

52.1 Introduction
The first mutations in Drosophila known to cause retinal degeneration were identified in the 1960s by the pioneering studies of Bill Pak and co-workers (Pak 1995). At that time, the clinical significance of the findings was not fully appreciated. The revolutionary finding that put flies into the spotlight was the one showing that genes controlling development in flies could also do so in humans. In the 1980s, homeodomain-containing transcription factors were found to be essential during development in Drosophila. Almost identical homeodomain-containing genes were found in the genomes of a wide range of organisms, including humans and mice (Lawrence 1992). This knowledge led to the conclusion that organisms as different as flies and humans contain nearly identical genes with similar functions.
At the turn of the twentieth century in the hands of Thomas Hunt Morgan, the founding father of Drosophila research, Drosophila emerged as a powerful genetic workhorse (Rubin and Lewis 2000). In 1910, Morgan identified the first mutation in Drosophila, which was a spontaneous mutation that caused a normally red-eyed fly to be white eyed (Fig. 52.1a). This first allele transformed our understanding of genetics and heredity. The white gene encodes a membrane-associated, adenosine triphosphate (ATP)-binding cassette, (ABC)-type multidrug transporter required for the transport of pigment precursors involved in eye pigment biosynthesis. In humans, mutations in the ABCA4 gene (also ABCR) account for approximately 3% of autosomal recessive retinitis pigmentosa (RP) and are linked to both recessive cone-rod dystrophy and recessive Stargardt macular dystrophy. The ABCA4 transporter serves as a flippase in the retinoid cycle. When the ABC4 gene is mutated, toxic detergent-like by-products accumulate in the retinal pigment epithelium (RPE) leading to severe pathology (Bok 2007). Therefore, the white gene, discovered at the turn of the century, was subsequently found to encode an ABC-type transporter required for eye pigment biosynthesis in Drosophila, and is related to another ABC-type transporter in the human eye that is involved in the retinoid cycle and several types of retinal diseases.
Fig. 52.1.

(a) Wild-type red-eyed fly, Canton S compared to a white-eyed mutant fly, w1118. (b) Cross section through the compound eye showing the R1-7 photoreceptor cells and their photosensitive rhabdomeres (R). The R8 photoreceptor cell is located below the plane of the section. (c) The adult Drosophila visual system showing the two compound eyes and the three simple eyes (ocelli) located on the top of the head (arrows). (d) A higher magnification of a rhabdomere showing the microvilli. The rhabdomeres are made up of about 60,000 microvilli and are 50 nm in diameter and 1-2 mm in length. (e) A newly eclosed ninaEI17 mutant fly, showing the reduced size of the rhabdomeres. ninaEI17 is a null allele, so the flies completely lack Rh1 rhodopsin expressed in the R1-6 photoreceptor cells. (f) Six-day-old ninaEI17 fly, showing that the rhabdomeres of the R1-6 photoreceptor cells are almost completely gone, but the R7 cell rhabdomere remains
52.2 The Compound Eye and Phototransduction
The Drosophila compound eye is composed of approximately 800 individual eye units called ommatidia (Figs. 52.1 c and 52.2). The major rhodopsin in the eye, Rh1, is encoded by the ninaE gene, and displays 22% amino acid identity with human rhodopsin and 37% identity with human melanopsin. Each Drosophila photoreceptor cell contains a photoreceptive rhabdomere, which is comprised of approximately 60,000 tightly packed microvilli, containing the rhodopsin photopigments and other components of the phototransduction cascade (Fig. 52.1b, d). The rhabdomeres are functionally equivalent to the vertebrate photoreceptor outer segments (Fig. 52.3).
Fig. 52.2.
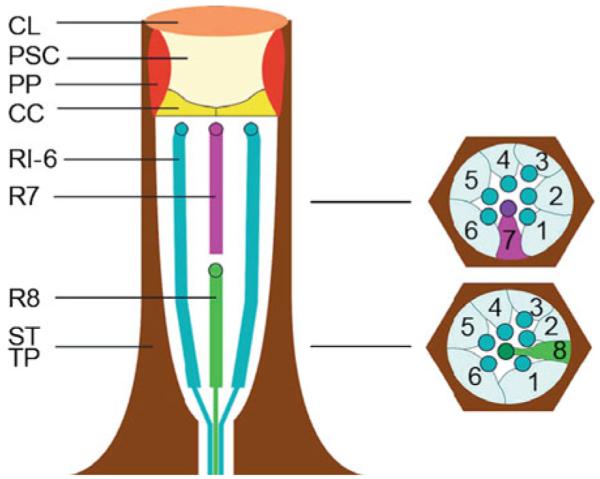
Schematic of an ommatidium. CL corneal lens; PSC pseudocone; PP primary pigment cells; CC cone cells; R1-6 R7, and R8 photoreceptor cells; SP and TP secondary and tertiary pigment cells (Tomlinson and Ready 1987)
Fig. 52.3.

The Drosophila photoreceptor cell compared with human rod and cone photoreceptor cells. In Drosophila, the pseudocone cone (also called the crystalline cone) and the corneal lens are the lens elements, and they are secreted by the underlying cone cells. The Drosophila lens is comprised of droscrystallin, which is similar to insect cuticular proteins. R rhabdomere; OS outer segments; I inner segments; N nucleus (Chang and Ready 2000)
Phototransduction in Drosophila utilizes a signaling cascade in which light stimulation of Rh1 leads to the activation of the G protein (Gq) and the stimulation of phospholipase C beta (PLC-beta), leading to the opening of the cation-selective transient receptor potential (TRP) and TRP-like (TRPL) channels. The photoreceptors depolarize as intracellular calcium dramatically rises. The Drosophila phototransduction cascade shares some similarities with the phototransduction cascade in mammalian rod and cone photoreceptor cells. Both cascades are initiated by light-activation of rhodopsin that in turn leads to the stimulation of G proteins. Phototransduction in Drosophila as well as in humans is terminated when arrestin binds to light-stimulated rhodopsin and prevents the binding of rhodopsin to the G-protein. However, notable differences are that rod and cone channels are gated by cyclic nucleotides and they close in response to light, leading to a hyperpolarizing response (Minke and Parnas 2006; Hardie and Postma 2008) .
Although certain features of phototransduction in Drosophila differ from rod and cone phototransduction, Drosophila phototransduction shares many common features with signaling in intrinsically photosensitive retinal ganglion cells (ipRGCs) (Panda et al. 2005). These cells function in circadian rhythm entrainment and pupil constriction. The light response in ipRGCs is initiated with absorption of light by melanopsin, which is more similar to Drosophila Rh1 than to the photopigments in rods and cones (Provencio et al. 1998). Light-stimulated melanopsin is thought to activate a phosphoinositide-cascade leading to the opening of DAG-sensitive TRPC6/7 channels (Panda et al. 2005; Sekaran et al., 2007; Graham et al. 2008). Therefore, Drosophila photoreceptor cells and ipRGCs share similar phototransduction cascades. Further, TRPM1 channels have been identified in ON-bipolar cells (Koike et al. 2010) and mutations in TRPM1 are implicated in autosomal-recessive congenital stationary night blindness (Audo et al. 2009). With the discovery of TRPC6/7 in ipRGCs and TRPM1 channels in ON-bipolar cells, findings in the fly may be relevant for understanding the mechanisms in the ipRGC and the ON-bipolar cells in the vertebrate retina.
52.3 Genetic Screens Identify Retinal Degeneration Loci
Several forward genetic screens in Drosophila led to an explosion in the identification of many genes involved in retinal degeneration and understanding their counterparts in human disease. The electroretinogram (ERG) genetic screening approach was pioneered by Bill Pak and co-workers in the 1960s and led to the isolation of over 200 ERG-defective mutants (Pak 1995). In the 1980s, the development of gene-cloning techniques for Drosophila made it possible to isolate the corresponding genes. For example, the major rhodopsin in Drosophila, Rh1, was cloned and determined to be encoded by the neither inactivation nor after potential E (ninaE) locus. The ninaE mutants isolated in the original ERG screen were found to harbor mutations in the structural gene for the major rhodopsin in Drosophila, Rh1. In addition, the first evidence that mutations in a rhodopsin gene led to retinal degeneration came from these elegant studies in the 1980s in Drosophila (Stephenson et al. 1983; O’Tousa et al. 1985, 1989; Zuker et al. 1985). The use of the ERG in Drosophila continues to be an effective strategy to identify mutants in phototransduction and also in loci in retinal degeneration.
The DPP is a sensitive phenotype in the eye that can be easily assessed in live flies. It is based on the precise packing of the photoreceptor cells. Any mutation leading to, even subtle, structural alterations in photoreceptor cells will cause attenuation in the DPP. For example, a reduction in rhodopsin levels in the R1-6 photoreceptor cells in ninaE mutants, leads to structural alterations in the photoreceptor cells (Fig. 52.1e, f) and attenuation in the DPP. A variety of mutants were isolated by this method, including dominant alleles of ninaE (rhodopsin), and alleles of two chaperones, ninaA (cyclophilin) and calnexin (Ondek et al. 1992; Colley et al. 1995; Rosenbaum et al. 2006). Both the ERG and the DPP screens accelerated the pace of identifying mutations that cause retinal degeneration in Drosophila.
52.4 Retinal Degenerations in Flies and Humans
Since the initial findings, that mutations in Drosophila rhodopsin lead to retinal degeneration, over 100 mutations in human rhodopsin have been found to cause autosomal dominant RP (adRP). The first mutation identified in adRP patients, published by Dryja et al. ( 1990), was a mutation that caused a proline residue located near the N-terminus of rhodopsin to be replaced by a histidine residue (Pro23His). A great majority of these mutants, including Pro23His, produce misfolded rhodopsin that is improperly transported through the secretory pathway (Roof et al. 1994). However, the mechanism by which the mutant rhodopsins cause dominant retinal degeneration was determined in Drosophila. It was found that rhodopsin mutations act dominantly to cause retinal degeneration by the mutant protein interfering with the maturation of normal rhodopsin (Colley et al. 1995; Kurada and O’Tousa 1995). These studies in Drosophila provided a mechanistic explanation for the cause of certain forms of adRP
It is now widely appreciated that retinal defects and retinal degeneration can be triggered by mutations in almost every component of the photoreceptor cells. One class of retinal degenerations involves defects in rhodopsin maturation. In Drosophila, as in humans, rhodopsin is synthesized and glycosylated in the ER, binds its vitamin-A-derived chromophore at a lysine residue in the seventh transmembrane domain, is transported through the various compartments of the Golgi, and is delivered to its final destination for phototransduction. The mechanisms that regulate rhodopsin maturation, such as its folding, glycosylation, chaperone interaction, chromophore attachment, and transport are key to photoreceptor survival in flies and humans.
In flies, the transport of Rh1 from the ER to the rhabdomere requires the cyclophilin, NinaA. Cyclophilins are known to display peptidyl-prolyl cis-trans isomerase and are thought to play a role in protein folding during biosynthesis. Consistent with a role in protein folding, NinaA resides in the ER. In addition, NinaA is detected in secretory transport vesicles together with Rh1, and forms a specific and stable complex with Rh1, consistent with a role as a chaperone in the secretory pathway (Colley et al. 1991; Baker et al. 1994). Similarly, in mammals a cyclophilin-like protein (RanBP2/Nup358) was found to act as a chaperone for red/cone opsin biogenesis (Ferreira et al. 1996). Another chaperone required for Rh1 biosynthesis in Drosophila is calnexin (Rosenbaum et al. 2006), Mutations in ninaA, ninaE, and calnexin all lead to severe retinal pathology in flies. In mammalian photoreceptors, calnexin is also expressed in the ER. Although calnexin is not required for the expression of rod rhodopsin, cone M-opsin, or melanopsin in the mouse, we have shown that it is required for proper retinal morphology (Kraus et al. 2010). Mechanisms that regulate rhodopsin maturation, such as its folding, chaperone interaction, and transport are essential for photoreceptor health in flies and humans (Colley 2010) .
52.5 Summary
Many genes are functionally equivalent between flies and humans. In addition, the same, or similar, mutations cause disease in both species. In fact, nearly three-fourths of all human disease genes have related sequences in Drosophila. The fly has a relatively small genome, made up of about 13,600 genes in four pairs of chromosomes. However, despite the dramatic differences in size and apparent complexity between humans and flies – we have less than twice as many genes as a fly – our genome is estimated to be made up of only 20,000-25,000 genes contained in 23 pairs of chromosomes. Therefore, despite the fly’s perceived simplicity, or our perceived complexity, our genetic makeup may not be all that different. Its versatility for genetic manipulation and convenience for unraveling fundamental biological processes continue to guarantee the fly a place in the spotlight for unraveling the basis of and therapeutic treatments for human eye diseases.
Acknowledgments
Our research, on retinal degeneration in Drosophila, is supported by funding from the National Eye Institute (R01 EY08768), the Retina Research Foundation, and the Retina Research Foundation/Walter H. Helmerich Research Chair. I gratefully acknowledge C. Vang, E. Rosenbaum, and B. Larson for assistance with preparing the manuscript and figures.
References
- Audo I, Kohl S, Leroy BP, et al. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet. 2009;85:720–729. doi: 10.1016/j.ajhg.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker EK, Colley NJ, Zuker CS. The cyclophilin homolog NinaA functions as a chaperone, forming a stable complex in vivo with its protein target rhodopsin. EMBO Journal. 1994;13:4886–4895. doi: 10.1002/j.1460-2075.1994.tb06816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bok D. Contributions of genetics to our understanding of inherited monogenic retinal diseases and age-related macular degeneration. Arch Ophthalmol. 2007;125:160–164. doi: 10.1001/archopht.125.2.160. [DOI] [PubMed] [Google Scholar]
- Chang HY, Ready DF. Rescue of photoreceptor degeneration in rhodopsin-null Drosophila mutants by activated Rac1. Science. 2000;290:1978–1980. doi: 10.1126/science.290.5498.1978. [DOI] [PubMed] [Google Scholar]
- Colley NJ. Retinal Degeneration through the Eye of the Fly. Encyclopedia of the Eye. 2010;4:54–61. [Google Scholar]
- Colley NJ, Baker EK, Stamnes MA, et al. The cyclophilin homolog ninaA is required in the secretory pathway. Cell. 1991;67:255–263. doi: 10.1016/0092-8674(91)90177-z. [DOI] [PubMed] [Google Scholar]
- Colley NJ, Cassill JA, Baker EK, et al. Defective intracellular transport is the molecular basis of rhodopsin-dependent dominant retinal degeneration. Proc Natl Acad Sci USA. 1995;92:3070–3074. doi: 10.1073/pnas.92.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryja TP, McGee TL, Reichel E, et al. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343:364–366. doi: 10.1038/343364a0. [DOI] [PubMed] [Google Scholar]
- Ferreira PA, Nakayama TA, Pak WL, et al. Cyclophilin-related protein RanBP2 acts as chap-erone for red/green opsin. Nature. 1996;383:637–640. doi: 10.1038/383637a0. [DOI] [PubMed] [Google Scholar]
- Graham DM, Wong KY, Shapiro P, et al. Melanopsin ganglion cells use a membrane-associated rhabdomeric phototransduction cascade. J Neurophysiol. 2008;99:2522–2532. doi: 10.1152/jn.01066.2007. [DOI] [PubMed] [Google Scholar]
- Hardie RC, Postma M. Phototransduction in Microvillar Photoreceptors of Drosophila and Other Invertebrates. Academic Press; San Diego, CA: 2008. [Google Scholar]
- Koike C, Numata T, Ueda H, et al. TRPM1: A vertebrate TRP channel responsible for retinal ON bipolar function. Cell Calcium. 2010;48:95–101. doi: 10.1016/j.ceca.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Kraus A, Groenendyk J, Bedard K, et al. Calnexin deficiency leads to dysmyelination. J Biol Chem. 2010;285:18928–18938. doi: 10.1074/jbc.M110.107201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurada P, O’Tousa JE. Retinal degeneration caused by dominant rhodopsin mutations in Drosophila. Neuron. 1995;14:571–579. doi: 10.1016/0896-6273(95)90313-5. [DOI] [PubMed] [Google Scholar]
- Lawrence PA. The Making of a Fly The Genetics of Animal Design. Blackwell Scientific Publications; Cambridge: 1992. [Google Scholar]
- Minke B, Parnas M. Insights on TRP channels from in vivo studies in Drosophila. Annu Rev Physiol. 2006;68:649–684. doi: 10.1146/annurev.physiol.68.040204.100939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Tousa JE, Leonard DS, Pak WL. Morphological defects in ora photoreceptors caused by mutation in R1-6 opsin gene of Drosophila. J Neurogenetics. 1989;6:41–52. doi: 10.3109/01677068909107099. [DOI] [PubMed] [Google Scholar]
- O’Tousa JE, Baehr W, Martin RL, et al. The Drosophila ninaE gene encodes an opsin. Cell. 1985;40:839–850. doi: 10.1016/0092-8674(85)90343-5. [DOI] [PubMed] [Google Scholar]
- Ondek B, Hardy RW, Baker EK, et al. Genetic dissection of cyclophilin function: saturation mutagenesis of the Drosophila cyclophilin homolog ninaA. Journal of Biological Chemistry. 1992;267:16460–16466. [PubMed] [Google Scholar]
- Pak WL. Drosophila in vision research. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1995;36:2340–2357. [PubMed] [Google Scholar]
- Panda S, Nayak SK, Campo B, et al. Illumination of the Melanopsin Signaling Pathway. Science. 2005;307:600–604. doi: 10.1126/science.1105121. [DOI] [PubMed] [Google Scholar]
- Provencio I, Jiang G, De Grip WJ, et al. Melanopsin: An opsin in melanophores, brain, and eye. Proc Natl Acad Sci USA. 1998;95:340–345. doi: 10.1073/pnas.95.1.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roof DJ, Adamian M, Hayes A. Rhodopsin accumulation at abnormal sites in retinas of mice with a human P23H rhodopsin transgene. Investigative Ophthalmology & Visual Science. 1994;35:4049–4062. [PubMed] [Google Scholar]
- Rosenbaum EE, Hardie RC, Colley NJ. Calnexin is essential for rhodopsin maturation, Ca2+ regulation, and photoreceptor cell survival. Neuron. 2006;49:229–241. doi: 10.1016/j.neuron.2005.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin GM, Lewis EB. A Brief History of Drosophila’s Contributions to Genome Research. Science. 2000;287:2216–2218. doi: 10.1126/science.287.5461.2216. [DOI] [PubMed] [Google Scholar]
- Sekaran S, Lall GS, Ralphs KL, Wolstenholme AJ, Lucas RJ, Foster RG, Hankins MW. 2-Aminoethoxydiphenylborane is an acute inhibitor of directly photosensitive retinal ganglion cell activity in vitro and in vivo. J Neurosci. 2007;27:3981–3986. doi: 10.1523/JNEUROSCI.4716-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson RS, O’Tousa J, Scavarda NJ, et al. Drosophila mutants with reduced rhodopsin content. Symposia of the Society for Experimental Biology. 1983;36:477–501. [PubMed] [Google Scholar]
- Tomlinson A, Ready DF. Cell fate in the Drosophila ommatidium. Developmental Biology. 1987;123:264–275. doi: 10.1016/0012-1606(87)90448-9. [DOI] [PubMed] [Google Scholar]
- Zuker CS, Cowman AF, Rubin GM. Isolation and structure of a rhodopsin gene from D. melanogaster. Cell. 1985;40:851–858. doi: 10.1016/0092-8674(85)90344-7. [DOI] [PubMed] [Google Scholar]