in proposed lung chemoprevention trials with long follow-up (12).
CONTEXT AND CAVEATS.
Prior knowledge
Retinoids have antiproliferative activity in vitro and have chemo-protective activity in the upper aerodigestive tract.
Study design
Three-arm, double-blind, placebo-controlled randomized chemo-prevention trial among former smokers to examine the ability of a 3-month treatment of 9-cis-retinoic acid (RA), 13-cis-RA and α-tocopherol, or placebo to restore expression of RA receptor β. The Ki-67 labeling index, a measure of cell proliferation, was a secondary endpoint. Analyses were done with the subject or the biopsy site being the analysis unit.
Contribution
In per-subject analyses, treatment with 13-cis-RA and α-tocopherol, compared with placebo, was statistically significantly associated with reduced bronchial epithelial cell proliferation; treatment with 9-cis-RA was not. In per-site analyses, statistically significant associations were obtained with both treatments.
Implications
Because the subject is the unit of chemopreventive treatment and the associations from per-subject analyses were weaker than those from the per-site analyses, more research is required before firm conclusions can be drawn about the chemopreventive effect of both treatments.
Limitations
The Ki-67 labeling index is an intermediate marker of response. Former smokers generally have low proliferation levels in their bronchial epithelium; in subjects with the lowest pretreatment proliferation indices, a statistically significant reduction in proliferation was especially difficult to detect.
Acknowledgments
Funding
National Cancer Institute, National Institutes of Health, Department of Health and Human Services (Public Health Services grants U19CA68437, P50CA70907, P30CA16672).
Footnotes
Notes
Dr W. N. Hittelman holds the Sophie Caroline Steves Distinguished Professorship in Cancer Research, Dr R. Lotan holds the Irving and Nadine Mansfield and Robert David Levitt Cancer research Chair, and Dr W. K. Hong holds the Samsung Distinguished University Chair in Cancer Medicine. The authors had full responsibility for the design of the study, the collection of the data, the analysis and interpretation of the data, the decision to submit the manuscript for publication, and the writing of the manuscript.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Carney DN. Lung cancer—time to move on from chemotherapy. N Engl J Med. 2002;346:126–7. doi: 10.1056/NEJM200201103460211. [DOI] [PubMed] [Google Scholar]
- 3.Burns DM. Primary prevention, smoking, and smoking cessation: implications for future trends in lung cancer prevention. Cancer. 2000;89:2506–9. doi: 10.1002/1097-0142(20001201)89:11+<2506::aid-cncr33>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- 4.Peto R, Darby S, Deo H, Silcocks P, Whitley E, Doll R. Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. BMJ. 2000;321:323–9. doi: 10.1136/bmj.321.7257.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Godrfredsen NS, Prescott E, Osler M. Effect of smoking reduction on lung cancer risk. JAMA. 2005;294:1505–10. doi: 10.1001/jama.294.12.1505. [DOI] [PubMed] [Google Scholar]
- 6.Lubin JH, Blot WJ. Lung cancer and smoking cessation: patterns of risk. J Natl Cancer Inst. 1993;85:422–3. doi: 10.1093/jnci/85.6.422. [DOI] [PubMed] [Google Scholar]
- 7.Tong L, Spitz MR, Fueger JJ, Amos CA. Lung carcinoma in former smokers. Cancer. 1996;78:1004–10. doi: 10.1002/(SICI)1097-0142(19960901)78:5<1004::AID-CNCR10>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 8.Cohen V, Khuri FR. Chemoprevention of lung cancer: concepts and strategies. Expert Rev Anticancer Ther. 2005;5:549–65. doi: 10.1586/14737140.5.3.549. [DOI] [PubMed] [Google Scholar]
- 9.Bach PB, Kattan MW, Thornquist MD, Kris MG, Tate RC, Barnett MJ, et al. Variations in lung cancer risk among smokers. J Natl Cancer Inst. 2003;95:470–9. doi: 10.1093/jnci/95.6.470. [DOI] [PubMed] [Google Scholar]
- 10.Kim ES, Hong WK, Khuri FR. Chemoprevention of aerodigestive tract cancers. Annu Rev Med. 2002;53:223–43. doi: 10.1146/annurev.med.53.082901.104015. [DOI] [PubMed] [Google Scholar]
- 11.Lippman SM, Lee JJ, Sabichi AL. Cancer chemoprevention: progress and promise. J Natl Cancer Inst. 1998;90:1514–28. doi: 10.1093/jnci/90.20.1514. [DOI] [PubMed] [Google Scholar]
- 12.Khuri FR, Cohen V. Molecularly targeted approaches to the chemoprevention of lung cancer. Clin Cancer Res. 2004;10:4249s–53s. doi: 10.1158/1078-0432.CCR-040019. [DOI] [PubMed] [Google Scholar]
- 13.Kelloff GJ, Lippman SM, Dannenberg AJ, Sigman CC, Pearce HL, Reid BJ, et al. Progress in chemoprevention drug development: the promise of molecular biomarkers for prevention of intraepithelial neoplasia and cancer—a plan to move forward. Clin Cancer Res. 2006;12:3661–97. doi: 10.1158/1078-0432.CCR-06-1104. [DOI] [PubMed] [Google Scholar]
- 14.Lee JS, Lippman SM, Benner SE, Lee JJ, Ro JR, Lukeman JM, et al. Randomized placebo-controlled trial of isotretoinin in chemoprevention of bronchial squamous metaplasia. J Clin Oncol. 1994;12:937–45. doi: 10.1200/JCO.1994.12.5.937. [DOI] [PubMed] [Google Scholar]
- 15.Shaipanich T, McWilliams A, Lam S. Early detection and chemoprevention of lung cancer. Respirology. 2006;11:366–72. doi: 10.1111/j.1440-1843.2006.00860.x. [DOI] [PubMed] [Google Scholar]
- 16.Kennedy TC, Franklin WA, Prindiville SA, Cook R, Dempsy EC, Keith RL, et al. High prevalence of occult endobronchial malignancy in high risk patients with moderate sputum atypia. Lung Cancer. 2005;49:187–91. doi: 10.1016/j.lungcan.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 17.Xu XC, Sozzi G, Lee JS, Lee JJ, Pastorino U, Pilotti S, et al. Suppression of retinoic acid receptor beta in non-small-cell lung cancer in vivo: implications for lung cancer development. J Natl Cancer Inst. 1997;89:624–9. doi: 10.1093/jnci/89.9.624. [DOI] [PubMed] [Google Scholar]
- 18.Lotan R, Dawson MI, Zou CC, Jong L, Lotan D, Zou CP. Enhanced effi cacy of combinations of retinoic acid- and retinoid X receptor-selective retinoids and alpha-interferon in inhibition of cervical carcinoma cell proliferation. Cancer Res. 1995;55:232–6. [PubMed] [Google Scholar]
- 19.Hong WK, Lippman SM, Itri LM, Karp DD, Lee JS, Byers RM, et al. Prevention of second primary tumors with isotretinoin in squamous-cell carcinoma of the head and neck. N Engl J Med. 1990;323:795–801. doi: 10.1056/NEJM199009203231205. [DOI] [PubMed] [Google Scholar]
- 20.Hong WK, Endicott J, Itri LM, Doos W, Batsakis JG, Bell R, et al. 13-cis-Retinoic acid in the treatment of oral leukoplakia. N Engl J Med. 1986;315:1501–5. doi: 10.1056/NEJM198612113152401. [DOI] [PubMed] [Google Scholar]
- 21.The Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N Engl J Med. 1994;330:1029–35. doi: 10.1056/NEJM199404143301501. [DOI] [PubMed] [Google Scholar]
- 22.Omenn GS, Goodman GE, Thornquist MD, Balmes J, Cullen MR, Glass A, et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N Engl J Med. 1996;334:1150–5. doi: 10.1056/NEJM199605023341802. [DOI] [PubMed] [Google Scholar]
- 23.van Zandwijk N, Dalesio O, Pastorino U, de Vries N, van Tinteren H. EUROSCAN, a randomized trial of vitamin and N-acetylcysteine in patients with head and neck cancer or lung cancer. For the European Organization for Research and Treatment of Cancer. J Natl Cancer Inst. 2000;92:977–86. doi: 10.1093/jnci/92.12.977. [DOI] [PubMed] [Google Scholar]
- 24.Lippman SM, Lee JJ, Karp DD, Vokes EE, Benner SE, Goodman GE, et al. Randomized phase III intergroup trial of isotretinoin to prevent second primary tumors in stage I non-small-cell lung cancer. J Natl Cancer Inst. 2001;93:605–18. doi: 10.1093/jnci/93.8.605. [DOI] [PubMed] [Google Scholar]
- 25.Kurie JM. The biological basis for the use of retinoids in cancer prevention and treatment. Curr Opin Oncol. 1999;11:497–502. doi: 10.1097/00001622-199911000-00011. [DOI] [PubMed] [Google Scholar]
- 26.Kurie JM, Lotan R, Lee JJ, Lee JS, Morice RC, Liu DD, et al. Treatment of former smokers with 9-cis-retinoic acid reverses loss of retinoic acid receptor-{beta} expression in the bronchial epithelium: results from a randomized placebo-controlled trial. J Natl Cancer Inst. 2003;95:206–14. doi: 10.1093/jnci/95.3.206. [DOI] [PubMed] [Google Scholar]
- 27.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–50. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 28.Lee JJ, Liu D, Lee JS, Kurie JM, Khuri FR, Ibarguen H, et al. Long-term impact of smoking on lung epithelial proliferation in current and former smokers. J Natl Cancer Inst. 2001;93:1081–8. doi: 10.1093/jnci/93.14.1081. [DOI] [PubMed] [Google Scholar]
- 29.Szabo E. Lung epithelial proliferation: a biomarker for chemoprevention trials? J Natl Cancer Inst. 2001;93:1042–3. doi: 10.1093/jnci/93.14.1042. [DOI] [PubMed] [Google Scholar]
- 30.Kurie JM, Lee JS, Griffin T, Lippman SM, Drum P, Thomas MP, et al. Phase I trial of 9-cis-retinoic acid in adults with solid tumors. Clin Cancer Res. 1996;2:287–93. [PubMed] [Google Scholar]
- 31.Miller VA, Rigas JR, Benedetti FM, Verret AL, Tong WP, Kris MG, et al. Initial clinical trial of the retinoid receptor pan agonist 9-cis retinoic acid. Clin Cancer Res. 1996;2:471–5. [PubMed] [Google Scholar]
- 32.Agresti A. Categorical data analysis. New York (NY): John Wiley and Sons; 1990. pp. 348–52. [Google Scholar]
- 33.Diggle PJ, Liang KY, Zeger SL, editors. Analysis of longitudinal data. New York (NY): Oxford University Press; 1994. pp. 143–59. [Google Scholar]
- 34.Khuri FR, Lee JS, Lippman SM, Lee JJ, Kalapurakal S, Yu R, et al. Modulation of proliferating cell nuclear antigen in the bronchial epithelium of smokers. Cancer Epidemiol Biomarkers Prev. 2001;10:311–8. [PubMed] [Google Scholar]
- 35.Meert AP, Feoli F, Martin B, Verdebout JM, Mascaux C, Verhest A, et al. Ki67 expression in bronchial preneoplastic lesions and carcinoma in situ defi ned according to the new 1999 WHO/IASLC criteria: a preliminary study. Histopathology. 2004;44:47–53. doi: 10.1111/j.1365-2559.2004.01748.x. [DOI] [PubMed] [Google Scholar]
- 36.Tan DF, Huberman J, Hyland A, Loewen G, Brooks J, Beck A, et al. MCM2—a promising marker for premalignant lesions of the lung: a cohort study. BMC Cancer. 2001;1:6. doi: 10.1186/1471-2407-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shin DM, Voravud N, Ro JY, Lee JS, Hong WK, Hittelman WN. Sequential increase in proliferating cell nuclear antigen in head and neck tumorigenesis: a potential biomarker. J Natl Cancer Inst. 1993;85:971–8. doi: 10.1093/jnci/85.12.971. [DOI] [PubMed] [Google Scholar]
- 38.Merrick DT, Kittelson J, Winterhalder R, Kotantoulas G, Ingeberg S, Keith RL, et al. Analysis of c-ErbB1/epidermal growth factor receptor and c-ErbB2/Her-2 expression in bronchial dysplasia: evaluation of potential targets for chemoprevention of lung cancer. Clin Cancer Res. 2006;12:2281–8. doi: 10.1158/1078-0432.CCR-05-2291. [DOI] [PubMed] [Google Scholar]
- 39.Dosaka-Akita H, Hommura F, Mishina T, Ogura S, Shimizu M, Katoh H, et al. A risk-stratifi cation model of non-small cell lung cancers using cyclin E, Ki-67, and ras p21: different roles of G1 cyclins in cell proliferation and prognosis. Cancer Res. 2001;61:2500–4. [PubMed] [Google Scholar]
- 40.Ramnath N, Hernandez FJ, Tan DF, Huberman JA, Natarajan N, Beck AF, et al. MCM2 is an independent predictor of survival in patients with non-small-cell lung cancer. J Clin Oncol. 2001;19:4259–66. doi: 10.1200/JCO.2001.19.22.4259. [DOI] [PubMed] [Google Scholar]
- 41.Hashimoto K, Araki K, Osaki M, Nakamura H, Tomita K, Shimizu E, et al. MCM2 and Ki-67 expression in human lung adenocarcinoma: prognostic implications. Pathobiology. 2004;71:193–200. doi: 10.1159/000078673. [DOI] [PubMed] [Google Scholar]
- 42.Yang J, Ramnath N, Moysich KB, Asch HL, Swede H, Alrawi SJ, et al. Prognostic signifi cance of MCM2, Ki-67, and gelsolin in non-small cell lung cancer. BMC Cancer. 2006;6:203. doi: 10.1186/1471-2407-6-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoshino H, Shibuya K, Chiyo M, Iyoda A, Yoshida S, Sekine Y, et al. Biological features of bronchial squamous dysplasia followed up by auto-fluorescence bronchoscopy. Lung Cancer. 2004;46:187–96. doi: 10.1016/j.lungcan.2004.04.028. [DOI] [PubMed] [Google Scholar]
- 44.Xu XC. Tumor-suppressive activity of retinoic acid receptor-beta in cancer. Cancer Lett. 2007;253:14–24. doi: 10.1016/j.canlet.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berard J, Gaboury L, Landers M, De Repentigny Y, Houle B, Kothary R, et al. Hyperplasia and tumours in lung, breast and other tissues in mice carrying a RAR beta 4-like transgene. EMBO J. 1994;13:5570–80. doi: 10.1002/j.1460-2075.1994.tb06894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu XC, Lee JJ, Wu TT, Hoque A, Ajani JA, Lippman SM. Increased retinoic acid receptor-beta4 correlates in vivo with reduced retinoic acid receptor-beta2 in esophageal squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 2005;14:826–9. doi: 10.1158/1055-9965.EPI-04-0500. [DOI] [PubMed] [Google Scholar]
- 47.Mao JT, Fishbein MC, Adams B, Roth MD, Goodglick L, Hong L, et al. Celecoxib decreases Ki-67 proliferative index in active smokers. Clin Cancer Res. 2006;12:314–20. doi: 10.1158/1078-0432.CCR-05-1440. [DOI] [PubMed] [Google Scholar]
- 48.Hittelman WN. Clones and subclones in the lung cancer field. J Natl Cancer Inst. 1999;91:1796–9. doi: 10.1093/jnci/91.21.1796. [DOI] [PubMed] [Google Scholar]