

Nelfinavir has anti-inflammatory effects directly upon human macrophages independent of antiviral activity.
Keywords: monocyte, lipopolysaccharide, microbial translocation, soluble CD14, tumor necrosis factor, interleukin-6, 20S proteasome, calyculin A, okadaic acid, protein phosphatase
Abstract
The HIV-1 PI NFV has off-target effects upon host enzymes, including inhibition of the 20S proteasome, resulting in activation of PP1. HIV-1-associated monocyte/macrophage activation, in part a result of systemically elevated levels of microbial products including LPS, is associated with risk of mortality, independent of viremia or CD4 T cell loss. This study tested the hypothesis that activation of protein phosphatases by NFV would reduce activation of monocytes/macrophages through dephosphorylation of signal transduction proteins. NFV uniquely blocked LPS-induced production by human monocyte-derived macrophages of the inflammatory cytokines TNF and IL-6, as well as sCD14. Although NFV failed to modulate NF-κB, NFV treatment reduced phosphorylation of AKT and MAPKs. Inhibition of PP2 with okadaic acid blocked the anti-inflammatory effect of NFV, whereas the PP1 inhibitor calyculin A failed to counter the anti-inflammatory effects of NFV. For in vivo studies, plasma sCD14 and LPS were monitored in a cohort of 31 pediatric HIV-1 patients for over 2 years of therapy. Therapy, including NFV, reduced sCD14 levels significantly compared with IDV or RTV, independent of ΔLPS levels, VL, CD4 T cell frequency, or age. The hypothesis was supported as NFV induced activation of PP2 in macrophages, resulting in disruption of inflammatory cell signaling pathways. In vivo evidence supports that NFV may offer beneficial effects independent of antiviral activity by reducing severity of chronic innate immune activation in HIV-1 infection.
Introduction
HIV-1 PIs and other antiretroviral drugs, including inhibitors of RT, integrase, or viral entry, continue to revolutionize treatment of HIV-1 infection and improve outcomes for patients [1]. In the absence of therapy, HIV-1 pathogenesis presents as chronic activation in adaptive and innate immune compartments [2–5]. Activation of CD4 and/or CD8 T cells is associated with reduced CD4 T cell frequency before therapy and impaired CD4 T cell recovery following therapy [6–9]. Suppression of viremia by cART markedly reverses T cell activation concomitant with rebound in CD4 T cells [2, 9–11]. Microbial translocation and resultant activation of monocytes and macrophages, measured by circulating plasma levels of LPS and sCD14, respectively, [12, 13], are elevated in untreated HIV infection and partially reduced following 2 years of cART [2, 3]. Although sCD14 levels remain significantly elevated compared with uninfected individuals [14], initiation of cART before decline in CD4+ T cells below 25% reduces the extent of monocyte/macrophage activation in the post-therapy patient [14]. Despite suppression of plasma viral levels by cART, inflammation-associated comorbidities, including HIV-associated neurocognitive impairment [15–20], cardiovascular disease/coagulopathy [21–24], endothelial dysfunction [25], and cancer [26, 27], continue to develop.
Partial reduction of HIV-1-associated innate immune activation by cART likely involves multiple factors, including reduced viral burden, resolution of opportunistic coinfections, and reduced microbial translocation. PIs have direct effects on host cells independent of anti-HIV-1 properties [28–33] and off-target effects related to drug toxicities or anticancer properties [34–38]. For example, functional studies have identified biological effects of PIs, including NFV, upon adipose tissue [39, 40], in macrophages derived from the monocyte tumor cell line THP-1, or in primary human osteoblasts [41]. NFV disrupts cell signaling and induces apoptosis in some tumor cells and is under evaluation as a chemotherapeutic agent for treatment of cancer [38, 42]. Several HIV PIs have been implicated in causing metabolic disorders related to glucose and/or lipid metabolism [43–46]. These clinical abnormalities are attributable, at least in part, to inhibitory effects of PIs on the 20S proteasome leading to ER stress and induction of an altered transcriptome profile characterized by modulation of stress and metabolism genes [47]. NFV, in particular, exhibits relatively potent activity against the 20S proteasome [47], which leads to accumulation of misfolded proteins, triggering the UPR and activation of a PP1 complex, which dephosphorylates AKT [48].
Overall, activity of PIs upon signaling pathways or inflammation is pleiotropic and seems context-dependent, so mechanisms of PIs in affecting HIV-induced inflammation need to be addressed. The current study tested the hypothesis that NFV reduces HIV-1-associated monocyte/macrophage activation through off-target effects on host cell signaling pathways. The mechanism by which NFV inhibits LPS-induced macrophage activation was determined using human MDMs in vitro, whereas effects of PIs on HIV-1-associated innate immune activation were determined in vivo in a cohort of subjects receiving therapy, including one of three first-generation HIV-1 PIs, IDV (Crixivan), RTV (Norvir), or NFV (Viracept).
MATERIALS AND METHODS
Monocyte/macrophage culture
Human monocytes, isolated from healthy donors by counter-current centrifugal elutriation [49], were obtained from Howard E. Gendelman at the University of Nebraska (Omaha, NE, USA). Monocytes were cultured 7 days in DMEM, supplemented with 10% human serum and 1 ng/ml M-CSF (Sigma-Aldrich, St. Louis, MO, USA) to differentiate MDMs. For flow cytometry (FACS), MDMs were cultured on UpCell Surface plates (Nalge Nunc, Rochester, NY, USA), incubated 20 min with ice-cold PBS, and gently washed to dissociate adherent MDMs without the use of enzymes. In some cases, freshly isolated monocytes were cultured in the absence of M-CSF, where determination of PI effects on undifferentiated monocytes was desired.
PI treatments and activation
PIs were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, U. S. National Institutes of Health (Bethesda, MD, USA). MDMs were pretreated with PI or vehicle control (100% ethanol for IDV, RTV, NFV, LPV, and TPV; 50/50v/v acetone/ethanol for ATV) for 30 min, followed by activation with LPS where indicated. Cells were cultured 6 h for analysis of cytokines or sCD14 or 18 h for analysis of cell surface proteins. To determine the effects of NFV on sCD14 production by MDM cultures, serum-free conditions were used for the final 6 h of culture to eliminate background as a result of the presence of sCD14 in human serum. TNF (TNF-α, TNFSF2; BD Biosciences, San Jose, CA, USA), IL-6 (BD Biosciences), or sCD14 (R&D Systems, Minneapolis, MN, USA) was determined by ELISA. Cell surface protein expression was determined by staining cells with panels of fluorochrome-tagged antibodies (CD14-AlexaFluor 488, CD14-PE, CD16-AlexaFluor 647, CD163-PE, CD80-PE, HLA-DR-FITC, CD4-allophycocyanin; BD Biosciences) and analysis with a BD FACSCalibur four-color cytometer. Viability of MDM was assessed by CellTiter 96 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium assay (Promega, Madison, WI, USA), according to the manufacturer's protocol. In some experiments, calyculin A (PP1 inhibitor), okadaic acid (PP2 inhibitor), wortmannin (PI3K inhibitor), or U0126 (MEK1/2 inhibitor) were included at indicated concentrations.
IL6 qPCR
MDMs were pretreated with 0–40 μM NFV and then cultured with media alone (untreated) or with 100 pg/ml LPS for 2 h. Total RNA was isolated, reverse-transcribed, and IL6 transcript was quantified by qPCR using SYBR green and the primers: IL-6, forward 5′-TACCCCCAGGAGAAGATTCC-3′, and IL-6, reverse 5′-TTTTCTGCCAGTGCCTCTTT-3′.
Western blot and phospho-MAPK array
For intracellular protein analyses, cells were pretreated with NFV or carrier for 30 min and then activated with LPS for 30 min as indicated. MDMs were washed with ice-cold PBS and then lysed to extract proteins with cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA) with 1% Triton, 1 mM PMSF, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and 1 μg/ml leupeptin. Proteins were separated by SDS-PAGE, transferred to PVDF membranes, which were then blocked with 5% BSA in TBS with 0.05% Tween 20 and probed with mAb (Cell Signaling Technology), including rabbit antiphospho-NF-κB p65 (Ser 536), rabbit antiphospho-IKKα/β (Ser 176/180), rabbit anti-NF-κB p65, rabbit anti-IKKα, mouse anti-IκBα, rabbit antiphospho-MEK1/2 (Ser 217/221), rabbit anti-MEK1/2, rabbit anti-p44/42 MAPK “phospho-ERK1/2” (Thr 202/Tyr 204), rabbit anti-p44/42 MAPK “ERK1/2”, goat anti-rabbit IgG-HRP, and goat anti-mouse IgG-HRP, and used according to the manufacturer's recommendation. A Proteome Profiler Phospho-MAPK Array (R&D Systems) was used for phospho-MAPK assays. For each condition, 18 million MDMs were pretreated for 30 min with 10 μM NFV or equivalent carrier, followed by treatment with 100 pg/ml LPS for 30 min. Cells were lysed with reagents provided in the kit, and 50 μg protein lysate was used for each analysis. Densitometry analyses were performed using ImageJ software (U.S. National Institutes of Health).
Phosphatase assay
To test the effect of NFV upon cellular PP1 and PP2 activity, 3 million MDMs were cultured in media alone or with NFV at 10, 20, or 40 μM for 30 min and lysed in RIPA buffer with PIs and 20 mM Na3VO4 to inhibit activity of tyrosine phosphatases but not serine/threonine phosphatases such as PP1 and PP2 [50, 51]. Cell lysates were then treated with PBS, calyculin A, or okadaic acid. Phosphatase activity was determined using a SensoLyte pNPP Protein Phosphatase Assay kit (AnaSpec, Fremont, CA, USA). Conversion of a pNPP colorimetric substrate was determined at 405 nm using a microplate reader.
Study subjects
Plasma was obtained from a cohort of HIV-infected children who received cART consisting of one PI (IDV, RTV, or NFV) plus two nucleotide RT inhibitors [stavudine (d4T) and lamivudine (3TC)], as described previously [2, 52–54]. The protocol was approved by the University of Florida Institutional Review Board (Gainesville, FL, USA); informed consent was obtained from guardians and assent from subjects over the age of 6. All HIV-1-infected subjects were infected perinatally, and viral and immune outcomes were known. Suppression of viremia was categorized as complete in 14 subjects (<400 copies of plasma viral RNA/ml) or incomplete in 17 subjects (>400 copies of plasma viral RNA/ml). Among incomplete suppressors, mean VL following therapy was 104.2 copies/ml. There was no significant difference in the frequency of incomplete suppression among subjects receiving IDV, RTV, or NFV. Whole blood samples were collected, using phlebotomy, in sterile Vacutainer (Becton Dickinson, San Diego, CA, USA) acid citrate dextrose tubes and processed within 12 h [55–57]. Plasma samples were stored at −80°C in nonpyrogenic polypropylene cyrovials.
Measurement of soluble markers for immune activation and microbial translocation
Plasma samples were handled in an endotoxin-free environment, diluted 1:4 in 0.15 M NaCl, heat-inactivated at 60°C for 30 min, and then used to measure sCD14 levels by ELISA and LPS levels by the LAL Chromogenic Endpoint Assay (Lonza Group, Allendale, NJ, USA), as described previously [2]. Controls were performed with LAL assays to rule out endotoxin contamination.
Statistical analysis
For longitudinal within-subject determination of change in any parameter, pretherapy values were subtracted from 96-week post-therapy values to indicate the magnitude and direction of change. Longitudinal comparison between pre-cART and post-cART values was performed by Wilcoxon matched pairs test, and cross-sectional comparisons between groups were compared by Mann-Whitney U test. Pearson's correlation and simple linear regression analyses were performed to determine the relationships between two variables. For comparison of ΔsCD14 levels among three PI treatment groups, a general linear model (univariate) was used to simultaneously determine independent effects of ΔLPS, antiviral efficacy, drug-treatment group, age, and CD4 T cell frequency upon ΔsCD14. A simple contrast was used to determine whether NFV (n=14), as the reference group, was different from the IDV (n=6), RTV (n=11), or IDV + RTV (n=17) group regarding ΔsCD14 while controlling for VL, ΔLPS, age, and CD4 T cell frequency as covariates. Analyses were performed with GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA, USA) and IBM SPSS Software (Armonk, NY, USA) and considered significant when P value was <0.05.
RESULTS
NFV inhibits LPS-induced macrophage activation
Effects of PIs upon human macrophage activation ex vivo were evaluated by measuring production of the inflammatory cytokines TNF or IL-6. Uninfected human MDMs were activated with LPS after initial treatment with one of six PIs. IDV, RTV, ATV, or LPV failed to inhibit LPS-induced TNF or IL-6 production within physiologically relevant or maximum concentrations (Fig. 1A and B). In contrast, NFV potently inhibited TNF secretion with an IC50 of 8.4 μM, a level within the physiological range of NFV in plasma [58] (Fig. 1A). Doses of NFV as low as 2.5 μM inhibited LPS-induced TNF production by 40%. NFV or TPV inhibited IL-6 secretion with IC50 concentrations of 5.5 μM or 25.4 μM, respectively (Fig. 1B). NFV displayed no cytotoxicity at four times the IC50 value (data not shown). LPS treatment resulted in increased levels of IL6 mRNA and IL-6 protein, whereas NFV mediated a dose-dependent suppression of steady-state mRNA and protein (Fig. 2).
Figure 1. NFV uniquely inhibits LPS-induced secretion of inflammatory cytokines by macrophages.

MDMs were cultured in the presence of 100 pg/ml LPS for 6 h, after which (A) TNF or (B) IL-6 concentrations in culture supernatants were determined by ELISA. Where indicated, increasing concentrations of PIs were used to pretreat MDMs before LPS treatment (percent inhibition is calculated as the percent reduction in cytokine secretion for each PI concentration compared with no PI). IC50 concentrations were calculated with BioDataFit 1.02. Shaded areas represent in vivo plasma drug concentration ranges for each PI [58–62], and vertical, dashed lines represent IC50 where 50% inhibition of TNF or IL-6 was achieved (error bars represent±1 sd among four donors).
Figure 2. NFV reduces LPS-induced IL-6 expression at the level of transcript.
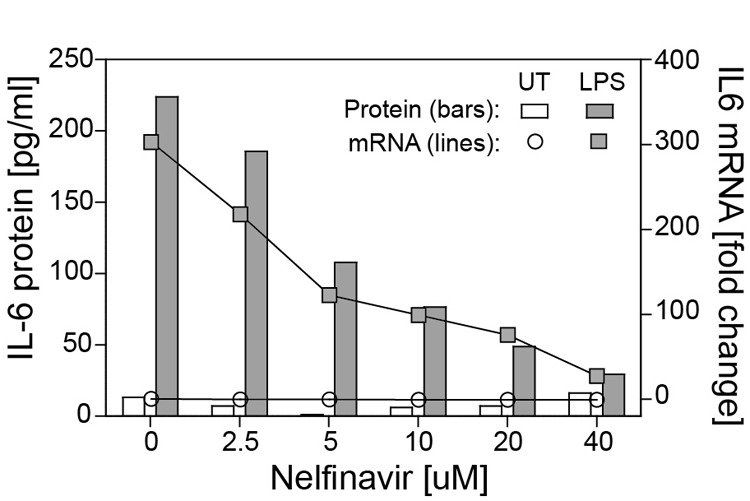
MDMs were pretreated with 0–40 μM NFV and then cultured with media alone [untreated (UT)] or with 100 pg/ml LPS for 2 h. Supernatant IL-6 protein was measured by ELISA. Total RNA was isolated and reverse-transcribed, and IL-6 transcripts were quantified by SYBR Green qPCR.
To evaluate the effects of NFV on LPS-induced macrophage activation phenotype, changes in cell surface markers were measured by flow cytometry. Untreated macrophages displayed a homogeneous resting phenotype characterized as CD14+ CD163+ CD16+ CD4+ CD80lo (Fig. 3A). Activation of macrophages with LPS produced no change in CD14 or CD16 expression (data not shown), increased expression of CD80 and HLA-DR, and reduced expression of CD163 and CD4 (Fig. 3B). Pretreatment with NFV had no effect on LPS-induced alterations to the cell surface phenotype. Thus, NFV suppression of macrophage activation/inflammation was selective rather than global.
Figure 3. NFV fails to alter LPS-induced macrophage surface protein phenotype.

Expression of cell surface proteins was determined by flow cytometry. (A) Representative phenotype of resting macrophages (left panel: gray, isotype control; black, anti-CD14; middle and right panels are gated on CD14+ cells). (B) Representative phenotype of resting MDMs (black), MDMs activated with 100 pg/ml LPS for 18 h (red), or MDMs pretreated with 10 μM NFV before activation with 100 pg/ml LPS for 18 h (blue). Isotype control antibody staining of resting MDM (gray).
NFV selectively inhibits LPS-induced Akt/MAPK activation in MDMs
To evaluate mechanisms by which NFV might suppress selective LPS-induced macrophage activation/inflammation, NF-κB and MAPK pathways, two main activation pathways of TLR4-induced signaling, were investigated. LPS treatment resulted in phosphorylation of IKKα/β and p65 and degradation of IκBα, key mediators of NF-κB signaling (Fig. 4). Pretreatment with NFV, even at 10 μM, failed to inhibit LPS-induced NF-κB activation.
Figure 4. LPS-induced NF-κ B signaling in macrophages is unchanged by NFV.

MDMs were pretreated with carrier alone or NFV (2.5 μM or 10 μM) for 30 min, followed by activation with 100 pg/ml LPS for 30 min. Cells were lysed to perform Western blots using 20 μg total protein/sample. phos, Phosphorylation.
Among a panel of 26 Akt/MAPK proteins, constitutive phosphorylation of five Akt and Erk proteins was detected in resting MDMs (Fig. 5A). LPS treatment increased phosphorylation of four Akt or Erk proteins/isoforms (Akt1, pan-Akt, ERK1, ERK2), as well as produced phosphorylation of eight additional proteins (CREB, GSK3α/β, HSP27, pan-Jnk, MSK2, p38δ, RSK1, and mTOR; Fig. 5B). Pretreatment with 10 μM NFV reduced constitutive or LPS-induced AKT phosphorylation, as well as LPS-induced phosphorylation of eight of 10 MAPK proteins (CREB, ERK1, ERK2, HSP27, pan-Jnk, MSK2, p38δ, and RSK1; Fig. 5C). Although NFV reduced phosphorylation of most proteins in the panel, GSK3α/β and mTOR phosphorylation were largely unaffected by NFV (Fig. 5D), indicating some selectivity. To investigate effects of NFV upon a proximal mediator of MAPK signaling (Mek1/2), phosphorylated versus total Mek1/2 and Erk1/2 proteins were measured by Western blot (Fig. 5E). Neither LPS nor NFV treatment altered steady-state levels of total Mek1/2 or Erk1/2 proteins. In contrast, LPS treatment produced phosphorylation of Mek1/2 and Erk1/2, which was reduced by NFV.
Figure 5. NFV disrupts LPS-induced phosphorylation of AKT and MAPK proteins in macrophages.

MDMs were treated for 30 min with carrier alone (A), 100 pg/ml LPS (B), or 10 μM NFV followed by activation with 100 pg/ml LPS for an additional 30 min (C), and cells were lysed to perform a phospho-MAPK array to detect 26 AKT/MAPK proteins/isoforms using 50 μg total protein/sample. (D) Densitometry analysis was performed using ImageJ software, and individual phopho-protein regions were considered detectable when signal was >1 sd above background signal. Graph represents fold change in phosphorylation for each protein relative to condition A (average of two individual monocyte donors). (E) MDMs were pretreated with carrier alone or NFV (2.5 μM or 10 μM) for 30 min, followed by activation with 100 pg/ml LPS for 30 min. Cells were lysed to perform Western blots using 20 μg total protein/sample.
Together with array results, the data indicate that NFV mediates selective disruption of AKT and MAPK pathways without perturbing the NF-κB pathway. Akt, MAPK, and NF-κB pathways diverge from proximal TLR4-induced signaling events to regulate macrophage activation in a coordinated effort that balances proactivation signals (MAPK and NF-κB) with antiactivation signals (Akt) [63–68] (Fig. 6). NFV mediates anticancer activity through inhibition of the 20S proteasome, activation of PP1, and dephosphorylation of Akt, implicating roles for protein phosphatases in activity by NFV on macrophages.
Figure 6. TLR4-induced macrophage activation regulated by NF-κB, PI3K/AKT, and MAPK signaling pathways.

A model for potential mechanisms of the anti-inflammatory effects of NFV. The scavenger molecule CD14 in membrane-associated or sCD14 form binds LPS and localizes to a TLR4 signaling complex. A phospho-protein cascade, including IL-1R-associated kinase (IRAK)1 and -4 and TRAF6, serves as a signaling hub that leads into multiple downstream pathways, including NF-κB, PI3K/AKT, and MAPK. NF-κB pathway is initiated by TGF-β-activated kinase 1 (TAK1)-dependent phosphorylation of IKKα/β, which subsequently phosphorylates the inhibitory protein IκBα, triggering its ubiquitination and degradation. Degradation of IκBα releases p50/p65, which is phophorylated and translocated to the nucleus to initiate transcription of proinflammatory genes. PI3K/AKT signaling is triggered by receptor-interacting serine/threonine-protein kinase (RIP) and culminates with inhibition of proinflammatory signaling/transcription and/or enhancement of anti-inflammatory gene expression. MAPK signaling is triggered by activation of proximal MAP3K proteins, followed by downstream activation of Mek1/2 and Erk1/2 and induction of proinflammatory genes. NFV inhibits proteasome activity, leading to accumulation of misfolded proteins and activation of the UPR, including activation of PP1 and possibly PP2, which can be inhibited chemically with calyculin A (CA) and okadaic acid (OA), respectively.
PP2 is required for the anti-inflammatory effects of NFV
To determine whether NFV induces activity of PP1 or the closely related PP2 in macrophages, MDMs were treated with 10 μM NFV and found to display overall twofold increased phosphatase activity above basal serine/threonine phosphatase activity (Fig. 7A). PP1 activity, either constitutive or NFV-induced, was inhibited by calyculin A in a dose-dependent manner. Okadaic acid markedly reduced PP2 serine/threonine phosphatase activity with greater effects than calyculin A upon NFV-induced phosphatase. Combination of inhibitors further reduced NFV-induced phosphatases, indicating that NFV causes activation of PP1 and PP2.
Figure 7. Inhibitory effects of NFV are mediated through activation of PP2.
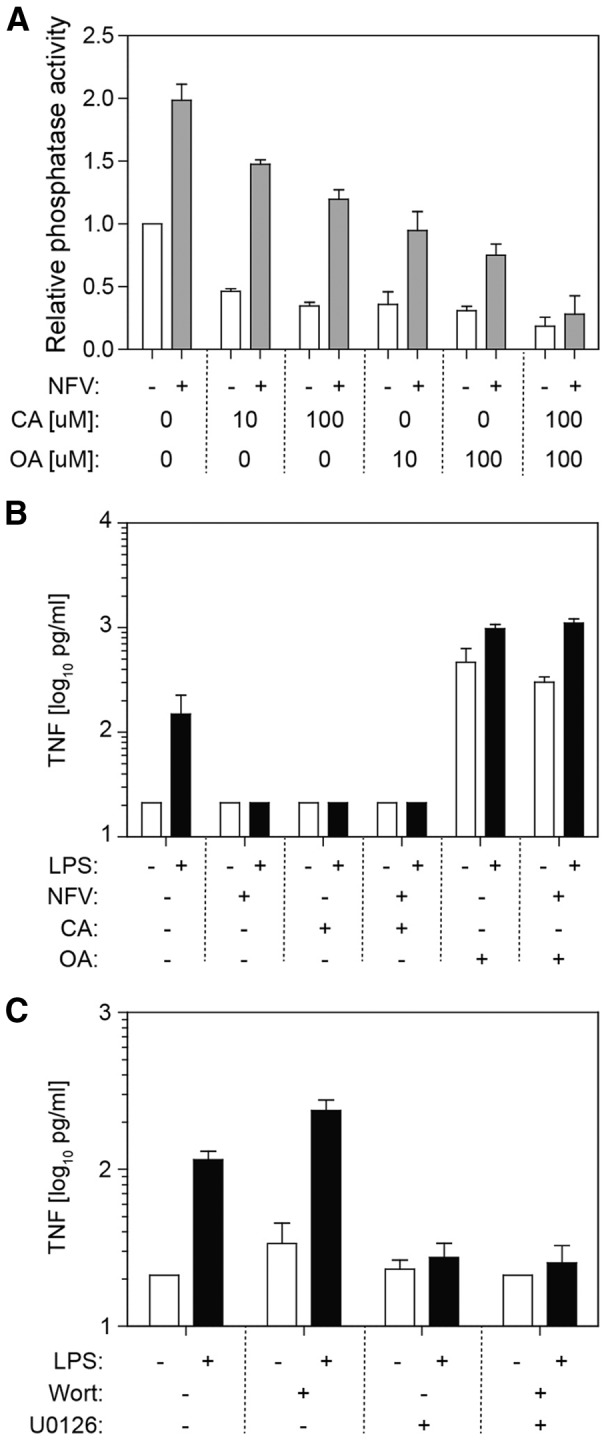
(A) MDMs were cultured in the presence (shaded bars) or absence (open bars) of NFV for 30 min. Cell lysates were prepared, and CA (PP1 inhibitor) or OA (PP2 inhibitor) was mixed with lysates before phosphatase assay. (B) MDMs were cultured in the presence or absence of NFV, CA, or OA for 30 min and then treated with media alone (open bars) or 1 ng/ml LPS (black bars). Supernatant TNF was measured by ELISA. (C) MDMs were cultured in the presence or absence of the PI3K/AKT inhibitor wortmannin (Wort) or the MEK1/2 inhibitor U0126 and then treated with media alone (open bars) or 1 ng/ml LPS (black bars) for 6 h. Supernatant TNF was measured by ELISA.
To determine if PP1 or PP2 contributes to the anti-inflammatory effect of NFV, MDMs were cultured with calyculin A, okadaic acid, or both inhibitors before addition of NFV (Fig. 7B). Following activation with LPS, NFV suppressed TNF production by approximately tenfold, whereas treatment with calyculin A inhibited LPS-induced TNF production, independent of NFV. In contrast, okadaic acid enhanced constitutive TNF production by almost 2 logs. Cotreatment with okadaic acid and LPS increased TNF production and abrogated the anti-inflammatory effect of NFV.
PP1 and PP2 mediate seemingly opposing effects. To determine whether PP2-induced dephosphorylation of MAPK by NFV (anti-inflammatory) was dominant over PP1-induced AKT dephosphorylation by NFV (proinflammatory), specific inhibitors for PI3K/AKT signaling (wortmannin) or Mek1/2 signaling (U0126) were used alone or in combination (Fig. 7C). Wortmannin enhanced LPS-induced TNF production by MDMs, whereas U0126 inhibited TNF production. When both inhibitors were applied, the net effect was inhibition of TNF production, indicating that inhibition of Mek1/2 and downstream MAPKs is dominant over inhibition of PI3K/AKT.
NFV inhibits sCD14 release from monocytes and macrophages
In vivo, levels of plasma sCD14 are indicative of systemic monocyte/macrophage activation in HIV-1-infected individuals, and sCD14 is an independent predictor of mortality [69]. To determine if NFV can modulate sCD14 release ex vivo, primary human monocytes or MDMs were cultured in media alone or in the presence of LPS (Fig. 8A). Monocytes spontaneously produced levels of sCD14 that were ∼20-fold greater than levels produced by MDMs. LPS treatment produced no change in sCD14 levels from monocytes but increased sCD14 by approximately fivefold from MDMs (P=0.02). When monocytes were treated with IDV, RTV, or NFV, spontaneous sCD14 release was reduced specifically and significantly by NFV (Fig. 8B). Similar to the effect on monocytes, only NFV reduced sCD14 production by macrophages (Fig. 8C), whereas IDV and RTV appeared to enhance LPS-induced sCD14 levels slightly. Potent inhibition by NFV of sCD14 release by monocytes and macrophages ex vivo provided a rationale to evaluate effects of NFV upon sCD14, a HIV disease biomarker in vivo.
Figure 8. NFV selectively inhibits sCD14 release from monocytes and macrophages in vitro.
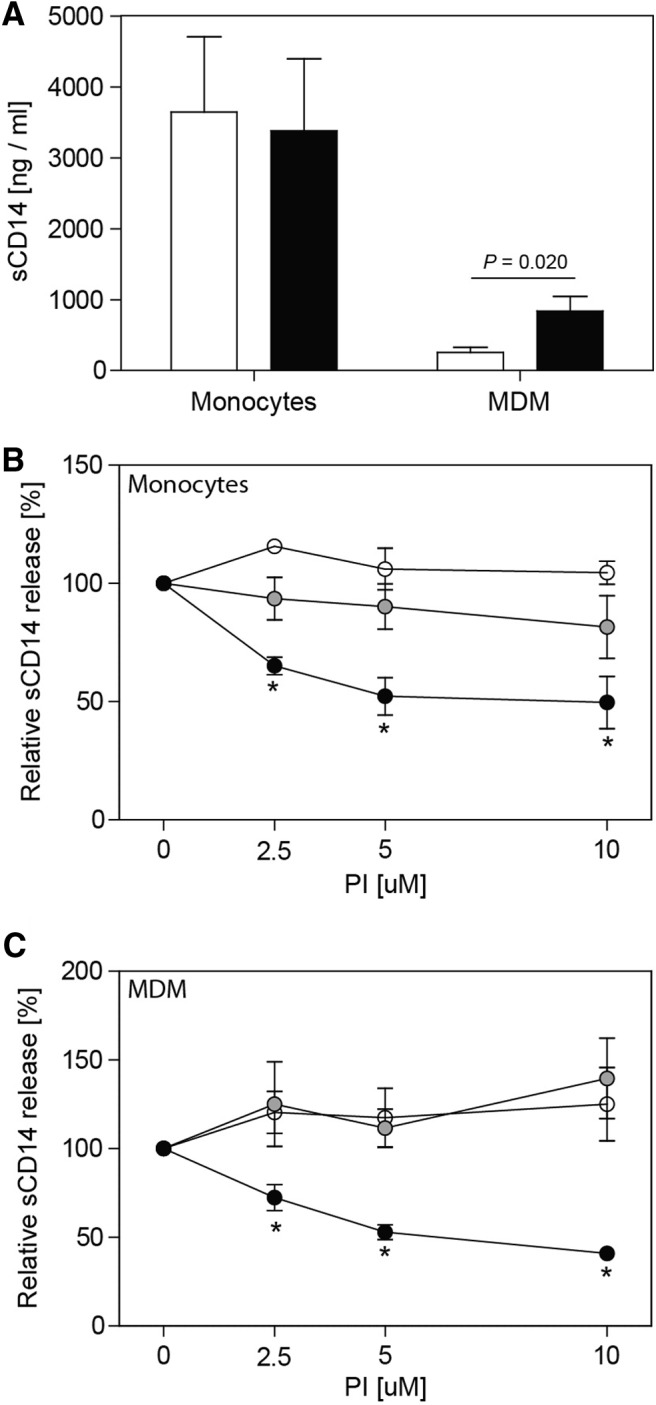
(A) Monocytes or macrophages were cultured in the presence (black bars) or absence (open bars) of 1 ng/ml LPS for 6 h, and supernatant sCD14 was measured by ELISA. (B) Monocytes were cultured with IDV (open circles), RTV (shaded circles), or NFV (black circles), and supernatant sCD14 was measured by ELISA. (C) MDMs were cultured with IDV (open circles), RTV (shaded circles), or NFV (black circles) for 30 min, followed by activation with 1 ng/ml LPS for 6 h, and supernatant sCD14 was measured by ELISA. Data represent mean ± sd from experiments with three donors. *P < 0.05.
Use of NFV in cART is independently associated with reduction in monocyte/macrophage activation in vivo
Monocyte/macrophage activation is evident in HIV-1-infected subjects before antiretroviral therapy, and sCD14 levels decline, albeit not to levels in uninfected individuals, with prolonged therapy (2 years) [2]. To determine whether NFV has anti-inflammatory effects in HIV-1-infected subjects, ΔsCD14 levels after 96 weeks of cART was evaluated. Those subjects receiving NFV, compared with IDV or RTV, had a markedly greater reduction in sCD14 (P<0.05; Fig. 9). Differences in age, LPS levels, VL, or CD4 T cell frequency could contribute to differences in ΔsCD14. To evaluate the independent effects of different PI regimens upon ΔsCD14 levels, a general linear model was assembled with PI group (IDV, RTV, or NFV), ΔLPS, age, CD4 T cell frequency, and viral outcomes as covariates. The model was well-supported (R2=0.539; P=0.001) with only the PI group (P=0.005) and ΔLPS (P=0.006) associated with ΔsCD14. Age (P=0.969), CD4 T cell frequency (P=0.759), or viral suppression (complete vs. incomplete as a nominal variable; P=0.448) was not associated with sCD14. The linear model was used to determine adjusted ΔsCD14 levels for each PI treatment group when adjusting for ΔLPS, age, CD4 T cell frequency, and viral success/failure (Table 1). Even when adjusting for covariates, reduction of sCD14 was greatest among subjects receiving NFV. A simple contrast test with NFV as the reference category showed that reduction of sCD14 was significantly greater for subjects receiving NFV compared with groups of subjects receiving IDV, RTV, or both IDV and RTV groups combined. A similar model substituted VL as a continuous variable in place of the nominal viral suppression variable (complete vs. incomplete) with no difference in results (data not shown). These data provide strong evidence that inclusion of NFV in cART reduces systemic monocyte/macrophage activation, independent of antiviral efficacy or effects on microbial translocation.
Figure 9. Greater reduction of plasma sCD14 is associated with inclusion of NFV in cART.
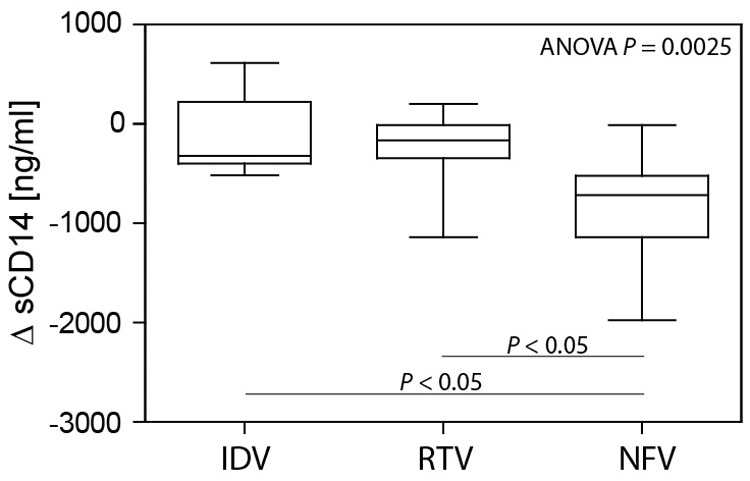
ΔsCD14, from immediately pretherapy to a time-point 96 weeks post-therapy, was compared among subjects receiving IDV (n=6), RTV (n=11), or NFV (n=14) using Kruskal-Wallis one-way ANOVA and Dunn's test.
Table 1. NFV Use Is Associated with Significantly Greater Reduction of Plasma sCD14 Levels.
| Treatment | Actual |
Adjusteda |
P valueb |
|||
|---|---|---|---|---|---|---|
| ΔsCD14 | ΔsCD14 | 95% Confidence interval | IDV versus NFV | RTV versus NFV | IDV + RTV versus NFV | |
| IDV (n=6) | −135.5 | −218 | −559, 123 | |||
| RTV (n=11) | −274.9 | −322 | −573, −70 | |||
| NFV (n=14) | −866.8 | −794 | −1,019, −569 | 0.008 | 0.009 | 0.008 |
Covariates in the model include ΔLPS, age, CD4 T cell frequency, and viral suppression (partial vs. complete).
Simple contrast.
DISCUSSION
Elucidation of HIV-1 protein structures and functions has led to a proliferation of anti-HIV drugs that target bioactivity of key viral enzymes involved in the HIV-1 life cycle. Yet, many HIV-1 drugs, including nRTI, non-nRTI, and PIs, demonstrate functional cross-reactivity with human enzymes. PIs, in particular, mediate pleiotropic effects on a vast array of cells, although most studies have focused on cancer cells, where NFV has as many as 17 unique effects on signaling proteins [38]. Indeed, as a result of its multifactorial anticancer properties, NFV has been used in at least six phase I/II clinical trials for cancer treatment [38]. Expansion of NFV use as an anti-inflammatory drug in HIV-1-infected individuals (in contrast to its initial role as an antiretroviral drug) may provide a means to reduce pathogenic innate immune activation associated with long-term morbidity/mortality. More significantly, delineation of signaling pathways disrupted by NFV may lead to development of new drugs, which mimic the anti-inflammatory effects of NFV with improved bioavailability and/or reduced toxicity.
Among the vast array of signaling proteins affected by NFV in cancer cells, a number of immune-related proteins, including inhibitory effects of NFV upon phosphorylation of AKT and STAT3, have been identified [38]. In hepatic cells, the HIV PIs, and NFV in particular, inhibit chymotrypsin activity of the 20S proteasome, leading to altered expression of several genes related to cell stress, lipid metabolism, and gluconeogenesis [47]. The 20S proteasome is the primary target of NFV in a signaling cascade that culminates with dephosphorylation of AKT [48] (Fig. 6). Inhibition of proteasome function by NFV [47] leads to accumulation of misfolded proteins that would normally be recycled through degradation by the proteasome but instead, activates the UPR. The UPR encompasses a range of enzyme activities, including activation of PP1, which dephosphorylates AKT [48]. Based on this discovery, we sought to determine the role of PP1 and PP2 in NFV-induced perturbations of macrophage activation and found that NFV treatment of MDM induced PP1 and PP2 activity and that PP2 activity is specifically required for anti-inflammatory effects of the drug. Related to induction of the UPR and resulting ER stress is induction of autophagy and/or apoptosis. In cancer cells, NFV, more potently than other HIV PIs, induces autophagy as a consequence of UPR-induced stress [42]. Interestingly, when autophagy was pharmacologically inhibited with 3-methyladenine, cancer cells underwent nonapoptotic cell death, indicating that autophagy serves to counteract cytotoxic effects of NFV [42, 70].
AKT activation, in innate immune cells, typically results in anti-inflammatory polarization characterized by reduced production of TNF and IL-12 and enhanced IL-10 production, whereas inhibition of AKT exacerbates proinflammatory activation [65–68]. Thus, it was not surprising that inhibition of PP1 by calyculin A to block the effect of NFV upon AKT failed to disrupt anti-inflammatory effects of NFV. In stark contrast, inhibition of PP2 by okadaic acid to block the effect of NFV upon MAPKs completely abrogated the anti-inflammatory effect of NFV. This finding implicates PP2-induced dephosphorylation of MAPK as the central mechanism by which NFV reduced LPS-induced macrophage activation, a result supported by the necessity of MAPK activation for transcription of cytokine genes in human macrophages [71]. Perhaps the most surprising finding of the current study was the precise selectivity of NFV for inhibition of specific cell-signaling pathways and functional outcomes, whereas other pathways/outcomes were left unaffected. Neither NF-κB signaling nor LPS-induced expression of cell-surface markers of activation was affected by NFV. Likewise, GSK3α/β and mTOR, both phosphorylated in response to LPS, were not affected by NFV. Such inhibition of selected signaling pathways may provide a means to specifically inhibit inflammation in vivo by targeting only certain outcomes such as expression of proinflammatory cytokines, an approach that may prove preferable to more broad immunosuppressive drugs such as corticosteroids.
Inhibition of LPS-induced MDM activation by NFV was unique, in that none of the other PIs tested inhibited TNF and IL-6 secretion within typical plasma levels of the drugs. NFV is a nonpeptidic, competitive inhibitor of HIV-1 protease similar to other HIV-1 PIs. Nonetheless, interactions between NFV and HIV-1 protease are unique, reflected in the occurrence of a NFV-specific drug-resistance point mutation at protease residue 30 (D30N) [72, 73]. We did observe that RTV, ATV, and TPV demonstrated mixed anti-inflammatory effects at superphysiologic doses in vitro. It is certainly plausible that free drug concentrations or effective intracellular drug concentrations in vivo are not accurately reflected by the concentrations of drug added in a cell-culture assay. Further study would be needed to directly assess biological activity of drugs in vivo, although reduced phosphorylation of AKT in PBMCs is observed in pancreatic cancer patients treated with NFV in a phase I clinical trial [74].
As chronic inflammation, even during suppressive antiretroviral therapy, is a manifestation of HIV-1 infection, assessment of therapy efficacy should include expanded evaluation of immunopathogenesis, including resolution of innate immune activation. For example, elevated plasma sCD14 provides a predictive marker for non-AIDS mortality in HIV-1-infected individuals [14, 69]. In the Strategies for Management of Anti-Retroviral Therapy study, HIV-1-infected subjects who fell in the third quartile of sCD14 levels had six times greater risk of death than those in the first sCD14 quartile [69]. Although our study was comprised by a limited number of individuals, use of NFV in cART reduced absolute sCD14 levels so that only one of 14 NFV-treated subjects fell within the third quartile. Our study differs from most studies of HIV-1-associated chronic inflammation in that the cohort consisted of perinatally infected children. It is possible that the long-term consequences of inflammation differ in children compared with adults. Furthermore, the effects of NFV may differ in adults as a result of differences in innate immunity or gut barrier functions, which could change the dynamics of microbial translocation and monocyte/macrophage activation as individuals age. Nonetheless, our cohort did span a large age range from 0.2 to 17 years of age, and when age was considered as a covariate in a model to assess factors contributing to ΔsCD14 levels, there was no significant association of age with outcome. Taken together, these findings provide a rationale for expanding clinical tests to include sCD14 as a measure of persistent innate activation in HIV-1 infection.
NFV demonstrates potent anti-inflammatory effects independent of its antiviral properties. This discovery may lead to novel applications for NFV or related compounds in the treatment of inflammatory illnesses, including but certainly not limited to HIV-1 infection. Although NFV is approved for use in HIV-1 combination therapy [75], Department of Health and Human Services guidelines for initial HIV-1 combination therapy do not recommend use of NFV as a result of inferior virological activity and high-incidence gastrointestinal adverse events [76]. We observed almost 50% inhibition of LPS-induced TNF and IL-6 at doses as low as 2.5 μM, and efficacy may be augmented by boosting strategies to improve bioavailability. As a supplemental medication, poor antiviral efficacy would not be a concern, as NFV could be used solely for anti-inflammatory effects in combination with more effective antiretroviral drugs. New trials are needed to assess tolerability and efficacy of supplementing current cART regimens with NFV for suppression of innate immune activation.
HIV PIs, especially early generation drugs, have been associated with a range of metabolic syndromes, including hyperlipidemia and insulin resistance [44]. As the metabolic consequences of NFV are intimately related to proteasome inhibition [47] and thus linked to anti-inflammatory effects of NFV, it may be challenging to achieve anti-inflammatory effects in vivo without detrimental side-effects. Identification of new signaling pathway targets that disrupt inflammatory activation of innate immune cells can provide a means to achieve selective therapeutic benefits, while minimizing detrimental effects of more pleiotropically active drugs such as NFV. Thus, new pharmacologic strategies for targeting MAPK signaling in macrophages may prove superior to NFV for reduction of chronic inflammation.
As our HIV-1-infected cohort included patients treated only with first-generation PIs, IDV, RTV, or NFV, assessment of PI effects was expanded ex vivo to include newer PIs, but only TPV displayed some anti-inflammatory activity. Investigation of all PIs used in current cART regimens, including combinations of PIs, should be performed to determine the best candidates for efficacy against pathogenic innate immune activation. For example, in a study of off-target effects of HIV PIs, amprenavir, saquinavir, and NFV inhibited AKT activation in vitro and in vivo using animal models; however, IDV and RTV failed to inhibit AKT activation [77].
In summary, NFV demonstrates unique anti-inflammatory effects upon monocytes and macrophages in vitro and in vivo. NFV may have additional clinical benefits, independent of antiviral efficacy, compared with other first-generation PIs. The most important finding is the specific pathway through which NFV mediates anti-inflammatory effects, which provides new targets for drug development. Finally, with expanded monitoring of innate immune biomarkers and inflammation-associated illnesses in HIV-1-infected patients, there is potential to improve outcomes by optimizing cART to target pathogenic inflammation, while still maintaining the primary clinical goal of suppressing viral replication and forestalling progression to AIDS.
ACKNOWLEDGMENTS
The following sources of funding were used. To M.A.W.: U.S. National Institutes of Health K22AI095015; Experimental Pathology Innovative grants from the University of Florida, Department of Pathology, Immunology and Laboratory Medicine; Thomas H. Maren Junior Investigator Postdoc Award from the University of Florida College of Medicine; and Laura McClamma fellowship for pediatric immune deficiency. To C.M.R.: Howard Hughes Medical Institutes Science for Life. To J.W.S.: U.S. National Institutes of Health R01AI47723 and Pediatric Clinical Research Center of All Children's Hospital and the University of South Florida, Maternal Child Health Bureau, R60 MC 00,003-01. To M.M.G.: U.S. National Institutes of Health R01DA031017 and R01AI28571; Stephany W. Holloway University Chair for AIDS Research; Center for Research in Pediatric Immune Deficiency; and Florida Center for AIDS Research. We thank Dr. Shannon Wallet for helpful comments on this manuscript.
Footnotes
- Δ
- change
- ATV
- atazanavir
- cART
- combination antiretroviral therapy
- IDV
- indinavir
- LAL
- limulus amoebocyte assay
- LPV
- lopinavir
- MSK2
- mitogen- and stress-activated protein kinase 2
- mTOR
- mammalian target of rapamycin
- NFV
- nelfinavir
- nRTI
- nucleoside RT inhibitor
- PI
- protease inhibitor
- pNPP
- p-nitrophenyl phosphate
- PP1/2
- protein phosphatase 1/2
- qPCR
- quantitative PCR
- RSK1
- ribosomal protein S6 kinase I
- RTV
- ritonavir
- sCD14
- soluble CD14
- TPV
- tipranavir
- UPR
- unfolded protein response
- VL
- viral load
AUTHORSHIP
M.A.W. conceived of the study, performed most experiments, analyzed data, and wrote the manuscript. C.M.R. performed cytokine ELISAs and contributed to the writing. J.C.W. determined sCD14 expression in freshly isolated monocytes and contributed to the writing. S.A. performed sCD14 ELISAs and contributed to the writing. G.L.G. performed NF-κB Western blots. B.G. managed the specimen database and provided clinical data for subjects. J.W.S. oversaw the project and contributed to the writing. M.M.G. was the principal investigator, oversaw the project, and contributed to the writing.
REFERENCES
- 1. Broder S. (2010) The development of antiretroviral therapy and its impact on the HIV-1/AIDS pandemic. Antiviral Res. 85, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wallet M., Rodriguez C., Yin L., Saporta S., Chinratanapisit S., Hou W., Sleasman J., Goodenow M. (2010) Microbial translocation induces persistent macrophage activation unrelated to HIV-1 levels or T-cell activation following therapy. AIDS 24, 1281–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brenchley J., Price D., Schacker T., Asher T., Silvestri G., Rao S., Kazzaz Z., Bornstein E., Lambotte O., Altmann D., Blazar B., Rodriguez B., Teixeira-Johnson L., Landay A., Martin J., Hecht F., Picker L., Lederman M., Deeks S., Douek D. (2006) Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 12, 1365–1371 [DOI] [PubMed] [Google Scholar]
- 4. Yin L., Rodriguez C., Hou W., Potter O., Caplan M., Goodenow M., Sleasman J. (2008) Antiretroviral therapy corrects HIV-1-induced expansion of CD8+ CD45RA+ CD2−) CD11a(bright) activated T cells. J. Allergy Clin. Immunol. 122, 166–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yin L., Kou Z., Rodriguez C., Hou W., Goodenow M., Sleasman J. (2009) Antiretroviral therapy restores diversity in the T-cell receptor Vβ repertoire of CD4 T-cell subpopulations among human immunodeficiency virus type 1-infected children and adolescents. Clin. Vaccine Immunol. 16, 1293–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deeks S. (2009) Immune dysfunction, inflammation, and accelerated aging in patients on antiretroviral therapy. Top. HIV Med. 17, 118–123 [PubMed] [Google Scholar]
- 7. Deeks S. G., Walker B. D. (2004) The immune response to AIDS virus infection: good, bad, or both? J. Clin. Invest. 113, 808–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hunt P., Brenchley J., Sinclair E., McCune J., Roland M., Page-Shafer K., Hsue P., Emu B., Krone M., Lampiris H., Douek D., Martin J., Deeks S. (2008) Relationship between T cell activation and CD4+ T cell count in HIV-seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. J. Infect. Dis. 197, 126–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hunt P. W., Martin J. N., Sinclair E., Bredt B., Hagos E., Lampiris H., Deeks S. G. (2003) T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J. Infect. Dis. 187, 1534–1543 [DOI] [PubMed] [Google Scholar]
- 10. Silvestri G., Munoz-Calleja C., Bagnarelli P., Piedimonte G., Clementi M., Montroni M. (1998) Early increase of CD4+ CD45RA+ and CD4+ CD95- cells with conserved repertoire induced by anti-retroviral therapy in HIV-infected patients. Clin. Exp. Immunol. 111, 3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Silvestri G., Munoz C., Butini L., Bagnarelli P., Montroni M. (1997) Changes in CD8 cell subpopulations induced by antiretroviral therapy in human immunodeficiency virus infected patients. Viral Immunol. 10, 207–212 [DOI] [PubMed] [Google Scholar]
- 12. Lien E., Aukrust P., Sundan A., Müller F., Frøland S., Espevik T. (1998) Elevated levels of serum-soluble CD14 in human immunodeficiency virus type 1 (HIV-1) infection: correlation to disease progression and clinical events. Blood 92, 2084–2092 [PubMed] [Google Scholar]
- 13. Nockher W., Bergmann L., Scherberich J. (1994) Increased soluble CD14 serum levels and altered CD14 expression of peripheral blood monocytes in HIV-infected patients. Clin. Exp. Immunol. 98, 369–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Papasavvas E., Azzoni L., Foulkes A., Violari A., Cotton M. F., Pistilli M., Reynolds G., Yin X., Glencross D. K., Stevens W. S., McIntyre J. A., Montaner L. J. (2011) Increased microbial translocation in ≤ 180 days old perinatally human immunodeficiency virus-positive infants as compared with human immunodeficiency virus-exposed uninfected infants of similar age. Pediatr. Infect. Dis. J. 30, 877–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ryan L., Zheng J., Brester M., Bohac D., Hahn F., Anderson J., Ratanasuwan W., Gendelman H., Swindells S. (2001) Plasma levels of soluble CD14 and tumor necrosis factor-α type II receptor correlate with cognitive dysfunction during human immunodeficiency virus type 1 infection. J. Infect. Dis. 184, 699–706 [DOI] [PubMed] [Google Scholar]
- 16. Burdo T., Ellis R., Fox H. (2008) Osteopontin is increased in HIV-associated dementia. J. Infect. Dis. 198, 715–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burdo T., Soulas C., Orzechowski K., Button J., Krishnan A., Sugimoto C., Alvarez X., Kuroda M., Williams K. (2010) Increased monocyte turnover from bone marrow correlates with severity of SIV encephalitis and CD163 levels in plasma. PLoS Pathog. 6, e1000842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tyor W., Glass J., Griffin J., Becker P., McArthur J., Bezman L., Griffin D. (1992) Cytokine expression in the brain during the acquired immunodeficiency syndrome. Ann. Neurol. 31, 349–360 [DOI] [PubMed] [Google Scholar]
- 19. Weis S., Neuhaus B., Mehraein P. (1994) Activation of microglia in HIV-1 infected brains is not dependent on the presence of HIV-1 antigens. Neuroreport 5, 1514–1516 [DOI] [PubMed] [Google Scholar]
- 20. Amor S., Puentes F., Baker D., van der Valk P. (2010) Inflammation in neurodegenerative diseases. Immunology 129, 154–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hakeem A., Bhatti S., Cilingiroglu M. (2010) The spectrum of atherosclerotic coronary artery disease in HIV patients. Curr. Atheroscler. Rep. 12, 119–124 [DOI] [PubMed] [Google Scholar]
- 22. Fichtenbaum C. (2010) Does antiretroviral therapy increase or decrease the risk of cardiovascular disease? Curr. HIV/AIDS Rep. 7, 92–98 [DOI] [PubMed] [Google Scholar]
- 23. Kuller L., Tracy R., Belloso W., De Wit S., Drummond F., Lane H., Ledergerber B., Lundgren J., Neuhaus J., Nixon D., Paton N., Neaton J., INSIGHT SMART Study Group (2008) Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 5, e203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Neuhaus J., Jacobs D. J., Baker J., Calmy A., Duprez D., La Rosa A., Kuller L., Pett S., Ristola M., Ross M., Shlipak M., Tracy R., Neaton J. (2010) Markers of inflammation, coagulation, and renal function are elevated in adults with HIV infection. J. Infect. Dis. 201, 1788–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cotter B. (2006) Endothelial dysfunction in HIV infection. Curr. HIV/AIDS Rep. 3, 126–131 [DOI] [PubMed] [Google Scholar]
- 26. Aggarwal B., Shishodia S., Sandur S., Pandey M., Sethi G. (2006) Inflammation and cancer: how hot is the link? Biochem. Pharmacol. 72, 1605–1621 [DOI] [PubMed] [Google Scholar]
- 27. Cadogan M., Dalgleish A. (2008) HIV induced AIDS and related cancers: chronic immune activation and future therapeutic strategies. Adv. Cancer Res. 101, 349–395 [DOI] [PubMed] [Google Scholar]
- 28. Rizza S. R., Tangalos E. G., McClees M. D., Strausbauch M. A., Targonski P. V., McKean D. J., Wettstein P. J., Badley A. D. (2008) Nelfinavir monotherapy increases naive T-cell numbers in HIV-negative healthy young adults. Front. Biosci. 13, 1605–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rizza S. A., Badley A. D. (2008) HIV protease inhibitors impact on apoptosis. Med. Chem. 4, 75–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chavan S., Kodoth S., Pahwa R., Pahwa S. (2001) The HIV protease inhibitor indinavir inhibits cell-cycle progression in vitro in lymphocytes of HIV-infected and uninfected individuals. Blood 98, 383–389 [DOI] [PubMed] [Google Scholar]
- 31. Sloand E. M., Kumar P. N., Kim S., Chaudhuri A., Weichold F. F., Young N. S. (1999) Human immunodeficiency virus type 1 protease inhibitor modulates activation of peripheral blood CD4(+) T cells and decreases their susceptibility to apoptosis in vitro and in vivo. Blood 94, 1021–1027 [PubMed] [Google Scholar]
- 32. Piccinini M., Rinaudo M., Anselmino A., Buccinnà B., Ramondetti C., Dematteis A., Ricotti E., Palmisano L., Mostert M., Tovo P. (2005) The HIV protease inhibitors nelfinavir and saquinavir, but not a variety of HIV reverse transcriptase inhibitors, adversely affect human proteasome function. Antivir. Ther. 10, 215–223 [PubMed] [Google Scholar]
- 33. Yang Y., Ikezoe T., Takeuchi T., Adachi Y., Ohtsuki Y., Takeuchi S., Koeffler H., Taguchi H. (2005) HIV-1 protease inhibitor induces growth arrest and apoptosis of human prostate cancer LNCaP cells in vitro and in vivo in conjunction with blockade of androgen receptor STAT3 and AKT signaling. Cancer Sci. 96, 425–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bruning A., Vogel M., Mylonas I., Friese K., Burges A. (2011) Bortezomib targets the caspase-like proteasome activity in cervical cancer cells, triggering apoptosis that can be enhanced by nelfinavir. Curr. Cancer Drug Targets 11, 799–809 [DOI] [PubMed] [Google Scholar]
- 35. Hudon S. E., Coffinier C., Michaelis S., Fong L. G., Young S. G., Hrycyna C. A. (2008) HIV-protease inhibitors block the enzymatic activity of purified Ste24p. Biochem. Biophys. Res. Commun. 374, 365–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Toschi E., Sgadari C., Malavasi L., Bacigalupo I., Chiozzini C., Carlei D., Compagnoni D., Bellino S., Bugarini R., Falchi M., Palladino C., Leone P., Barillari G., Monini P., Ensoli B. (2011) Human immunodeficiency virus protease inhibitors reduce the growth of human tumors via a proteasome-independent block of angiogenesis and matrix metalloproteinases. Int. J. Cancer 128, 82–93 [DOI] [PubMed] [Google Scholar]
- 37. André P., Groettrup M., Klenerman P., de Giuli R., Booth B. L., Cerundolo V., Bonneville M., Jotereau F., Zinkernagel R. M., Lotteau V. (1998) An inhibitor of HIV-1 protease modulates proteasome activity, antigen presentation, and T cell responses. Proc. Natl. Acad. Sci. USA 95, 13120–13124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chow W. A., Jiang C., Guan M. (2009) Anti-HIV drugs for cancer therapeutics: back to the future? Lancet Oncol. 10, 61–71 [DOI] [PubMed] [Google Scholar]
- 39. Lagathu C., Eustace B., Prot M., Frantz D., Gu Y., Bastard J. P., Maachi M., Azoulay S., Briggs M., Caron M., Capeau J. (2007) Some HIV antiretrovirals increase oxidative stress and alter chemokine, cytokine or adiponectin production in human adipocytes and macrophages. Antivir. Ther. 12, 489–500 [PubMed] [Google Scholar]
- 40. Leroyer S., Vatier C., Kadiri S., Quette J., Chapron C., Capeau J., Antoine B. (2011) Glyceroneogenesis is inhibited through HIV protease inhibitor-induced inflammation in human subcutaneous but not visceral adipose tissue. J. Lipid Res. 52, 207–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Malizia A. P., Cotter E., Chew N., Powderly W. G., Doran P. P. (2007) HIV protease inhibitors selectively induce gene expression alterations associated with reduced calcium deposition in primary human osteoblasts. AIDS Res. Hum. Retroviruses 23, 243–250 [DOI] [PubMed] [Google Scholar]
- 42. Gills J., Lopiccolo J., Tsurutani J., Shoemaker R., Best C., Abu-Asab M., Borojerdi J., Warfel N., Gardner E., Danish M., Hollander M., Kawabata S., Tsokos M., Figg W., Steeg P., Dennis P. (2007) Nelfinavir, a lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin. Cancer Res. 13, 5183–5194 [DOI] [PubMed] [Google Scholar]
- 43. Woerle H. J., Mariuz P. R., Meyer C., Reichman R. C., Popa E. M., Dostou J. M., Welle S. L., Gerich J. E. (2003) Mechanisms for the deterioration in glucose tolerance associated with HIV protease inhibitor regimens. Diabetes 52, 918–925 [DOI] [PubMed] [Google Scholar]
- 44. Calza L., Manfredi R., Chiodo F. (2004) Dyslipidaemia associated with antiretroviral therapy in HIV-infected patients. J. Antimicrob. Chemother. 53, 10–14 [DOI] [PubMed] [Google Scholar]
- 45. Purnell J. Q., Zambon A., Knopp R. H., Pizzuti D. J., Achari R., Leonard J. M., Locke C., Brunzell J. D. (2000) Effect of ritonavir on lipids and post-heparin lipase activities in normal subjects. AIDS 14, 51–57 [DOI] [PubMed] [Google Scholar]
- 46. Noor M. A., Parker R. A., O'Mara E., Grasela D. M., Currie A., Hodder S. L., Fiedorek F. T., Haas D. W. (2004) The effects of HIV protease inhibitors atazanavir and lopinavir/ritonavir on insulin sensitivity in HIV-seronegative healthy adults. AIDS 18, 2137–2144 [DOI] [PubMed] [Google Scholar]
- 47. Parker R. A., Flint O. P., Mulvey R., Elosua C., Wang F., Fenderson W., Wang S., Yang W. P., Noor M. A. (2005) Endoplasmic reticulum stress links dyslipidemia to inhibition of proteasome activity and glucose transport by HIV protease inhibitors. Mol. Pharmacol. 67, 1909–1919 [DOI] [PubMed] [Google Scholar]
- 48. Gupta A. K., Li B., Cerniglia G. J., Ahmed M. S., Hahn S. M., Maity A. (2007) The HIV protease inhibitor nelfinavir downregulates Akt phosphorylation by inhibiting proteasomal activity and inducing the unfolded protein response. Neoplasia 9, 271–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gendelman H., Orenstein J., Martin M., Ferrua C., Mitra R., Phipps T., Wahl L., Lane H., Fauci A., Burke D. (1988) Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J. Exp. Med. 167, 1428–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huyer G., Liu S., Kelly J., Moffat J., Payette P., Kennedy B., Tsaprailis G., Gresser M. J., Ramachandran C. (1997) Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J. Biol. Chem. 272, 843–851 [DOI] [PubMed] [Google Scholar]
- 51. Gordon J. A. (1991) Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods Enzymol. 201, 477–482 [DOI] [PubMed] [Google Scholar]
- 52. Ghaffari G., Passalacqua D., Caicedo J., Goodenow M., Sleasman J. (2004) Two-year clinical and immune outcomes in human immunodeficiency virus-infected children who reconstitute CD4 T cells without control of viral replication after combination antiretroviral therapy. Pediatrics 114, e604–e11 [DOI] [PubMed] [Google Scholar]
- 53. Perez E., Rose S., Peyser B., Lamers S., Burkhardt B., Dunn B., Hutson A., Sleasman J., Goodenow M. (2001) Human immunodeficiency virus type 1 protease genotype predicts immune and viral responses to combination therapy with protease inhibitors (PIs) in PI-naive patients. J. Infect. Dis. 183, 579–588 [DOI] [PubMed] [Google Scholar]
- 54. Ho S., Perez E., Rose S., Coman R., Lowe A., Hou W., Ma C., Lawrence R., Dunn B., Sleasman J., Goodenow M. (2009) Genetic determinants in HIV-1 Gag and Env V3 are related to viral response to combination antiretroviral therapy with a protease inhibitor. AIDS 23, 1631–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Weinberg A., Zhang L., Brown D., Erice A., Polsky B., Hirsch M., Owens S., Lamb K. (2000) Viability and functional activity of cryopreserved mononuclear cells. Clin. Diagn. Lab. Immunol. 7, 714–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Weinberg A., Song L., Wilkening C., Sevin A., Blais B., Louzao R., Stein D., Defechereux P., Durand D., Riedel E., Raftery N., Jesser R., Brown B., Keller M., Dickover R., McFarland E., Fenton T. (2009) Optimization and limitations of use of cryopreserved peripheral blood mononuclear cells for functional and phenotypic T-cell characterization. Clin. Vaccine Immunol. 16, 1176–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. (2009) ACTG/IMPAACT Laboratory Manual. HIV/AIDS Network Coordination, Seattle, WA, USA [Google Scholar]
- 58. Pellegrin I., Breilh D., Montestruc F., Caumont A., Garrigue I., Morlat P., Le Camus C., Saux M., Fleury H., Pellegrin J. (2002) Virologic response to nelfinavir-based regimens: pharmacokinetics and drug resistance mutations (VIRAPHAR study). AIDS 16, 1331–1340 [DOI] [PubMed] [Google Scholar]
- 59. Dickinson L., Khoo S., Back D. (2008) Differences in the pharmacokinetics of protease inhibitors between healthy volunteers and HIV-infected persons. Curr. Opin. HIV AIDS 3, 296–305 [DOI] [PubMed] [Google Scholar]
- 60. MacGregor T., Sabo J., Norris S., Johnson P., Galitz L., McCallister S. (2004) Pharmacokinetic characterization of different dose combinations of coadministered tipranavir and ritonavir in healthy volunteers. HIV Clin. Trials 5, 371–382 [DOI] [PubMed] [Google Scholar]
- 61. Hsu A., Granneman G., Witt G., Locke C., Denissen J., Molla A., Valdes J., Smith J., Erdman K., Lyons N., Niu P., Decourt J., Fourtillan J., Girault J., Leonard J. (1997) Multiple-dose pharmacokinetics of ritonavir in human immunodeficiency virus-infected subjects. Antimicrob. Agents Chemother. 41, 898–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stein D., Fish D., Bilello J., Preston S., Martineau G., Drusano G. (1996) A 24-week open-label phase I/II evaluation of the HIV protease inhibitor MK-639 (indinavir). AIDS 10, 485–492 [DOI] [PubMed] [Google Scholar]
- 63. Aggarwal B. (2003) Signalling pathways of the TNF superfamily: a double-edged sword. Nat. Rev. Immunol. 3, 745–756 [DOI] [PubMed] [Google Scholar]
- 64. Kyriakis J., Avruch J. (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 81, 807–869 [DOI] [PubMed] [Google Scholar]
- 65. Weichhart T., Säemann M. D. (2008) The PI3K/Akt/mTOR pathway in innate immune cells: emerging therapeutic applications. Ann. Rheum. Dis. 67 (Suppl. 3), iii70–iii74 [DOI] [PubMed] [Google Scholar]
- 66. Hazeki K., Nigorikawa K., Hazeki O. (2007) Role of phosphoinositide 3-kinase in innate immunity. Biol. Pharm. Bull. 30, 1617–1623 [DOI] [PubMed] [Google Scholar]
- 67. Bhattacharyya S., Sen P., Wallet M., Long B., Baldwin A. J., Tisch R. (2004) Immunoregulation of dendritic cells by IL-10 is mediated through suppression of the PI3K/Akt pathway and of IκB kinase activity. Blood 104, 1100–1109 [DOI] [PubMed] [Google Scholar]
- 68. Sen P., Wallet M., Yi Z., Huang Y., Henderson M., Mathews C., Earp H., Matsushima G., Baldwin A. J., Tisch R. (2007) Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-κB activation in dendritic cells. Blood 109, 653–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sandler N. G., Wand H., Roque A., Law M., Nason M. C., Nixon D. E., Pedersen C., Ruxrungtham K., Lewin S. R., Emery S., Neaton J. D., Brenchley J. M., Deeks S. G., Sereti I., Douek D. C., INSIGHT SMART Study Group (2011) Plasma levels of soluble CD14 independently predict mortality in HIV infection. J. Infect. Dis. 203, 780–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gills J. J., Lopiccolo J., Dennis P. A. (2008) Nelfinavir, a new anti-cancer drug with pleiotropic effects and many paths to autophagy. Autophagy 4, 107–109 [DOI] [PubMed] [Google Scholar]
- 71. Carter A. B., Monick M. M., Hunninghake G. W. (1999) Both Erk and p38 kinases are necessary for cytokine gene transcription. Am. J. Respir. Cell Mol. Biol. 20, 751–758 [DOI] [PubMed] [Google Scholar]
- 72. Clemente J., Hemrajani R., Blum L., Goodenow M., Dunn B. (2003) Secondary mutations M36I and A71V in the human immunodeficiency virus type 1 protease can provide an advantage for the emergence of the primary mutation D30N. Biochemistry 42, 15029–15035 [DOI] [PubMed] [Google Scholar]
- 73. Markowitz M., Conant M., Hurley A., Schluger R., Duran M., Peterkin J., Chapman S., Patick A., Hendricks A., Yuen G., Hoskins W., Clendeninn N., Ho D. (1998) A preliminary evaluation of nelfinavir mesylate, an inhibitor of human immunodeficiency virus (HIV)-1 protease, to treat HIV infection. J. Infect. Dis. 177, 1533–1540 [DOI] [PubMed] [Google Scholar]
- 74. Brunner T. B., Geiger M., Grabenbauer G. G., Lang-Welzenbach M., Mantoni T. S., Cavallaro A., Sauer R., Hohenberger W., McKenna W. G. (2008) Phase I trial of the human immunodeficiency virus protease inhibitor nelfinavir and chemoradiation for locally advanced pancreatic cancer. J. Clin. Oncol. 26, 2699–2706 [DOI] [PubMed] [Google Scholar]
- 75. Perry C., Frampton J., McCormack P., Siddiqui M., Cvetković R. (2005) Nelfinavir: a review of its use in the management of HIV infection. Drugs 65, 2209–2244 [DOI] [PubMed] [Google Scholar]
- 76. (2011) Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. U.S. Department of Health and Human Services, Washington, DC, USA, from http://www.aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf [Google Scholar]
- 77. Gupta A. K., Cerniglia G. J., Mick R., McKenna W. G., Muschel R. J. (2005) HIV protease inhibitors block Akt signaling and radiosensitize tumor cells both in vitro and in vivo. Cancer Res. 65, 8256–8265 [DOI] [PubMed] [Google Scholar]