

Abstract
The most important function of the intestinal mucosa is to form a barrier that separates luminal contents from the intestine. Defects in the intestinal epithelial barrier have been observed in several intestinal disorders such as inflammatory bowel disease (IBD). Recent studies have identified a number of factors that contribute to development of IBD including environmental triggers, genetic factors, immunoregulatory defects and microbial exposure. The current review focuses on the influence of the farnesoid X receptor (FXR) on the inhibition of intestinal inflammation in patients with IBD. The development and investigation of FXR agonists provide strong support for the regulatory role of FXR in mucosal innate immunity. Activation of FXR in the intestinal tract decreases the production of proinflammatory cytokines such as interleukin (IL) 1-beta, IL-2, IL-6, tumour necrosis factor-alpha and interferon-gamma, thus contributing to a reduction in inflammation and epithelial permeability. In addition, intestinal FXR activation induces the transcription of multiple genes involved in enteroprotection and the prevention of bacterial translocation in the intestinal tract. These data suggest that FXR agonists are potential candidates for exploration as a novel therapeutic strategy for IBD in humans.
Keywords: Bile acids, Farnesoid X receptor, Inflammation, Inflammatory bowel desease, Intestinal permeability
Abstract
La principale fonction de la muqueuse intestinale est de former une barrière qui sépare de l’intestin le contenu de la lumière. On a observé des anomalies de la barrière épithéliale de l’intestin dans plusieurs troubles intestinaux, telles que les maladies inflammatoires de l’intestin (MII). Des études récentes ont permis de repérer un certain nombre de facteurs qui contribuent à l’apparition d’une MII, y compris des déclencheurs environnementaux, des facteurs génétiques, des anomalies immunorégulatrices et une exposition microbienne. La présente analyse aborde l’influence du récepteur farnésoïde X (RFX) sur l’inflammation intestinale chez les patients atteints d’une MII. L’apparition et l’exploration des agonistes du RFX étayent fortement le rôle régulateur du RFX dans l’immunité innée des muqueuses. L’activation du RFX dans le tube digestif réduit la production de cytokines proinflammatoires telles que l’interleukine (IL) 1-bêta, l’IL-2, l’IL-6, le facteur de nécrose tumorale alpha et l’interféron gamma, contribuant ainsi à une réduction de l’inflammation et de la perméabilité épithéliale. En outre, l’activation du RFX intestinal induit la transcription de multiples gènes qui participent à l’entéroprotection et à la prévention de la translocation bactérienne dans le tube digestif. Ces données laissent croire que les agonistes du RFX sont des candidats potentiels à l’exploration en vue de devenir une nouvelle stratégie thérapeutique des MII chez des humains.
Chronic inflammatory disorders of the gastrointestinal tract, collectively termed inflammatory bowel disease (IBD), include Crohn disease (CD) and ulcerative colitis (UC). It is estimated that 1.5 million Americans suffer from UC and CD. The etiology of UC and CD are unknown, although both are believed to arise from a disordered immune response to gut contents in genetically predisposed individuals (1). The onset of IBD peaks between 15 and 25 years of age (2). Multiple studies have evaluated the epidemiology of IBD in adults from various geographical regions. In the United States, estimates of CD incidence varies between six and eight per 100,000, with a prevalence of 100 to 200 per 100,000 (1).
The intestinal epithelium represents the largest and most important barrier between the host and the luminal contents of the intestine. It acts as a barrier to prevent the passage of harmful intraluminal entities including foreign antigens, microorganisms and their toxins (3). At the same time, the intestinal barrier must be permeable to essential dietary nutrients, electrolytes and water absorbed from the intestinal lumen into the circulation because of their importance to growth and development (4). It has been shown that the function of the intestinal barrier is modulated by the immune system, ingestion of alcohol or nonsteroidal anti-inflammatory drugs, enteric pathogens and their toxins, and proteases (3). Altered intestinal barrier function results in altered epithelial permeability and altered intestinal innate immunity seen in various diseases including IBD. Dysregulation of the immune response to intestinal bacteria in patients with IBD occurs because of a shift in the balance of the secretion of anti-inflammatory mediators toward proinflammatory molecules (5). Recently, the nuclear farnesoid X receptor (FXR) has been implicated in immune modulation and barrier function in the intestine (6). FXR is activated by bile salts and regulates the transcription of genes involved in bile salt synthesis, transport and metabolism in the liver and intestine, as well as genes involved in many different functions by binding FXR response elements (FXREs) in promoters of target genes as a heterodimer with the retinoid X receptor (RXR) (7). Thus, immune cell modulation by the FXR signalling pathway could lead to improvement in intestinal inflammation (8). Because bile acids and FXR play an important role in modulating a range of inflammatory responses, barrier function and the prevention of bacterial translocation in the intestinal tract, the current review focuses on the influence of FXR on the inhibition of intestinal inflammation in patients with IBD.
IBD AND FACTORS INVOLVED IN INTESTINAL INFLAMMATION
IBD comprises primarily two disorders: UC and CD. It is likely that a number of factors contribute to the development of mucosal inflammation. Recent experimental studies have identified a role for several factors such as environmental triggers, genetic factors, immunoregulatory defects and microbial exposure (9,10). The generally accepted theory is that a combination of environmental agents and a dysfunctional mucosal immune system in genetically susceptible individuals leads to the development of either CD or UC (11,12). The hallmark of IBD is chronic, uncontrolled inflammation of the intestinal mucosa, with potentially severe complications and even mortality (13). CD can involve any part of the gastrointestinal tract, while UC is limited to the rectum through to the large intestinal tract (12). The epithelial phenotype in active IBD is very similar in CD and UC. It is characterized by increased secretion of water and chloride, leading to diarrhea, increased permeability via both the transcellular and paracellular routes, and increased apoptosis of epithelial cells (14). Increased permeability of the epithelial lining of the gut results in continuous stimulation of the mucosal immune system. It has been suggested that this may be the primary defect in individuals with IBD. Animal studies have shown a tendency for the development of severe inflammation in areas of the intestine lying beneath the permeability defect (15). Multiple molecular mechanisms for increased intestinal permeability in patients with IBD have been reported including reduction of tight junction (TJ) strands, strand breaks, alterations of TJ protein content and composition, and increased epithelial apoptosis (16). Several cytokines are known to increase permeability in the intestinal epithelial monolayer by modulating TJ protein expression and localization, which in turn facilitates the recruitment of neutrophils to the mucosa from the peripheral blood (17,18). CD is associated with a T helper cell (Th) 1 type immune response, with excessive production of tumour necrosis factor-alpha (TNF-α), interferon-gamma (IFN-γ) and interleukin (IL)-12, whereas UC is the result of a mainly Th2 response with abundant IL-5, IL-10 and IL-13 production (19). In IBD patients, nuclear factor kappa B (NF-κB) was identified as a key factor in the proinflammatory response (20), resulting in strongly enhanced expression of proinflammatory genes and recruitment of excess inflammatory cells to the intestinal wall. Additionally, pathogens and bacterial toxins influence epithelial permeability by modulating TJ proteins (21). Probiotics may functionally modulate the intestinal epithelial barrier of the host by different mechanisms, including prevention of pathogenic bacterial growth, blocking of pathogen binding to or penetration of mucosal surfaces, stimulation of mucosal barrier function and altering immunoregulation (22–24). Although permeability defects could conceivably be due to the marked apoptosis that occurs during the inflammatory process, numerous studies have clearly shown that epithelial cell apoptosis alone does not entirely account for permeability deficits (11).
CURRENT AND EMERGING DRUGS FOR THE TREATMENT OF IBD
The treatment of chronic inflammatory gastrointestinal conditions, such as UC and CD, is difficult due to the ambiguity surrounding their precise etiology. Treatment of IBD includes conservative measures as well as surgical approaches in individuals who do not respond to medical treatment (25,26). The overall treatment goals are to reduce disease severity and prolong periods of disease-free remission by suppression of inflammation (27). Treatment strategies are aimed at maintaining the epithelial barrier, and inhibiting innate proinflammatory cells and effector T cells within the lamina propria, ultimately leading to control of the secondary effects without correcting the primary genetic susceptibility factors that are involved in the development of IBD (10,28). Pharmaceutical treatment of IBD includes six major categories: anti-inflammatory drugs (mesalazine, corticosteroids); immunosuppressives (methotrexate, cyclosporine, azathioprine and 6-mercaptopurine); biological agents (TNF-α blocking strategies, eg, infliximab, adalimumab); antibiotics (ciprofloxacin, metronidazole, ornidazole, clarithromycin); probiotics; and drugs for symptomatic relief (25). Recently, a large number of agents have been studied in patients with IBD. Apart from TNF-α inhibitors, other molecules, such as unfractionated or low molecular-weight heparin, omega-3 polyunsaturated fatty acids, microbes and microbial products, have been studied (25). The agents that are currently used for the treatment of IBD vary in their ability to maintain symptom control, and in their tolerability and toxicity (29). Despite the efficacy of medical therapy, significant side effects (eg, sepsis, opportunistic infections, tuberculosis, lymphoma, diabetes and osteoporosis) and treatment failures can occur, emphasizing the need for novel treatment options in IBD (8).
THE STRUCTURE OF FXR AND ITS MECHANISM OF ACTION
FXR is a member of the family of nuclear receptors involved in many aspects of mammalian physiology including development, reproduction and metabolism (30). One of the unique characteristics of nuclear receptors distinguishing them from other classes of receptors is that they can directly interact with DNA and control transcription (31). FXR is most abundantly expressed in the tissues commonly exposed to bile acids in normal physiology, including the liver, intestine and kidneys, but is also expressed in the adrenal gland, pancreas and reproductive tissues. Along the intestinal tract, higher FXR levels can be found in the ileal epithelium, the main site of intestinal bile acid absorption (32).
There are two FXR genes (FXRα [NR1H4] and FXRβ [NR1H5]) in mammals. FXRβ is a functional receptor in mice, rats, rabbits and dogs, but constitutes a pseudogene in humans and primates. The functional role of FXRβ remains unclear. The single FXRα gene in humans and primates encodes four FXRα isoforms (FXRα1, FXRα2, FXRα3 and FXRα4) as a result of the use of different promoters and alternative splicing of RNA (7). FXRα is expressed mainly in the liver, intestine, kidney and adrenal glands, and at much lower levels in adipose tissue (33).
Similar to nuclear receptors in general, the structure of FXR has been well characterized (Figure 1A) and includes a DNA-binding domain in the N-terminal region and a ligand-binding domain in the C-terminal region. The ligand-independent N-terminal transactivation domain (AF1) and the ligand-induced transactivation domain (AF2) are the regions in the FXR molecule responsible for interactions with regulatory proteins (34).
Figure 1).
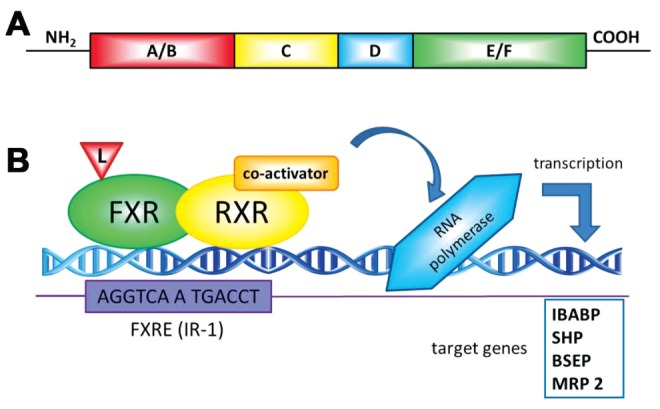
A General structure of nuclear receptors. A typical nuclear receptor contains several domains: the N-terminal region (A/B), which contains the ligand-independent AF1 transactivation domain; the DNA binding domain (C) contains the conserved DNA binding domain; a flexible hinge region (D) that connects the DNA- and ligand-binding domain and the C-terminal ligand-binding domain (E/F) containing the ligand-dependent AF2 activation domain. B Mechanisms of farnesoid X receptor (FXR)-mediated transcriptional regulation. On ligand binding, FXR alters the transcription of target genes by interacting with FXR response elements (FXRE) as a heterodimer with the retinoid X receptor (RXR, 9-cis-retinoic acid receptor). Examples of consensus sequences are shown. After recruiting the coregulators and the RNA polymerase machinery, gene transcription is induced. BSEP Bile salt export pump; IBABP Intestinal bile acid binding protein; IR Inverted repeat; L Ligand; MRP 2 Multidrug resistance protein 2; SHP Small heterodimer partner
Ligand-activated FXR binds to DNA sequences on target genes (Figure 1B) known as FXREs, either as a heterodimer with RXR or as a monomer, and regulates the expression of a wide variety of target genes involved in bile acid, lipid and glucose metabolism (31). FXRE is composed of two inverted repeats (IRs) of the core hexanucleotidic AGGTCA sequence (or closely related sequences) separated by one nucleotide, IR-1, and can be activated by ligands for both receptors (bile acids and/or 9-cis retinoic acid) (33). This FXRE has been detected in many FXR target genes (34). The FXR/RXRα heterodimer also binds to and activates a variety of other FXREs, such as IR-0, IR-8, ER-8 or DR-1, but binds to the consensus IR-1 sequence with the highest affinity (35). It is commonly considered that without ligand binding, a corepressor complex may be associated with the FXR/RXR dimer, which prevents the recruitment of the transcriptional activation machinery to access FXR target genes (34). On ligand binding, FXR undergoes conformational changes to release corepressors such as nuclear corepressor (NCor) and recruit coactivators such as steroid receptor coactivator (SRC)-1, protein arginine methyl transferase (PRMT)-1, coactivator-associated arginine methyltransferase (CARM)-1, peroxisome proliferator-activated receptor-γ coactivator (PGC)-1α and vitamin D receptor-interacting protein (DRIP)-205 (33), consequently affecting transcriptional rates of target genes (36). The mechanism(s) that regulate recruitment of these coactivators by FXR ligands and the relevance of these molecules in the regulation of specific genes by FXR are unknown.
NATURAL AND SYNTHETIC LIGANDS OF FXR
Most nuclear receptors are activated by small lypophilic ligands such as bile acids, fatty acids, lipophilic vitamins and steroidal hormones (37). FXR was originally proposed to be a receptor for an intermediary metabolite known as farnesol (32). However, the supraphysiological concentrations required to activate FXR preclude the use of farnesol as a ligand. The major breakthrough in FXR biology was the discovery that bile acids are endogenous ligands for this nuclear receptor (7). Of the two most important primary bile acids in humans, the more hydrophobic chenodeoxycholic acid (CDCA) is clearly a more potent FXR activator than the hydrophilic cholic acid. Secondary bile acids, such as lithocholic acid and deoxycholic acid, also activate FXR, but to a lesser extent. Ursodeoxycholic acid, a hydrophilic bile acid used therapeutically in cholestatic diseases, was shown to function as a very weak FXR agonist (37). It has been reported that some natural extracts contain FXR modulators. Guggulsterone, the active moeity of guggulipid, may represent an example of a gene-selective modulator for FXR (38). Stigmasterols, components of soy-derived lipids, were able to antagonize the activity of FXR target genes in HepG2 cells (39). Additionally, cafestol, a diterpene isolated from unfiltered coffee brew, has been shown to have agonistic effects on FXR (40).
Exploiting the knowledge of the structure-activity relationship of bile acids for the FXR, semisynthetic and synthetic molecules have been formulated to obtain more potent FXR activators. Because bile acids can activate multiple signalling pathways, the development of specific synthetic FXR agonists, including GW4064 (41), fexaramine (42), AGN34 (43) and a semisynthetic agonist, 6α-ethyl-CDCA (6-ECDCA, INT-747) (44), have provided powerful tools to dissect FXR-specific transcriptional signalling (35). The most widely used FXR ligand is the nonsteroidal isoxazole analogue GW4064, but the uncertain bioavailability and potential cytotoxic effects limit its further use. Instead, 6-ECDCA, a novel compound derived from the natural FXR ligand CDCA, has become an alternative agonist ligand for FXR (7). It is expected that several new specific synthetic agonists of FXR will emerge in the future.
FXR REGULATION OF BILE ACID METABOLISM
It has long been suspected that bile salts have immunosuppressive actions and that inflammation affects bile homeostasis. Although additional mechanisms may exist, some insights into these mechanisms through which bile salts interact with the immune system via the FXR have been revelaed (45).
Bile acids, the end products of hepatic cholesterol catabolism, have a significant role in nascent bile formation, biliary cholesterol solubilization and intestinal absorption of lipids and lipid-soluble molecules (46). Apart from these important roles, they function as signalling molecules that coordinately regulate a network of metabolic pathways including lipid, glucose, drug and energy metabolism (47). Bile salts, together with gastric acid, pancreatic secretions, intestinal motility and local immunity, are known to maintain the physiological balance of the gut microflora and the integrity of the intestinal epithelial barrier (46). A major underlying pathway responsible for these effects is bile acid-mediated activation of FXR. On activation by bile salt ligand binding, FXR regulates the transcription of genes involved in bile salt synthesis, transport and metabolism in the liver and intestine (7) (Figure 2).
Figure 2).
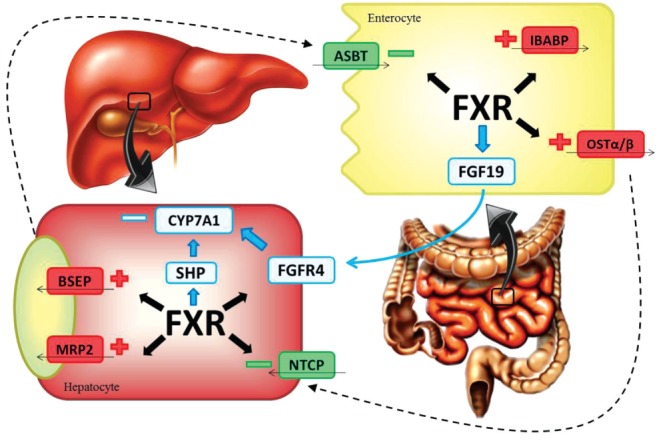
Mechanisms of farnesoid X receptor (FXR) regulation of bile acids homeostasis. FXR negatively regulates bile acid production by repressing CYP7A1. FXR modulates CYP7A1 expression by induction of fibroblast growth factor (FGF)-19 expression. On its secretion, FGF19 activates the hepatic FGF receptor FGFR-4, which in turn downregulates CYP7A1. FXR induces the expression of a small heterodimer partner (SHP), which in turn interacts with two other receptors that decrease the transcription of CYP7A1. FXR activates the expression of the bile acid export transporters multidrug resistance protein 2 (MRP2) and bile salt export pump (BSEP), and simultaneously represses bile acid import by downregulation of the sodium-dependent cotransporting polypeptide (NTCP). At the intestinal level, FXR induces the expression of intestinal bile acid binding protein (IBABP) and organic solute transporter (OST)α–OSTβ, and influences the import of bile acids by interfering with the transcription factor network controlling the apical sodium-dependent bile acid transporter (ASBT)
Bile acids function as homeostatic regulators and signalling molecules to adjust their own intracellular levels (36). One FXR target gene is the small heterodimer partner (SHP), an atypical nuclear receptor that lacks a DNA-binding domain and dimerizes with and inactivates both liver receptor homologue 1 and liver receptor homologue α, resulting in a decrease in CYP7A1 expression and inhibition of bile acid synthesis through a neutral pathway (33). In enterocytes, FXR activation by bile salts induces the production/secretion of fibroblast growth factor (FGF) 15/19 (in mice and humans, respectively), which is involved in liver and intestinal homeostasis. FGF 15/19 activates hepatic FGFR4, inducing the negative feedback regulation of de novo synthesis of bile acids in the liver (48). Various transport proteins in the liver and intestine involved in hepatic uptake, biliary excretion and intestinal reabsorption of bile acids and salts are regulated by nuclear FXR and the liver X receptor (49). FXR negatively regulates bile acid uptake systems. In conditions of elevated hepatocellular bile acid concentrations, bile acids, via FXR activation, suppress the expression of the major bile acid uptake system, Na+-taurocholate cotransporting protein, which localizes to the basolateral membrane of hepatocytes (36). In the liver and intestine, bile acid-activated FXR induces cellular bile acid efflux and detoxification. Thus, in conditions of increased bile acid load in hepatocytes, bile acids enhance their own efflux into bile by activating FXR and consequently increasing ABCB11 (bile salt export pump [BSEP]) and ABCB4 expression (50). Although BSEP is responsible for the efflux of monovalent bile acids from hepatocytes into bile, the multidrug resistance-associated protein 2 (MRP2, ABCC2), which is also upregulated by FXR, contributes to the overall canalicular bile acid efflux by exporting divalent and sulphated or glucuronidated bile acids into the bile (51). At the intestinal level, FXR activation reduces the expression of the apical sodium-dependent bile acid transporter (ASBT), which mediates bile acid transport from the intestinal lumen into the enterocytes, while also increasing the expression of intestinal bile acid binding protein (IBABP), thus eliciting intracellular trafficking of bile salts from the apical to the basolateral membrane. At the same time, FXR directly activates the expression of organic solute transporter α/β (OSTα/β), which mediates bile acid efflux into the portal blood at the basolateral membrane (52).
Consistent with the role of FXR in bile homeostasis, FXR knockout mice were initially reported to experience dysregulation of bile acid and lipid homeostasis, with elevated cholesterol levels in both liver and serum (53). Interestingly, when these mice were followed-up for longer periods, hepatic levels of the proinflammatory cytokines IFN-γ, TNF-α and IL-6 were increased, suggesting an additional immunoregulatory role for FXR (54).
FXR ACTIVATION IN IBD
Several studies have emphasized the role of FXR-bile acid interaction in the pathophysiology of a wide range of diseases of the gastrointestinal tract. At the intestinal level, FXR activity alleviates inflammation and preserves the integrity of the intestinal epithelial barrier in many ways by regulating the extent of the inflammatory response, maintaining the integrity and function of the intestinal barrier, and preventing bacterial translocation into the intestinal tract (50).
The impact of FXR on intestinal inflammation and immunoregulation
FXR is expressed in epithelial cells, which play an essential role in the mucosal immune response, thus exerting strong influence on immuno-regulation (55). FXR ligands exert anti-inflammatory activities through their ability to antagonize other signalling pathways, in part through the interaction with other transcription factors, including activator protein-1, and signal transducers and activators of transcription (33).
Vavassori et al (6) recently provided supportive evidence for the involvement of FXR in IBD due to counter-regulatory effects on cells of innate immunity. They noticed that FXR-deficient mice responded to intestinal inflammation with an uncontrolled immune reaction and inflammation-driven fibrosis in the colon. FXR activation by natural and synthetic ligands represses the expression of a set of toll-like receptor 4-regulated genes, including proinflammatory cytokines, chemokines and their receptors (6). Several of the intestinal macrophage genes inhibited by FXR agonists are established targets for NF-κB genes (TNF-α, IL-1β, IL-6, cyclooxygenase-1, cyclooxygenase-2) and activator protein-1, which are the most important transcriptional regulators of innate and adaptive immunity in cells (56).
In two complementary experimental murine models (intrarectal administration of trinitrobenzensulfonic acid and oral administration of dextrane sodium sulphate), concurrent administration of the potent synthetic FXR ligand (6-ECDCA, INT-747) protected against colitis in wild type mice, but not in FXR-knockout mice (6). Consistent with this, Gadaleta et al (8) also showed that administration of the FXR agonist INT-747 alleviated intestinal inflammation in two models of murine colitis. Furthermore, it has been shown that not only does FXR inhibit inflammation in the gut, but also that FXR activation is inhibited by proinflammatory stimuli in different model systems (45). Decreased FXR activity may lead to altered enterohepatic bile salt circulation and potentially contribute to cholestatic liver disease, which often coexists in patients with IBD (57).
It has been reported that FXR activation results in a decrease of epithelial permeability and proinflammatory cytokine messenger RNA expression in the murine intestinal mucosa (8). Many cytokines that contribute to the pathophysiology of IBD, such as the interleukins IL-0, IL-1, IL-3 and IL-4, as well as TNF-α and IFN-β, influence epithelial and endothelial TJ function, and the actin cytoskeleton both in vivo and in vitro (58), leading to disruption of intestinal epithelial tight TJ and intestinal hyperpermeability (the so-called ‘leaky gut’), which may result in bacterial translocation, systemic inflammatory response and multiple organ dysfunction syndrome (16). Decreasing the level of proinflammatory cytokines, FXR activation has indirect influence on intestinal permeability and transport. It has been demonstrated that proinflammatory cytokines initiate many changes in gene expression by activating transcription factors as well as many transporters and drug metabolizing enzymes that are suppressed during inflammation, such as multidrug resistance (MDR)-1 protein, MRP2, MRP3, BSEP, organic anion-transporting polypeptide-2 and CYP3A (59).
There is increasing evidence that changes in MDR1 (also known as P-glycoprotein or ABCB1) function and/or expression contribute to the pathogenesis of inflammatory disorders of the gastrointestinal tract (60). P-glycoprotein, encoded by the ABCB1/MDR1 gene located on chromosome 7q, is highly expressed on the apical side of the intestinal epithelium (61,62). Its function is to mediate efflux of compounds from the mucosa to the gut lumen and, in that manner, protect the body against xenobiotics from nutrients. MDR1 can also limit the absorption of hydrophobic drugs by transporting them back into the lumen, and protect against bacterial infection (63). Decreased expression of P-glycoprotein was identified in IBD patients compared with controls (64). Consistent with that result, Panwala et al (65) showed that MDR1 null mice developed spontaneous intestinal inflammation. It was suggested that the MDR1 C3435T polymorphism, which is associated with decreased MDR1 expression, may confer a genetic predisposition to UC (66).
There is strong evidence supporting the influence of FXR agonists on inflammation processes at other levels of the gastrointestinal tract. Lian et al (67) noticed that FXR protects human and murine gastric epithelial cells against TNF-α-induced inflammatory cell damage. They have also identified keratin 13, an antiapoptotic protein of desmosomes, as a novel CDCA-regulated FXR-target gene. The most recent published study (68) emphasized the role of FXR in mucosal protection in models of gastrointestinal injury caused by nonsteroidal anti-inflammatory drugs, exhibiting its effect on a gastrointestinal expression of cystathionine-γ-lyase, an enzyme required for generation of hydrogen sulphide. At the hepatic level, FXR is a negative modulator of NF-kB-mediated inflammation, reducing the expression of inflammatory mediators both in vivo and in vitro (69).
The role of FXR in intestinal antibacterial genes
It was previously known that bile acids have antimicrobial activity in the small intestine, thus helping to maintain the integrity of the intestinal barrier. Lorenzo-Zúñiga et al (70) provided strong evidence for this hypothesis, demonstrating that the feeding of bile or conjugated bile acids in conditions of bile acid deficiency in the intestine abolished bacterial overgrowth and reduced bacterial translocation to intestinal lymph nodes. Based on studies involving mice, Inagaki et al (71) presented compelling evidence that the antibacterial effect of conjugated bile acids in the distal small intestine was mediated by a cellular pathway involving the FXR. They showed that mice lacking FXR experienced bacterial overgrowth, increased intestinal permeability and large amounts of bacteria in mesenteric lymph nodes, as well as significant inflammation of the intestinal wall. FXR activation by the synthetic ligand GW4064 alleviated these adverse effects in wild type, but not in FXR knockout mice. They demonstrated that FXR activation promoted the expression of several intestinal genes that are involved in enteroprotection (71). Perhaps the most notable of these is the gene encoding inducible nitric oxide synthase, given the antimicrobial properties of nitric oxide, as well as its role in different aspects of the innate immune response, including mucus secretion, vascular tone and epithelial barrier function (71,72). Another gene identified as GW4064-inducible is the gene encoding angiogenin, a part of the acute phase response to infection, which has potent antibacterial and antimycotic actions (71,73). The proinflammatory cytokine IL-18 is also induced by FXR stimulation. IL-18 stimulates resistance to an array of pathogens, including intracellular and extra-cellular bacteria and mycobacteria, and appears to have a protective role during the early, acute phase of mucosal immune response (71,74). Activity of this nuclear receptor induces messenger RNA expression of carbonic anhydrase 12. This enzyme participates in antibacterial defense by maintaining appropriate intestinal pH and ion balance, which is important for the homeostasis of intestinal luminal contents and epithelial barrier integrity (71).
These results are entirely consistent with the proposal that FXR is critical for controlling intestinal bacterial growth, which has significant implications for maintaining a competent barrier and, thus, contributing to the prevention of intestinal inflammation.
Other mechanisms of FXR enteroprotection
It has been shown that activation of FXR by agonists leads to upregulation of angiotensin type II receptor (AT2R) in vascular smooth muscle cells by binding to an IR2 FXRE in the AT2R promoter (75). Activation of AT2R resulted in an inhibition of AT1R signalling, which in turn decreases the activity of Th1 and Th17 cells. Because both of these T cell subsets have a critical role in IBD pathogenesis, this may be an additional mechanism by which FXR signalling suppresses intestinal inflammation (76,77). Furthermore, some of the FXR functions in the intestine are probably mediated by induction of FGF15 due to fact that FGF15 knockout mice show altered intestinal morphology, suggesting that activation of FGF15 by FXR could also have protective effects in the intestine (48). Additionally, it has been demonstrated that FXR activation interferes with Wnt/β-catenin signalling, an important regulator of intestinal homeostasis. Increased levels of Wnt/β-catenin activation, which is observed in FXR deficiency, may contribute to tumour formation. Given that patients with IBD are at increased risk of developing colorectal cancer, FXR agonists may reduce risk in a dual fashion, both by directly inhibiting Wnt/β-catenin tumorigenesis and by decreasing local inflammation (77).
It is clear that the genetic basis for IBD is focused on genetic factors that regulate the components of the innate and adaptive immune responses, as well as the regulation of intestinal epithelial cell barrier function and, interestingly, the composition of the normal commensal microbiota, within the intestines (78–80). It is estimated that known genetic associations account for only 20% of the genetic variance underlying susceptibility to IBD, leaving ample space for additional genetic factors. Nijmeijer et al (81) recently evaluated whether genetic variation in FXR is associated with IBD. In that study, none of the functional or common tagging single nucleotide polymorphisms proved to be significantly associated with CD or UC.
The impact of bile salt-FXR interactions in intestinal inflammation is an interesting observation with respect to surgical intervention in IBD. In normal conditions, the terminal ileum functions as the conduit by which bile acids from the intestinal lumen are reclaimed into the enterohepatic circulation. In a murine model of ileocaecal resection, the remnant colon is able to adapt to the loss of ileal bile acid uptake by upregulating the molecular machinery necessary to import, transport and export bile acids from the lumen, decreasing the expression of IBABP, ASBT and FGF15. This upregulation is partially FXR-dependent and requires colonic bacteria for initiation. It is possible that ileocecal resection is not only involved in removing local disease, but also contributes to immunomodulation in the remaining colon (82).
PROSPECTS
These new findings suggest that FXR has much broader roles than previously believed. By regulating the expression of genes involved in diverse metabolic pathways, FXR is becoming an attractive drug target for different diseases including IBD. We expect that further investigation of FXR function in these new areas will provide novel insights into the complex mechanism of FXR action. FXR agonists are currently being evaluated in phase I/II trials as treatment for hepatic and metabolic disorders. The number of synthetic FXR agonists is expected to grow in the future. Recent studies have shown that FXR activation affects both immune cells and intestinal epithelium, contributing to intestinal immunomodulation at various levels, thus providing a rationale to extend these clinical trials to patients with IBD. The therapeutic benefits or risks of synthetic FXR ligands require careful consideration in light of differences between mice and humans. With an in-depth understanding of FXR function and regulation at the cell-, gene- and tissue-specific levels, these agonists may emerge as novel drugs to prevent and treat hepatic, gastrointestinal and systemic diseases.
Acknowledgments
This work was supported by the Ministry of Science and Technological Development, Republic of Serbia, project number 41012.
REFERENCES
- 1.Talley NJ, Abreu MT, Achkar JP, et al. An evidence-based systematic review on medical therapies for inflammatory bowel disease. Am J Gastroenterol. 2011;106:S2–25. doi: 10.1038/ajg.2011.58. [DOI] [PubMed] [Google Scholar]
- 2.Habal FM, Kapila V. Inflammatory bowel disease and pregnancy: Evidence, uncertainty and patient decision-making. Can J Gastroenterol. 2009;23:49–53. doi: 10.1155/2009/531638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Groschwitz KR, Hogan SP. Intestinal barrier function: Molecular regulation and disease pathogenesis. J Allergy Clin Immunol. 2009;124:3–20. doi: 10.1016/j.jaci.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blikslager AT, Moeser AJ, Gookin JL, Jones SL, Podle J. Restoration of barrier function in injured intestinal mucosa. Physiol Rev. 2007;87:545–64. doi: 10.1152/physrev.00012.2006. [DOI] [PubMed] [Google Scholar]
- 5.Hooper LV, Bry L, Falk PG, et al. Host-microbial symbiosis in the mammalian intestine: Exploring an internal ecosystem. Bioessays. 1998;20:336–43. doi: 10.1002/(SICI)1521-1878(199804)20:4<336::AID-BIES10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 6.Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol. 2009;183:6251–61. doi: 10.4049/jimmunol.0803978. [DOI] [PubMed] [Google Scholar]
- 7.Wang YD, Chen WD, Moore DD, Huang W. FXR: A metabolic regulator and cell protector. Cell Res. 2008;18:1087–95. doi: 10.1038/cr.2008.289. [DOI] [PubMed] [Google Scholar]
- 8.Gadaleta RM, van Erpecum KJ, Oldenburg B, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. 2011;60:463–72. doi: 10.1136/gut.2010.212159. [DOI] [PubMed] [Google Scholar]
- 9.McGuckin MA, Eri R, Simms LA, Florin TH, Radford-Smith G. Intestinal barrier disfunction in inflammatory bowel deseases. Inflamm Bowel Dis. 2009;15:100–13. doi: 10.1002/ibd.20539. [DOI] [PubMed] [Google Scholar]
- 10.Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117:514–21. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laukoetter MG, Nava P, Nusrat A. Role of the intestinal barrier in inflammatory bowel disease. World J Gastroenterol. 2008;14:401–7. doi: 10.3748/wjg.14.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Narula N, Fedorak RN. Exercise and inflammatory bowel disease. Can J Gastroenterol. 2008;22:497–504. doi: 10.1155/2008/785953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanauer SB. Inflammatory bowel disease: Epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis. 2006;12:S3–9. doi: 10.1097/01.mib.0000195385.19268.68. [DOI] [PubMed] [Google Scholar]
- 14.Salim SY, Söderholm JD. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17:362–81. doi: 10.1002/ibd.21403. [DOI] [PubMed] [Google Scholar]
- 15.Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol. 2003;3:521–33. doi: 10.1038/nri1132. [DOI] [PubMed] [Google Scholar]
- 16.Schulzke JD, Ploeger S, Amasheh M, et al. Epithelial tight junctions in intestinal inflammation. Ann N Y Acad Sci. 2009;1165:294–300. doi: 10.1111/j.1749-6632.2009.04062.x. [DOI] [PubMed] [Google Scholar]
- 17.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–29. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 18.Ceponis PJ, Botelho F, Richards CD, McKay DM. Interleukins 4 and 13 increase intestinal epithelial permeability by a phosphatidylinositol 3-kinase pathway. Lack of evidence for STAT 6 involvement. J Biol Chem. 2000;275:29132–7. doi: 10.1074/jbc.M003516200. [DOI] [PubMed] [Google Scholar]
- 19.Papadakis KA, Targan SR. Role of cytokines in the pathogenesis of inflammatory bowel disease. Annu Rev Med. 2000;51:289–98. doi: 10.1146/annurev.med.51.1.289. [DOI] [PubMed] [Google Scholar]
- 20.Rogler G, Brand K, Vogl D, et al. Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology. 1998;115:357–69. doi: 10.1016/s0016-5085(98)70202-1. [DOI] [PubMed] [Google Scholar]
- 21.Fasano A, Nataro JP. Intestinal epithelial tight junctions as targets for enteric bacteria-derived toxins. Adv Drug Deliv Rev. 2004;56:795–807. doi: 10.1016/j.addr.2003.10.045. [DOI] [PubMed] [Google Scholar]
- 22.Mikov M, Al-Salami H, Golocorbin-Kon S. Liu Chih-Pin., editor. Potentials and limitations of bile acids and probiotics in diabetes mellitus. Type 1 Diabetes – Complications, Pathogenesis, and Alternative Treatments. < www.intechopen.com/articles/show/title/potentials-and-limitations-of-bile-acids-and-probiotics-in-diabetes-mellitus> (Accessed November 22, 2011) [DOI] [PubMed]
- 23.Sartor RB. Probiotic therapy of intestinal inflammation and infections. Curr Opin Gastroenterol. 2005;21:44–50. [PubMed] [Google Scholar]
- 24.Mack DR, Lebel S. Role of probiotics in the modulation of intestinal infections and inflammation. Curr Opin Gastroenterol. 2004;20:22–6. doi: 10.1097/00001574-200401000-00006. [DOI] [PubMed] [Google Scholar]
- 25.Triantafillidis JK, Merikas E, Georgopoulos F. Current and emerging drugs for the treatment of inflammatory bowel disease. Drug Des Devel Ther. 2011;5:185–210. doi: 10.2147/DDDT.S11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McLeod RS. Surgery for inflammatory bowel diseases. Dig Dis. 2003;21:168–79. doi: 10.1159/000073248. [DOI] [PubMed] [Google Scholar]
- 27.Ardizzone S, Porro GB. Inflammatory bowel disease: New insights into pathogenesis and treatment. J Intern Med. 2002;252:475–96. doi: 10.1046/j.1365-2796.2002.01067.x. [DOI] [PubMed] [Google Scholar]
- 28.Siegmund B. Targeted therapies in inflammatory bowel disease. Dig Dis. 2009;27:465–9. doi: 10.1159/000233284. [DOI] [PubMed] [Google Scholar]
- 29.Targan SR. Current limitations of IBD treatment: Where do we go from here? Ann N Y Acad Sci. 2006;1072:1–8. doi: 10.1196/annals.1326.032. [DOI] [PubMed] [Google Scholar]
- 30.Modica S, Gadaleta RM, Moschetta A. Deciphering the nuclear bile acid receptor FXR paradigm. Nucl Recept Signal. 2010;8:e005. doi: 10.1621/nrs.08005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang K, Wan YJ. Nuclear receptors and inflammatory diseases. Exp Biol Med. 2008;233:497–506. doi: 10.3181/0708-MR-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forman BM, Goode E, Chen J, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81:687–93. doi: 10.1016/0092-8674(95)90530-8. [DOI] [PubMed] [Google Scholar]
- 33.Fiorucci S, Cipriani S, Mencarelli A, Renga B, Distrutti E, Baldelli F. Counter-regulatory role of bile acid activated receptors in immunity and inflammation. Curr Mol Med. 2010;10:579–95. doi: 10.2174/1566524011009060579. [DOI] [PubMed] [Google Scholar]
- 34.Zhu Y, Li F, Guo GL. Tissue specific function of farnesoid X receptor in liver and intestine. Pharmacol Res. 2011;63:259–65. doi: 10.1016/j.phrs.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kemper JK. Regulation of FXR transcriptional activity in health and disease: Emerging roles of FXR cofactors and post-translational modifications. Biochim Biophys Acta. 2011;1812:842–50. doi: 10.1016/j.bbadis.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eloranta JJ, Kullak-Ublick GA. The role of FXR in disorders of bile acid homeostasis. Physiology. 2008;23:286–95. doi: 10.1152/physiol.00020.2008. [DOI] [PubMed] [Google Scholar]
- 37.Lew JL, Zhao A, Yu J, et al. The farnesoid X receptor controls gene expression in a ligand- and promoter- selective fashion. J Biol Chem. 2004;279:8856–61. doi: 10.1074/jbc.M306422200. [DOI] [PubMed] [Google Scholar]
- 38.Cui J, Huang L, Zhao A, et al. Guggulsterone is a farnesoid X receptor antagonist in coactivator association assays but acts to enhance transcription of bile salt export pump. J Biol Chem. 2003;278:10214–20. doi: 10.1074/jbc.M209323200. [DOI] [PubMed] [Google Scholar]
- 39.Carter BA, Taylor OA, Prendergast DR, et al. Stigmasterol, a soy lipid-derived phytosterol, is an antagonist of the bile acid nuclear receptor FXR. Pediatr Res. 2007;62:301–6. doi: 10.1203/PDR.0b013e3181256492. [DOI] [PubMed] [Google Scholar]
- 40.Ricketts ML, Boekschoten MV, Kreeft AJ, et al. The cholesterol-raising factor from coffee beans, cafestol, as an agonist ligand for the farnesoid and pregnane X receptors. Mol Endocrinol. 2007;21:1603–16. doi: 10.1210/me.2007-0133. [DOI] [PubMed] [Google Scholar]
- 41.Maloney PR, Parks DJ, Haffner CD, et al. Identification of a chemical tool for the orphan nuclear receptor FXR. J Med Chem. 2000;43:2971–4. doi: 10.1021/jm0002127. [DOI] [PubMed] [Google Scholar]
- 42.Downes M, Verdecia MA, Roecker AJ, et al. A chemical, genetic, and structural analysis of the nuclear bile acid receptor FXR. Mol Cell. 2003;11:1079–92. doi: 10.1016/s1097-2765(03)00104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dussault I, Beard R, Lin M, et al. Identification of gene-selective modulators of the bile acid receptors FXR. J Biol Chem. 2003;278:7027–33. doi: 10.1074/jbc.M209863200. [DOI] [PubMed] [Google Scholar]
- 44.Pellicciari R, Fiorucci S, Camaioni E, et al. 6Alpha-ethylchenodeoxycholic acid (6-EDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002;45:3569–72. doi: 10.1021/jm025529g. [DOI] [PubMed] [Google Scholar]
- 45.Gadaleta RM, Oldenburg B, Willemsen EC, et al. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-κB signaling in the intestine. Biochim Biophys Acta. 2011;1812:851–8. doi: 10.1016/j.bbadis.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 46.Mikov M, Fawcett JP, Kuhajda K, Kevresan S. Pharmacology of bile acids and their derivatives: Absorption promoters and therapeutic agents. Eur J Drug Metab Pharmacokinet. 2006;31:237–51. doi: 10.1007/BF03190714. [DOI] [PubMed] [Google Scholar]
- 47.Chiang JYL. Bile acids: Regulation of synthesis. J Lipid Res. 2009;50:1955–66. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Inagaki T, Choi M, Moschetta A. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–25. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 49.Klaassen CD, Aleksunes LM. Xenobiotic, bile acid, and cholesterol transporters: Function and regulation. Pharmacol Rev March. 2010;62:1–96. doi: 10.1124/pr.109.002014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gadaleta RM, van Mil SW, Oldenburg B, Siersema PD, Klomp LW, van Erpecum KJ. Bile acids and their nuclear receptor FXR: Relevance for hepatobiliary and gastrointestinal disease. Biochim Biophys Acta. 2010;1801:683–92. doi: 10.1016/j.bbalip.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 51.Konig J, Nies AT, Cui Y, Leier I, Keppler D. Conjugate export pumps of the multidrug resistance protein (MRP) family: Localization, substrate specificity, and MRP2-mediated drug resistance. Biochim Biophys Acta. 1999;1461:377–94. doi: 10.1016/s0005-2736(99)00169-8. [DOI] [PubMed] [Google Scholar]
- 52.Modica S, Moschetta A. Nuclear bile acid receptor FXR as pharmacological target: Are we there yet? FEBS Lett. 2006;580:5492–9. doi: 10.1016/j.febslet.2006.07.082. [DOI] [PubMed] [Google Scholar]
- 53.Sinal CJ, Tohkin M, Miyata M, et al. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–44. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 54.Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007;67:863–7. doi: 10.1158/0008-5472.CAN-06-1078. [DOI] [PubMed] [Google Scholar]
- 55.Schote AB, Turner JD, Schiltz J, Muller CP. Nuclear receptors in human immune cells: Expression and correlations. Mol Immunol. 2007;44:1436–45. doi: 10.1016/j.molimm.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 56.Wagner EF, Eferl R. Fos/AP-1 proteins in bone and the immune system. Immunol Rev. 2005;208:126–40. doi: 10.1111/j.0105-2896.2005.00332.x. [DOI] [PubMed] [Google Scholar]
- 57.Fiorucci S, Clerici C, Antonelli E, et al. Protective effects of 6-ethyl chenodeoxycholic acid, a farnesoid X receptor ligand, in estrogen-induced cholestasis. J Pharmacol Exp Ther. 2005;313:604–12. doi: 10.1124/jpet.104.079665. [DOI] [PubMed] [Google Scholar]
- 58.Förster C. Tight junctions and the modulation of barrier function in disease. Histochem Cell Biol. 2008;130:55–70. doi: 10.1007/s00418-008-0424-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petrovic V, Teng S, Piquette-Miller M. Regulation of drug transporters: During infection and inflammation. Mol Interv. 2007;7:99–111. doi: 10.1124/mi.7.2.10. [DOI] [PubMed] [Google Scholar]
- 60.Blokzijl H, Borght SV, Bok LI, et al. Decreased P-glycoprotein (P-gp/MDR1) expression in inflamed human intestinal epithelium is independent of PXR protein levels. Inflamm Bowel Dis. 2007;13:710–20. doi: 10.1002/ibd.20088. [DOI] [PubMed] [Google Scholar]
- 61.Potocnik U, Ferkolj I, Glavac D, Dean M. Polymorphisms in multidrug resistance 1 (MDR1) gene are associated with refractory Crohn disease and ulcerative colitis. Genes Immun. 2004;5:530–9. doi: 10.1038/sj.gene.6364123. [DOI] [PubMed] [Google Scholar]
- 62.Thiebaut F, Tsuruo T, Hamada H, et al. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci USA. 1987;84:7735–8. doi: 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCole DF, Barrett KE. Epithelial transport and gut barrier function in colitis. Curr Opin Gastroenterol. 2003;19:578–82. doi: 10.1097/00001574-200311000-00011. [DOI] [PubMed] [Google Scholar]
- 64.Lawrance IC, Fiocchi C, Chakravarti S. Ulcerative colitis and Crohn’s disease: Distinctive gene expression profiles and novel susceptibility candidate genes. Hum Mol Genet. 2001;10:445–56. doi: 10.1093/hmg/10.5.445. [DOI] [PubMed] [Google Scholar]
- 65.Panwala CM, Jones JC, Viney JL. A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J Immunol. 1998;161:5733–44. [PubMed] [Google Scholar]
- 66.Schwab M, Schaeffeler E, Marx C, et al. Association between the C3435T MDR1 gene polymorphism and susceptibility for ulcerative colitis. Gastroenterology. 2003;124:26–33. doi: 10.1053/gast.2003.50010. [DOI] [PubMed] [Google Scholar]
- 67.Lian F, Xing X, Yuan G, et al. Farnesoid X receptor protects human and murine gastric epithelial cells against inflammation-induces damage. Biochem J. 2011;315:23. doi: 10.1042/BJ20102096. 438; [DOI] [PubMed] [Google Scholar]
- 68.Fiorucci S, Mencarelli A, Cipriani S, Renga B, Palladino G, Santucci L, Distrutti E. Activation of the farnesoid-X receptor protects against gastrointestinal injury caused by non-steroidal anti-inflammatory drugs in mice. Br J Pharmacol. 2011;164:1929–38. doi: 10.1111/j.1476-5381.2011.01481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor κB in hepatic inflammatory response. Hepatology. 2008;48:1632–43. doi: 10.1002/hep.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lorenzo-Zúñiga V, Bartolí R, Planas R, et al. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology. 2003;37:551–7. doi: 10.1053/jhep.2003.50116. [DOI] [PubMed] [Google Scholar]
- 71.Inagaki T, Moschetta A, Lee YK, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci USA. 2006;103:3920–5. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wallace JL, Miller MJ. Nitric oxide in mucosal defense: A little goes a long way. Gastroenterology. 2000;119:512–20. doi: 10.1053/gast.2000.9304. [DOI] [PubMed] [Google Scholar]
- 73.Hooper LV, Stappenbeck TS, Hong CV, et al. Angiogenins: A new class of microbicidal proteins involved in innate immunity. Nat Immunol. 2003;4(3):269–73. doi: 10.1038/ni888. [DOI] [PubMed] [Google Scholar]
- 74.Biet F, Locht C, Kremer L. Immunoregulatory functions of interleukin 18 and its role in defense against bacterial pathogens. J Mol Med. 2002;80:147–62. doi: 10.1007/s00109-001-0307-1. [DOI] [PubMed] [Google Scholar]
- 75.Zhang Q, He F, Kuruba R, et al. FXR-mediated regulation of angiotensin type 2 receptor expression in vascular smooth muscle cells. Cardiovasc Res. 2008;77:560–9. doi: 10.1093/cvr/cvm068. [DOI] [PubMed] [Google Scholar]
- 76.Platten M, Youssef S, Hur EM, et al. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc Natl Acad Sci USA. 2009;106:14948–53. doi: 10.1073/pnas.0903958106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wildenberg ME, van den Brink GR. FXR activation inhibits inflammation and preserves the intestinal barrier in IBD. Gut. 2011;60:432–3. doi: 10.1136/gut.2010.233304. [DOI] [PubMed] [Google Scholar]
- 78.Blumberg RS. Inflammation in the intestinal tract: Pathogenesis and treatment. Dig Dis. 2009;27:455–64. doi: 10.1159/000235851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Govern DM, Ahmad T. New IBD genes? Gut. 2005;54:1060–1. doi: 10.1136/gut.2004.048918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fiocchi C. Susceptibility genes and overall pathogenesis of inflammatory bowel disease: Where do we stand? Dig Dis. 2009;27:226–35. doi: 10.1159/000228554. [DOI] [PubMed] [Google Scholar]
- 81.Nijmeijer RM, Gadaleta RM, van Mil SWC. Farnesoid X receptor (FXR) activation and FXR genetic variation in inflammatory bowel disease. PLoS ONE. 2011;6:e23745. doi: 10.1371/journal.pone.0023745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dekaney CM, von Allmen DC, Garrison AP, et al. Bacterial-dependent up-regulation of intestinal bile acid binding protein and transport is FXR-mediated following ileo-cecal resection. Surgery. 2008;144:174–81. doi: 10.1016/j.surg.2008.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]