

Abstract
In injured neurons, “leaky” voltage-gated sodium channels (Nav) underlie dysfunctional excitability that ranges from spontaneous subthreshold oscillations (STO), to ectopic (sometimes paroxysmal) excitation, to depolarizing block. In recombinant systems, mechanical injury to Nav1.6-rich membranes causes cytoplasmic Na+-loading and “Nav-CLS”, i.e., coupled left-(hyperpolarizing)-shift of Nav activation and availability. Metabolic injury of hippocampal neurons (epileptic discharge) results in comparable impairment: left-shifted activation and availability and hence left-shifted INa-window. A recent computation study revealed that CLS-based INa-window left-shift dissipates ion gradients and impairs excitability. Here, via dynamical analyses, we focus on sustained excitability patterns in mildly damaged nodes, in particular with more realistic Gaussian-distributed Nav-CLS to mimic “smeared” injury intensity. Since our interest is axons that might survive injury, pumps (sine qua non for live axons) are included. In some simulations, pump efficacy and system volumes are varied. Impacts of current noise inputs are also characterized. The diverse modes of spontaneous rhythmic activity evident in these scenarios are studied using bifurcation analysis. For “mild CLS injury”, a prominent feature is slow pump/leak-mediated EIon oscillations. These slow oscillations yield dynamic firing thresholds that underlie complex voltage STO and bursting behaviors. Thus, Nav-CLS, a biophysically justified mode of injury, in parallel with functioning pumps, robustly engenders an emergent slow process that triggers a plethora of pathological excitability patterns. This minimalist “device” could have physiological analogs. At first nodes of Ranvier and at nociceptors, e.g., localized lipid-tuning that modulated Nav midpoints could produce Nav-CLS, as could co-expression of appropriately differing Nav isoforms.
Author Summary
Nerve cells damaged by trauma, stroke, epilepsy, inflammatory conditions etc, have chronically leaky sodium channels that eventually kill. The usual job of sodium channels is to make brief voltage signals –action potentials– for long distance propagation. After sodium channels open to generate action potentials, sodium pumps work harder to re-establish the intracellular/extracellular sodium imbalance that is, literally, the neuron's battery for firing action potentials. Wherever tissue damage renders membranes overly fluid, we hypothesize, sodium channels become chronically leaky. Our experimental findings justify this. In fluidized membranes, sodium channel voltage sensors respond too easily, letting channels spend too much time open. Channels leak, pumps respond. By mathematical modeling, we show that in damaged channel-rich membranes the continual pump/leak counterplay would trigger the kinds of bizarre intermittent action potential bursts typical of injured neurons. Arising ectopically from injury regions, such neuropathic firing is unrelated to events in the external world. Drugs that can silence these deleterious electrical barrages without blocking healthy action potentials are needed. If fluidized membranes house the problematic leaky sodium channels, then drug side effects could be diminished by using drugs that accumulate most avidly into fluidized membranes, and that bind their targets with highest affinity there.
Introduction
In any healthy sodium channel (Nav)-rich plasma-membrane, the bilayer is a far-from-equilibrium nanostructure that degrades wherever mechanical or chemical insult causes the inner leaflet to detach from adherent cortical cytoskeleton [1]–[4]. Severe insults cause readily-visualized rounded blebs of disordered, fluidized bilayer (see Figure 1), while milder damage causes intermediate degrees of disordered “bleb-like” injury [2], [5], [6]. Though many membrane proteins would be affected, Nav channels are overwhelmingly the key players in many excitable membranes, including nodes of Ranvier [1]. Positive-feedback Nav currents yield action potentials (APs) that dissipate Na/K gradients maintained by Na/K-ATPases so not surprisingly, membrane-damaging conditions (e.g., trauma, ischemia, muscular dystrophy) that render Nav channels chronically leaky trigger excitotoxic cellular demise [3], [7].
Figure 1. Schematic of a mechanically-injured node of Ranvier.

depicted with a mix of intact-looking and severely-blebbed axolemma (as labelled) such as seen in transmission electromicrographs of stretch-injured optic nerve nodes [2]. In pipette aspiration bleb injury, the cortical actomyosin-spectrin skeleton progressively detaches [6]. Our model considers a node as one equipotential compartment in which actual spatial arrangements of pumps and channels are irrelevant. However, the fraction of Nav channels in the injured portion of the membrane, along with the severity of their gating abnormality, are model parameters.
Pipette aspiration electrophysiology studies on Nav1.6-rich membranes (patch-clamped Xenopus oocyte patches) [8] showed that the aspiration-induced bleb-like injury causes “left-shift Nav-leak”; progressive aspiration damage irreversibly and progressively shifts the voltage midpoint of gNa(V) in the hyperpolarizing direction. When maximal disorder is reached, the irreversible shifting process “saturates” and any further aspiration-induced left-shifts are reversible (see also [9], [10]). Nav-availability depends on fast inactivation, a transition limited by fast activation [10]. Accordingly, damage causes equal-magnitude left-shifts for gNa(V) and availability(V), thereby left-shifting the window conductance. The shifted steady-state (window) current constitutes a Nav-leak or abnormal “subthreshold persistent current”, the entity we describe as coupled left-shift (CLS) Nav-leak [11]. Pipette aspiration also causes irreversible Nav-CLS for Nav1.4 and Nav1.7 [12], [13] (preparation: oocytes) and Nav1.5 channels [14] (preparation: HEK cells).
In hippocampal neurons, a Nav-CLS based window current left-shift occurs following metabolic insult from prolonged epilepsy-like stimulation [15]. Hippocampal neurons exposed to the industrial compound, melamine, also show Nav-CLS and exhibit hyperexcitability [16]. In neurons and muscle fibers, left-shift of Nav availability (i.e., steady-state inactivation) [17] is reported for many damaging and/or toxic conditions (see Table 1 of [3]) and for membrane fluidizing conditions [18]; fast activation also becomes left-shifted in muscles (e.g. [19]) and neurons (e.g. [20]). As predicted when membrane injury causes Nav-CLS [11], sick excitable cell pathologies range from hyperexcitability through ectopic excitation to depolarizing block. Sucrose-gap voltage clamp of nodes of Ranvier subjected to ischemic and other insults could, we have suggested [3], be used to test to what extent the initial Nav-leak of injured Nav-rich native membranes is explained by Nav-CLS. Injured-bilayer ICLS-window could help explain [3] why, invariably, clinical Nav inhibitors protective against excitotoxicity are lipophilic [21]; moreover, if bilayer structure matters, then retuning therapeutic strategies to better target channels in damaged bilayer could pay dividends [3].
Table 1. Parameters for node of Ranvier with pump.
| Membrane capacitance | C = 1 µF/cm2 |
| Maximal Na+conductance |
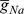 = 120 mS/cm2 = 120 mS/cm2
|
| Maximal K+ conductance |
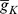 = 36 mS/cm2 = 36 mS/cm2
|
| Faraday constant | F = 96485.3399 |
| Temperature | T = 293.15 K |
| Volume of inside compartment | Voli = 10−15 m3 |
| Volume of outside compartment | Volo = 10−15 m3 |
| Surface area | A = 6×10−8 cm2 |
| K+ pump leak conductance |
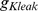 = 0.1 mS/cm2 = 0.1 mS/cm2
|
| Na+ pump leak conductance |
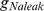 = 0.2 mS/cm2 = 0.2 mS/cm2
|
| Leak conductance |
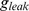 = 0.5 mS/cm2 = 0.5 mS/cm2
|
| Leak reversal potential |
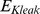 = −59.9 mV = −59.9 mV |
| Maximum pump current range used |
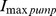 = 15 to 156 µA/cm2 = 15 to 156 µA/cm2
|
| (standard value, except as noted) | = 95 µA/cm2 |
| Pump K+-dissociation constant |
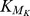 = 3.5 mM = 3.5 mM |
| Pump Na+-dissociation constant |
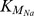 = 10 mM = 10 mM |
Here, our goal is to understand in dynamical terms the mechanisms of abnormal steady-state excitability in membrane modeled with CLS-type injuries of various intensities. In particular, we wish to determine if – in conjunction with axolemmal Na/K pumps – CLS-injury predicts forms of abnormal excitability reported for “mildly” injured excitable cells, i.e., cells that, with appropriate remediation, could be salvaged. In healthy unstimulated axons, quiescence is the norm and spontaneous firing is neuropathic and ectopic.
Several recent studies involving simulation and dynamical modeling addressed abnormal or paroxysmal discharge in injured neurons. Injured DRG neuron simulations [22] showed that subthreshold oscillations (STOs) resembling ones recorded in vivo and relevant to neuropathic pain can be generated via depolarizing stimuli to a membrane with three distinctive gNas. Importantly, though, for STOs, the slower two gNas required first order, rather than the usual third order activation kinetics. We address this later. Similarly, for two Nav-type and two Kv-type conductances, Choi and Waxman [23] showed responses to current injection (mimicking sensory input) that included STOs with the spikes; there too, for STOs to occur, one of the gNas required first-order activation kinetics.
STOs typically occur in conjunction with bursts of spikes, appearing in healthy neurons, (e.g., entorhinal cortex [24]) as small Vm oscillations for a discrete range of conditions. Dynamically-speaking, STOs emerge where there is bistability between tonic firing and quiescence [25]. For allodynia-related neuropathic pain, a dynamical study of firing patterns showed EAnion changes having a key stability role [26]. Firing abnormalities of demyelinated axon were mimicked in a dynamical modeling study with fixed Nernst potentials and gNa/gleak ratios varied [27]. Regarding paroxysmal spiking, the same group [28] explored initiation and termination of afterdischarges and spontaneous activity; there, a non-inactivating gNa induced bistability and to mimic [ion] homeostasis following transient Na+-loading, an exponential decay provided a slow relaxation toward initial [Na +]intracellular. Käger et al [29] investigated epilepsy-like spontaneous bursting patterns and elevated [K +]extracellular, and Barreto, Cressman and colleagues [30], [31] performed a dynamical study for oscillating [ion]s connected with epileptic seizures.
Our Nav-CLS-based model of membrane injury uses a Hodgkin-Huxley axon without or with a Na/K pump as developed previously in [11]. That model focused on transitory responses to abrupt CLS “injury” (left-shift intensity and the fraction of total gNa affected were varied) and revealed how CLS-injury affects Vm(t) and ion homeostasis. Once the injured system (no pumps) stabilized, depolarizing current was applied, and based on spontaneous and stimulated behaviors, Nav-CLS injury activity was classified under various regimes (Figures 5 and 9 in [11]). CLS-injury caused hyperexcitability, ectopic excitation or depolarizing block, and interfered with saltatory propagation of normal action potential (AP) traffic. Varying extracellular volume can significantly alter the quantitative measures of the bursting, consistent with a major role for pump activity. Unexpectedly, it was discovered that Nav-CLS in conjunction with pumps could generate STOs. Since injured neuron neuropathic pain discharge is characterized by diverse STO phenomena, we undertook the dynamical analysis of Nav-CLS injury presented here.
Figure 5. Burst dynamics explained with three-dimensional Vm trajectories and bifurcation diagrams.

(for bursts in Figure 4A with Imaxpump = 95 µA/cm2). Note: x-axis scales are different is each plot. (A) The first burst of spikes in Figure 4A plotted as a function of time are now plotted in 3-D as a function of ENa and EK (blue arrow: direction of Vm trajectory). (B) Bifurcation diagram for fixed EK and Ipump as per Figure 4A pink star; ENa is the slowly varying parameter. Lines and points are labeled (see also abbreviation list) and have the same meanings in (C). For ENa at its Figure 4A pink star value (40.2 mV), the only stable solution is a periodic orbit (pink oval), with each cycle corresponding to a spike (in a burst). The oval-loop symbolizes one cycle of Vm oscillation at fixed ENa. During a burst the ENa decline shifts orbits leftward into the bistability regime (pink area) where there exist two stable solutions: a limit cycle and a stable fixed point. (C) Bifurcation diagram for EK and Ipump fixed as per Figure 4A green dot. For ENa at its Figure 4A green dot value (37.27 mV), the system (large green dot) is within the bistability regime (green area). Vm, attracted by this fixed point, has STOs (drawn as the green loop) until ENa increases and superthreshold-oscillations (spikes) return. The PD region corresponds to period-doubling bifurcations. (D) Two-parameter phase diagram for EK and ENa. Pink solid and dashed curves represent LP (saddle-node bifurcation) and HB, respectively, when Ipump is fixed as in B. The green solid, dashed, and dash-dotted curves represent LP, HB and PD, respectively, when Ipump is fixed as in C. With varying Ipump the bistability regime shifts from the pink to the green area (the zone with both colors is the overlap of these two areas). The gray area between two green dash-dotted curves is a zone with PD bifurcations. The black loop shows EK and ENa orbits during a burst.
Figure 9. Increased Imaxpump shortens bursting periods (BP) and burst duration (BD).

(A)–(E) Five values of Imaxpump as labeled elicited activities as shown with LS: Gaussian distribution, 2.5±1 mV (steady-state values in the absence of CLS injury are listed in Table 2). Note the different time scales in each panel. For a bursting node, Ipump is periodic with a period equal to BP. Inset in (A) shows the effect of injecting a 12 µA/cm2 stimulating current. (F) For direct comparison of BPs and burst durations, Ipump traces from panels A–E (color coding preserved) are plotted together.
We model acute injury: even in egregiously blebbed native membranes, Nav channels retain their ability to respond to voltage [32], [33]. Keeping gNa-max constant corresponds to an absence of de novo Nav channel expression or membrane remodelling. Immunochemistry confirms an abundance of axolemmal Nav channels at damaged nerve-end neuromas, sites considered to be loci of ectopic excitation [34]. Our CLS-injury model is consistent with present views of voltage-gated channel structure/function [35]–[37]. Nav-CLS injury in situ would likely be of non-uniform intensity, a point we address computationally in several ways. Except for some instances of modified pump intensity, “impairment” refers exclusively to Nav-CLS.
Na/K pump activity is sensitive to ATP levels, injury and bilayer lipid characteristics [38]–[40], but how mild membrane injury would affect pump rates is unclear. So here we vary the maximal pump rate to explore its effect on tonic firing and bursting (in this paper, both firing patterns are spontaneous).
The diverse spontaneous steady-state rhythmic activity patterns that became evident in our injury scenarios are studied using bifurcation analyses. For mild CLS injury, a prominent and robust feature was the emergent slow process of pump/leak-mediated EIon oscillations [11]. As we will illustrate, these slowly oscillating EIon values cause time-dependent changes in the firing thresholds, which in turn will lead to complex spontaneous STO and bursting behaviors; the complexity of the observed patterns is further exacerbated by current noise.
In summary, we study, via dynamical analysis, a simple, robust, and biophysically explicable model of mild axonal injury in which pumps remain functional, i.e., we examine a collection of mild Nav-CLS injury scenarios. The major finding is that interactions between the window current based Nav-leak and Na/K pump activity engender slow oscillations of the Na+ and K+ driving forces that spontaneously trigger a plethora of known neuropathological excitability patterns. The following results are based on simulations of a Nav-CLS model of injured node described in the Methods section and in Table 1 which is a list of parameters.
Results
Membrane potential oscillations in CLS injured nodes for fixed Eion
Simulated nodes with Nav-CLS injury were shown [11] to exhibit activity regimes that include tonic and burst firing, plus a quiescent steady state. In this section we explore the node with simulated more nuanced versions of Nav-CLS injury and find that rhythmic firing combined with small-amplitude Vm oscillations are commonplace. We start with cases of constant [Ion] (pump turned off), approximating axonal situations over brief times. We choose cases that provide snapshots of dynamics at selected fixed reversal potentials that could occur at different stages of injury. For fixed reversal potentials (EIon) we take three sets of values corresponding to different transmembrane Na+ and K+ gradients: (i) ENa = 50 mV, EK = −77 mV; (ii) ENa = 42 mV, EK = −77 mV when initial injury has elicited a change in INa thence [Na +]s; and (iii) ENa = 42 mV, EK = −71 mV. In this latter case, IKpump, because of its dependence on [Na +]s, changes, which results in EK changes.
Nav-CLS intensities induced by membrane damage would vary within and between axons, as suggested in Figure 8 Aiii in [8]. To reflect this, three sets of LSi and fi are applied. To incorporate the idea of “smeared-out LS”, the fraction fi of the population are given a Gaussian distribution of LS with mean±SD of 1.3±0.4 mV, 8±1 mV and 15±1 mV. The numerical results are plotted in Figure 2. For the first two EIon pairs (i) and (ii) (left and middle columns), Vi dynamics in the injured node vary from quiescence to repetitive spiking, with the firing rate increasing and the spike amplitude decreasing as mean(LS) increases. However, for EIon pair (iii) (right column), this node shows changes from STF at high amplitude (again, with increasing firing rate and decreasing amplitude) to a very low amplitude oscillation (Figure 2I) that shares features of subthreshold oscillations (STOs).
Figure 8. Classes of Nav-CLS-induced activities.

(A) Nav-CLS of Gaussian distributions with mean±SD(LS) = [0.5±0.2, 1±0.4, 2.5±1.0, 5±5, 10±5, 20±5, 27±5, 30±5, 20±10]mV. (B)–(I) various activities of a node are demonstrated for increased injury (i.e. > mean(LS)): quiescence (B), burst (C,D), tonic firing (E,F,G), burst (H) and quiescence (I). Insets in (B) and (I) show the effect of injecting a 12 µA/cm2 stimulating current. (J) Doubling the SD(LS) in G from 5 to 10 mV slightly increases the tonic firing rate. (K) Instantaneous firing rate (inverse of the interval between two spikes) for tonic or within-burst spikes, versus mean(LS), corresponding to firing patterns C–H (see color coding). (L) Spike (tonic or intraburst) voltage excursions begin to decrease for mean(LS) >10 mV.
Figure 2. Nav-CLS induced spontaneous activities of injured node: steady state with fixed EIon, tonic spiking, tonic subthreshold oscillations (STOs).

By setting Imaxpump, gNaleak, and gKleak to zero and eliminating Eqs (11) and (12), the EIon are artificially maintained at fixed values. Three sets of values are considered: left column, ENa = 50 mV, EK = −77 mV; middle column: ENa = 42 mV, EK = −77 mV; right column: ENa = 42 mV, EK = −71 mV. For the first 3 rows, Nav-CLS have Gaussian distributions (mean±SD): (A,B,C) 1.3±0.4 mV; (D,E,F) 8±1 mV; (G,H,I) 15±1 mV. For the last rows,(J,K,L) bifurcation diagrams (solution of Vm in terms of LS) are plotted, and there, for computational tractability, single gNa populations (i.e. f = 1, no Gaussian “smears”) are used. As labeled, the solid line, dashed line, filled dots, open dots and “HB” respectively denote: stable fixed point (i.e. resting potential (RP)), unstable fixed point, stable limit cycle (SLC, i.e., tonically firing APs), unstable limit cycle (ULC) and Hopf bifurcation point (HB). When ENa alone changes (J to K), the bifurcation structure shows only slight changes in the amplitude of SLC and the locations of subcritical HB on both ends. When EK changes (K to L), the HBs on both ends shift to the left (i.e. towards relatively smaller ENa) and the previously subcritical HB (right side) becomes supercritical. Across the 3 columns, note that if the system did have pumps, interactions between the EIon and Ipump would continually and slowly change EIon thereby repeatedly evoking these activity patterns.
The question of what happens to the threshold for STF for these changes in EIon is raised by these results. The bottom row, panels J-K-L, displays the bifurcation diagram of the voltage excursions as a function of left-shift; the EIon values for each panel are those used in the three panels above it. For these diagrams, the left shift is the bifurcation parameter; all channels (f = 1) assume this shift (there is no spread, i.e. SD (standard deviation) = 0). Each diagram is constructed by choosing a value of LS, running a simulation, letting transient behavior die out, then measuring various characteristics of the steady-state voltage solution. In particular, here we measure the value of a fixed point (resting potential, RP) or the minimum and maximum values of a Vm oscillation. This procedure is repeated over a range of LS values, and the characteristic values are plotted against LS.
As LS increases, we see in panels J, K, and L a Hopf bifurcation (HB) i.e. a transition between a stable resting potential (RP), or in the diagram, stable fixed point, and tonic firing. Further, there is a narrow range of LS where both RP and tonic firing coexist; this is known as a “subcritical” HB. Subcritical HBs are seen on the left part of each diagram, and on the right part of diagrams J and K. In panel L however, the right part displays no such coexistence, and the HB is “supercritical”. This means that as LS increases further, there is an abrupt (although continuous) transition from tonic firing to RP. Upon decreasing ENa (panels J to K), the threshold moves to higher LS values, and the spike height is reduced. However, when EK is reduced (panel L), the threshold moves to a lower LS, which explains the onset of firing in panel C. The decrease of the repolarizing current also continuously raises the minima and lowers the maxima of the voltage excursions. This underlies the trend to low amplitude spiking behavior (cf. panels C→F→I). Thus, in axons where injury has caused Nav-CLS and is fostering gradual rundown (depletion) of the ion gradients, a range of abnormal excitability patterns could be expected to appear then disappear.
Note that the mean LS in Figure 2I corresponds to the limit cycle regime (Figure 2L) – although it is very close to the fixed point (quiescent) regime which is to the right of the supercritical Hopf bifurcation in Figure 2L. This is why the spike amplitude is so small – but technically they are still spikes, not STOs; this would be the case even if the Hopf bifurcation were subcritical (as in the right part of Figures 2J and K). There is no injected current here, and the system can generate tonic firing of low amplitude spikes, so we cannot speak of depolarization block. However, depolarization block can be produced by injecting a current of sufficient magnitude (not shown).
Since the EIon are controlled by intra- and extracellular [Ion]s (Eq (13)) which in turn depend on pump currents via Eq (10), these results imply: a) Ipump will substantially affect excitability at injured nodes [41], and b) in the presence of a small rundown of the K+ gradient, Nav-CLS is sufficient to produce STO-like voltage excursions. We will see in the two sections on bursting below (Bursting in injured nodes and Bifurcation analysis of bursting) that these excursions and STOs share a common origin.
Vm oscillations in a CLS injured node with the pumps on
Whereas the transition from quiescence to tonic firing when EK moves closer to zero (Figure 2B to 2C) is expected, since the move causes a small depolarization, the appearance of STO-like excursions (Figure 2H to 2I) is less intuitively obvious. To better understand how pump current affects the emergence of small oscillations, we simplify the above multiple Gaussian-distributed gNa populations by forming two subpopulations: LSi = [0,15]mV and fi = [0.5,0.5], signifying that half of Nav channels have no shift, and the rest have a 15 mV CLS. This choice for gNa populations is taken from the tonic firing regime (Figure 9B in [11]) that occupies the largest part (∼40%) portion of the LS/f plane depicting five spontaneous activity regimes of injured nodes. We turn on the Na/K pump and mimic injury to pump function by varying the parameter Imaxpump in Eq (10). Imaxpump is a functional maximum, a rate that, as per Eq 13 would only be attained for [K+]o = 0 and [Na+]i = 0 (unrealistic in living systems). It differs among axons [42] and will vary with the cellular surface area, Na/K-ATPase density, bilayer lipid composition [39], [42] and fluidity [43], and temperature [44] among other factor [45]. Importantly, Imaxpump can be reversibly decreased experimentally by specific inhibitors (e.g. strophanthidin, ouabain).
Figure 3 demonstrates that when Imaxpump falls from 95 to 30 µA/cm2, repetitive AP activity (dotted line) is replaced by tonic low-amplitude Vm oscillations (solid line). While the Vm -oscillation amplitude increases as Imaxpump increases (Figure 3D), the firing rate decreases. This suggests that, if Ipump were to decrease, repetitive APs could change to STOs. Inhibiting the Na/K pump can further lead to a complete cessation of firing. This is in general agreement with experimental results [42]. It is noteworthy that because [Na+]i and [K+]o (Eq (10)) limit maximal pump current, the upper values of Ipump are considerably less than Imaxpump. In Figure 3B, e.g., with Imaxpump at 95 µA/cm2, Ipump fluctuates around 22.7±0.5 µA/cm2. The section Effects of varying maximal pump current discusses the impact of Imaxpump on the bursting regime.
Figure 3. Ectopic steady-state Vm excursions are tuned by Ipump.

(A) From the spontaneous tonic firing regime of Figure 9 in [11], a random point (LSi = [0,15]mV, fi = [0.5,0.5]) is selected; as illustrated, Vm oscillation amplitude varies as Imaxpump is set at 30 and 95 µA/cm2. (B) Ipump changes from a constant 10 µA/cm2 (associated with a small periodic Vm fluctuation) to a fluctuating 23±0.5 µA/cm2 (associated with a train of APs). (C) Corresponding total INa. (D) With growing Imaxpump, voltage excursions (blue) increase and their oscillation frequencies (red) decrease.
Bursting in injured nodes
After repetitive firing activity, the electrogenic Na/K pump mediates the restoration of the ion gradients on a relatively slower time scale. Figures 2E and 2F suggest that pump current, by driving EIon changes, could also induce patterns of bursting in which volleys of high-amplitude spikes are separated by periods of low-amplitude STOs.
In this section we pursue this idea by simulating nodes with three gNa populations (LSi = [26.5,2,0]mV and fi = [0.2,0.08,0.72]) and Imaxpump set at 95 µA/cm2. In Figure 4A a node injured this way produces bursts of spikes separated by STOs. The silent and active phases of such bursts are associated with pump current dynamics and the left-shifted window conductance (m3 h). In the silent phase where Vm is near the RP (−60 mV), the Nav-CLS channels are a “leaky” Nav component (Figure 4B and 4C) flowing into the injured node, but without depolarizing it above firing threshold, because outward INapump slightly exceeds the inward sodium current (the sum of INa and INaleak). The result: Vm is destabilized and STOs develop. INapump keeps decreasing (Figure 4Aii), becoming less than inward sodium current at about the time marked (pink star) in Figure 4Ai. Therefore Na+ accumulates inside and K+ outside the node, resulting in ENa and EK changes, based on Eq (13).
Figure 4. Transitions between STO and burst behaviors.

(A) Upper: Vm (black solid line) at an injured node and the varying EIon (ENa and EK; blue dotted lines). Three gNa populations are used: LSi = [0,2,26.5]mV and fi = [0.72,0.08,0.2] with Voli = Volo = 10−15 m3. Initiation and termination times of a burst of spikes (pink star, green dot, respectively) are used in Figure 5. Lower: corresponding Na+ currents, as labelled. (B) Equilibrium values of Nav channel activation and inactivation variables, mi and hi. (C) The steady-state open probabilities, 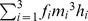 : with a mild injury gwindow(V) magnitude is slightly less at 0 mV (vertical line and circles) but much enlarged at voltages near the normal RP (−65.5 mV for fixed [ion] condition). (D) Another example: LSi = [0,2,20]mV and fi = [0.72,0.08,0.2] and Voli = Volo = 3×10−15 m3, with expanded detail showing STOs. Note the difference of time scales in (A) and (D), reflecting the fact that a 3-fold lower axonal surface-to-volume ratio in (D) slows the rate of [ion] changes.
: with a mild injury gwindow(V) magnitude is slightly less at 0 mV (vertical line and circles) but much enlarged at voltages near the normal RP (−65.5 mV for fixed [ion] condition). (D) Another example: LSi = [0,2,20]mV and fi = [0.72,0.08,0.2] and Voli = Volo = 3×10−15 m3, with expanded detail showing STOs. Note the difference of time scales in (A) and (D), reflecting the fact that a 3-fold lower axonal surface-to-volume ratio in (D) slows the rate of [ion] changes.
When changes in EIon further reduce the firing threshold (system dynamics will be explained in the following section) the injured node would cross the diminished threshold and produce APs. With the consequent [Na+]i increase, INapump starts increasing. It eventually prevents the node from crossing the threshold and APs cease (green dot in Figure 4Aii) and Vm falls back to the STO state around the RP. Repeated switching between clusters of spikes and STO intervals constitutes a form of bursting behavior that, given the involvement of the pump, should be sensitive to the surface to volume ratio, as per the numerical results of Käger et al [29]. With intracellular and extracellular nodal volumes tripled (Voli = Volo = 3×10−9 m3) and the surface area fixed (6×10−8 m3) (Figure 4D), bursting period (BP – from the beginning of one burst to the beginning of the next) and burst duration (BD) (Figure 4Ai) are prolonged. This confirms that, for the Nav-CLS model of injury, both the burst parameters are indeed sensitive to the surface-to-volume ratio.
Interestingly, the solutions in Figures 4A and 4D do not exactly repeat. The number of spikes during the active phase of the burst varies slightly among bursts. Additionally, missed spikes at the end of the burst phase relate to the existence of period-doubled solutions (PD) as we will see in Figure 5C. Even though below we characterize them by an approximate bursting “period”, BP, these solutions are likely chaotic.
Bifurcation analysis of bursting
As seen above, bursting is followed by quiescence and STO's are observed at the onset and offset of bursting. We now expose the dynamical structures underlying these behaviors. During firing, the ion gradients are slowly depleted due to the window current in the injured node. The pumps cannot compensate for the loss, and ENa and EK move towards zero. During quiescence the pumps replenish the concentration gradients, and the absolute values of ENa and EK increase. Spontaneous rhythmic bursts in Figure 4A are shown three-dimensionally in Figure 5A with dynamic (i.e. time-varying) EIon. A bifurcation analysis based on a decomposition of the full dynamics into slowly and rapidly changing variables (e.g. EIon and Vm respectively, Figure 5B and 5C) helps identify the burst mechanism. This is known as a slow-fast decomposition ([24] and references therein). We first focus on the initial phase of the burst (e.g. pink star, Figure 4A). For now, EK and Ipump are regarded as fixed, i.e. as constants taking their (arbitrarily chosen) values at the pink star (−83.17 mV,14.96 µA/cm2). We pick a value of ENa, run a simulation, wait until transients die out, and measure the minimum and maximum values of Vm. This procedure is repeated for the range of ENa values on the abscissa of Figure 5B. On the ordinate we plot the corresponding minima and maxima of Vm. A striking feature is that in certain regions of the diagram there are more than two ordinate values; this is a hallmark of bistability as discussed further below. Since ENa = 40.2 mV at the pink star, the diagram demonstrates that the CLS injury places the system in a superthreshold oscillation (i.e. spiking) regime. The spiking trajectory at the pink star in Figure 4A and 5A corresponds to the stable limit cycle (i.e., stable periodic oscillation) in Figure 5B.
At the onset of firing (pink star in Figure 5A), the pumps are still driving ENa upwards and past (i.e. to the right of) the Hopf bifurcation point (see Figure 5B). In that region, the fixed point, around which the STO oscillates, has become unstable; the trajectory moves towards the only possible stable state, a stable limit cycle corresponding to repetitive firing (APs).
During firing ENa is decreasing and consequently this limit cycle shifts to the left and approaches the bistability regime. During this process, both amplitude and frequency of the limit cycle decrease. In the bistability region of Figure 5B, the system has two stable states: a steady fixed point or resting potential (i.e. RP, marked by solid line) and a superthreshold oscillation (i.e. APs, or tonic firing). Between these states is an unstable limit cycle. The system phase space is then split into two sets of points. In the first set, typically near the RP, phase space solutions converge to the RP. In the other, typically further from the RP, solutions converge to the tonically firing solution. In both these sets, initial conditions close to the unstable limit cycle yield solutions that diverge away from it in an oscillatory manner: in the first set the oscillation grows into the tonic solution, and below, oscillations damp out towards the RP. Further, a perturbation from the RP must “jump over” this unstable limit cycle in order to lead to tonic firing, and vice-versa.
Near the offset of firing (green dot), with ENa still decreasing, the dynamics move toward a region of period doubling (PD). Sometimes the trajectory falls off the limit cycle on the upper branch before reaching this region such as in Figure 5C. Other times during the same solution the trajectory enters briefly into the period doubling regime. This manifests itself as a longer interspike interval at the end of the firing phase (see Figure 4A, second burst). For parameter choices corresponding to bursting the trajectories are not periodic, i.e. they appear to be chaotic unless the transients exceed our simulation times. The precise dynamics in this region warrant more extensive investigation beyond the scope of this paper. Note also that the AP amplitude decreases together with ENa. This is expected as the ion concentration gradients drive the action potential. This suggests that the upper branch of the PD regime is not reached by the trajectories near firing offset.
Let us inspect Figure 5 in more detail. Figure 5A shows that both ENa and EK are dynamic, changing slowly during a bursting period. Figures 5B and 5C capture the behavior of the system near the two transitions (pink star and green dot) in Figure 5A. The pink bistability region in Figure 5B – corresponding to the pink star – sits between a limit point (LP: where a “saddle-node of limit cycles” bifurcation occurs), and a Hopf bifurcation point (HB: where a transition between a stable fixed point and a stable limit cycle occurs). The green region in Figure 5C lies between (at left) an area (PD) of complex dynamics where many period-doubling bifurcations occur and (at right) a HB. The unstable limit cycle solution (marked by open dots) between the stable fixed point and stable limit cycle is the aforementioned threshold boundary. The arrow in Figure 5B shows the direction in which ENa changes during the firing phase (EK changes as well). Likewise, the blue arrow in Figure 5C shows how ENa changes during the STO phase. During the bursting period, the bifurcation diagram in Figure 5B slowly morphs into the Figure 5C diagram and back. These two diagrams, snapshots of the system's possible steady state behaviors at the two ends of the bursting cycle, help us understand how the whole solution plays out during an entire bursting cycle.
Interestingly, from Figure 5B one might think spiking would continue until the point LP is reached, where ENa is almost nil. However, the pump's dependence on [Ion]s limits this decay, such that a PD region appears and truncates the spiking phase at a relatively large value of ENa (note the difference in ENa scales in Figure 5B and 5C).
The two-parameter phase diagram in Figure 5D provides a view of the burst dynamics from the EIon perspective. Here the pink star and green dot have the same meaning as in Figure 5A–C (see caption). The black loop connecting pink star to green dot is the projection of the 3D trajectory in Figure 5A onto the ENa−EK plane, and displays the ENa and EK trajectories during bursting. The solid and dashed curves are, respectively, sets of all the LP and HB points for the range of EIon of interest (exemplars of these points were seen in Figure 5B,C). The area bounded by them is the bistability regime. This involves computation of many bifurcation diagrams along the entire bursting trajectory, choosing from the trajectory the two extremal pump values (pink for the lowest and green for the highest). One can imagine that, when the system state moves from pink star to green dot, the periodically varying Ipump pushes the bistability regime between the pink-curve-bounded and green-curve-bounded areas. Thus, injured node dynamics switch between different modes, resulting in bursting behavior.
Nav-CLS phase diagram: 3 gNa subpopulations
Various spontaneous activities of an injured node with three gNa subpopulations (LSi = [26.5,2,0]mV) comprising different fractions are presented in Figure 6A. The fractions (f1 and f2) corresponding to the Nav-CLS populations form the x- and y- axes; the fraction of intact channels is simply f3 = 1−f1−f2. When the percentage of channels with deeper injury (i.e. LSi = 26.5 mV) increases, the state of the injured node goes from RP→bursting→bistability (APs or RP, depending on the initial condition)→ tonic firing→bistability→RP. For two random points from the burst and bistability regimes, Vm is plotted in Figures 6B and 6C, respectively. Overall, STOs can arise from various combinations involving decay to or growth away from RP, but the particular behavior seen depends sensitively on the percentage and degree of injury.
Figure 6. Classes of Nav-CLS-induced activities with two injured and one intact gNa population.

(A) Membrane excitability map of injured node with LSi = [26.5,2,0]mV. White, pink, green and blue regions represent different stable state activities: quiescence, bursts, tonic firing, and bistability between quiescence and tonic firing. Here the fraction of intact gNa is f3 = 1−f1−f2. (B) and (C), typical Vm trajectories when the system is in bursting and bistability regimes, respectively.
Nav-CLS and susceptibility to noise-induced bursts
Neuronal noise arises naturally from a wide variety of sources including channel noise, synaptic noise, electrogenic ion pumps, and thermal noise. In this subsection we investigate the influence of current noise on spontaneous electric activity of a node with Nav-CLS injury by including a simplified Gaussian white noise σξ on the right side of Eq 1 as an additive term. This additive noise ξ has zero mean, and its autocorrelation function is <ξ(t)ξ(s)> = δ(t−s) where δ(·) is the Dirac delta function; σ is the noise strength (in µA/cm2). For a node, the white noise can be taken as describing conductance and thermal noise. By definition it has equal power at all time scales, in particular beyond the fastest time scales present in our deterministic (i.e. noiseless) node model. Furthermore, because the central limit theorem applies to the sum of different independent fluctuation sources acting on Vm, its amplitude distribution is assumed to be Gaussian (see for instance, [46]). For a deterministic bursting activity with burst period BP = 10.2 s illustrated in Figure 6B, additive noise dramatically decreases BP (Figure 7A). It also makes BP irregular because the crossing of the threshold by the Vm trajectory is now a probabilistic process, especially in the vicinity of the threshold [47].
Figure 7. Noise induces or modifies bursting.

(A) Without noise, bursts are as shown in Figure 6B but here, under the influence of noise (σ = 0.1 µA/cm2), the bursting periods are random. Right (here and in B): an expanded section showing STOs induced by noise via coherence resonance. (B) Without noise the system is in the bistability regime as shown in Figure 6C, but here, noise (σ = 0.4 µA/cm2) causes Vm to randomly visit quiescent or repetitive firing states, hence producing noisy bursts.
When the node operates in a bistable regime, as in Figure 6C, this additive noise causes stochastic switching between the quiescent and tonic firing modes (Figure 7B); noise-induced bursts appear. In fact, the injured node produces bursting in both the burst and bistability regimes of Figure 6A. With higher noise intensity, the mean BPs of noise-induced bursts is shorter. Such bursting is also referred to as noise-induced mixed-mode oscillations [48]. This phenomenon has been found in experimental settings, for example the bistable squid giant axon [49], electroreceptors of paddlefish [50] and entorhinal cortex neurons [51], as well as in excitable neuron models such as the Hodgkin-Huxley model [49], the FitzHugh-Nagumo model [48], and an excitable spine model [52].
The expanded sections in the right column of Figure 7 demonstrate that, upon introduction of noise, the silent phase of the burst behaviour in Figure 6B and the steady state in Figure 6C turn into STOs. This is observed in injured primary sensory neurons [53], and constitutes coherence resonance (CR) (or autonomous stochastic resonance) wherein noise alone excites subthreshold oscillatory responses in excitable systems (see [47], [54] and references therein). Noise continually modifies the firing pattern because it randomly pushes the system into and out of different regions of phase space, - similar behavior has been described for thermoreceptors [46]. In terms of Figure 5C, when the system is on the lower STO branch, the noise induces the behavior seen at the nearby Hopf bifurcation. In summary, for our Nav-CLS system, noise can elicit a random mixture of the behaviors seen in the phase diagram in Figure 6.
Nav-CLS variance and diverse patterns of ectopic excitation
Next, we explored the spontaneous activity of an injured node with different Nav-CLS variances. Gaussian distributions of Nav-CLS were imposed with different means and standard deviations (SDs). As illustrated in Figure 8A, we tested mean(LS) = [0.5,1,2.5,5,10,20,27,30,20]mV with corresponding SD(LS) = [0.2,0.4,5,5,5,5,5,5,10]mV. With increased injury intensity (greater mean(LS)), the Vm changes are dramatic (Figure 8). There is a progression from quiescence (B)→bursting (C–D)→tonic firing (E–G)→bursting (H)→quiescence (I). For a smeared Nav-CLS injury as small as LS = 1±0.5 mV, the node generates bursts (BP = 35 s), indicating this system's extreme susceptibility to even small changes in window current. (Left-shifting Kv activation by an equivalent amount [11] does, however, provide some stabilizing protection for Vm). A slightly larger trauma (mean(LS) = 2.5 mV) shrinks BP dramatically to 0.78 s, then further increasing mean(LS) leads to tonic firing. If, however, mean(LS) is taken to 27 mV (Figure 8H), bursting with STOs (not visible) returns. Then, for mean(LS)≥30 mV no spiking occurs. In Figures 8C, D, H the within-burst firing rate and Vm excursions decrease with time. In Figures 8E–G the instantaneous within-burst and tonic firing rates gradually increase with mean(LS) (Figure 8K). The corresponding mean Vm excursions decrease with increased mean(LS) (Figure 8L). With a smear of Nav-CLS that encompasses a wider range of LS values (SD = 10 mV: Figure 8J; see Figure 8A)), the injured node fires tonically, as in the lower SD case for the same mean(LS) (Figure 8G). Evidently the larger LS components of the SD = 10 mV distribution do not sufficiently dominate to yield the type of quiescence associated with a (smaller SD) 30 mV LS (Figure 8I).
Effects of varying maximal pump current
In Figure 3D we saw that decreased Imaxpump suppresses spontaneous tonic firing with CLS injury. In Figure 9 we look at the effect of varying the pump activity on bursting patterns in a mildly injured node (LS = 2.5±0.5 mV). Imaxpump = 95 µA/cm2 (the value used in Figure 9C) is our standard or healthy cell reference. Imaxpump depends on the density of pump expression in the membrane, which will differ among axons and also for different regions of a given axon. It also varies with a multitude of factors, including injury conditions that affect the turnover rate at individual pump molecules. To explore these possibilities in a mildly injured node, we varied Imaxpump over a wide range below and above the standard value in Figure 9C.
Figure 9B shows that a lower Imaxpump is associated with longer interburst intervals (IBI: see Figure 4) than for the standard case (Figure 9C). Figure 9A, an extreme example in which excitability is lost, has an infinite IBI. Increasing Imaxpump, by contrast, initiates burst of spikes and shortens both burst duration (BD) and burst period (BP) (Figure 9D,E), reflecting the hyperpolarization produced by the electrogenic Na/K pump. The differences between Figure 9B and 9C are consistent with experimental observations in rat spinal interneurons where, during the course of pump blockade, the burst rate decreases (larger BP) [42]. It is noteworthy that for the uninjured system, varying maximal pump current had little impact on the steady- state Vm (see Table 2). What vary substantially are the steady state EIon. This is consistent with the mechanistic interpretation that Ipump oscillations responding to chronic Nav-CLS-based Nav-leak underlie the array of bursting patterns in the many simulations above. The basic phenomena (ectopic APs and STOs) are extremely robust properties of nodal membranes with mild Nav-CLS and a pump, and appear in various manifestations without the need to invoke novel conductances or Nav channels with peculiar kinetics.
Table 2. Steady-states for intact nodes (Vm = −59.9 mV) as Imaxpump varies (units as in Table 1).
| Fig. 9 | Imaxpump | ENa | EK | Ipump |
| A | 15 | 3.5 | −73.7 | 5.1 |
| B | 55 | 42.8 | −82.3 | 8.3 |
| C | 95 | 54.3 | −84.6 | 9.2 |
| D | 120 | 58.6 | −85.4 | 9.6 |
| E | 156 | 63.1 | −86.3 | 9.9 |
Nav-CLS, pseudo-first order activation and subthreshold persistent current
Unlike nodes, somata express multiple Nav isoforms, and in recent modeling work of injured somata, Kovalsky et al [21] showed that a system with three distinctive gNa sub-types (but no Kv and no Na/K pumps) yields STOs, provided the two slow gNa sub-types are given 1st rather than 3rd order activation. This provided the system with the “several delayed components” needed to mimic the repertoire of STO-triggered phenomena of live neurons. To simulate Nav1.7 and Nav1.8 interacting in a fashion that elicits STOs, Choi and Waxman [23] also depicted one of the gNa sub-types with 1st order activation. Interestingly, in our system, left-shifting a fraction of the m3 h-based Iwindow acts like introducing a lower order activation process (see Figure 10). What physical change in injured axons that could yield such pseudo-first order m∞(V) curves for total gNa, even though individual channels continue to exhibit their normal m3 activation? Heterogeneity in the extent of bilayer damage could cause this by fluidizing a fraction of the nodal bilayer. This would yield a subthreshold persistent INa. Differences in bilayer packing in different parts of the membrane [3], [36] also arise developmentally in some circumstances. In a given region of plasma membrane, a membrane remodeling process that places some Nav channels in (say) cholesterol-poor [55] and others in cholesterol-rich subdomains could, in principle, create or “tune” physiologically useful subthreshold persistent currents.
Figure 10. Partial Nav-CLS in a population and apparently first order activation kinetics.

Equilibrium activation values (m3, 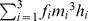 and m ) of co-existing intact Nav and CLS Nav channels plotted as a function of Vm. Black solid and black dashed line represent m3 for intact and 20 mV CLS channels with f = 1. For a membrane with large Nav-CLS injury given by: LSi = [0,20]mV and fi = [0.3,0.7], the gray bold line denotes
and m ) of co-existing intact Nav and CLS Nav channels plotted as a function of Vm. Black solid and black dashed line represent m3 for intact and 20 mV CLS channels with f = 1. For a membrane with large Nav-CLS injury given by: LSi = [0,20]mV and fi = [0.3,0.7], the gray bold line denotes 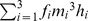 . Red line: m of the intact channels.
. Red line: m of the intact channels.
Discussion
Here we demonstrated that different patterns of spontaneous activity can be expected from mildly damaged nodes of Ranvier in which some or all the Hodgkin-Huxley type Nav channels have undergone a coupled left-shift in their activation and inactivation kinetics. This is called Nav-CLS [11] and as a form of Nav-leak, could result in the ectopic activity that emanates from injured nodes. As detailed in the Introduction, there is growing evidence that activation and inactivation are left-shifted in various chemical and mechanical injuries in different cell types and Nav isoforms [10], [12]–[19]. In more focused studies, it has been demonstrated that these left-shifts are coupled [8]–[10], and this CLS in Nav1.6 (the nodal isoform) is the basis of our study [11]. In diverse sick excitable cells, it has been hypothesized that Nav-CLS occurs wherever channel-embedding bilayer becomes structurally degraded and fluidized as a result of mechanical or chemical insults [3], [4].
The following are the main findings of this study which is, essentially, a dynamical analysis of a standard node of Ranvier model with Na/K pumps, under conditions of coupled-left-shift (CLS) injury to some or all of the Nav channels and injury to the Na/K pump in terms of lowered output. The analysis uses simulations and bifurcation theory.
With EIon values fixed (i.e. pump activity irrelevant) at standard or mildly depleted values (10–15%), LS injury levels up to 20 mV (including for “smeared”, i.e., Gaussian distributions of CLS) elicit either quiescence or tonic firing. With increasing LS, a transition from quiescence to tonic firing occurs via a subcritical Hopf bifurcation, then at yet higher LS, tonic firing is replaced by quiescence via another subcritical Hopf bifurcation. If ENa and EK both approach zero, that bifurcation is supercritical; in its vicinity, tonic APs of arbitrarily small amplitude occur.
Maximal pump current is an important determinant of nodal excitability. When increased above a certain value, it yields an abrupt transition from quiescence to a relatively high rate of tonic firing; this is a supercritical Hopf bifurcation.
There exist CLS injury conditions where the system is bistable, exhibiting either quiescence or tonic firing. In this bistability regime, behavior depends on initial conditions in a complicated manner not explored here. Stimuli, like spikes arriving from an upstream node, could thus toggle the system these two behaviors.
Spontaneous bursting activities occur for small and for large LS injuries, while intermediate intensity injuries elicit tonic firing. In the case of bursting, time-varying EIon values due to pump activity provide a slow almost periodic element in the system; EIon values approach zero during the burst and grow during the interburst interval. Slow-fast decomposition of the full dynamics reveals that these slow EIon bring the fast spiking dynamics into and out of a limit cycle (tonic firing) regime.
STOs can appear with single injured Nav population or for complex CLS injuries. STOs that bracket bursts of firing are associated with the complex eigenvalues governing dynamics in the vicinity of the lower branch of fixed point equilibria. Burst onset is associated with a subcritical Hopf bifurcation while offset is organized around a saddle-node bifurcation of limit cycles. Period-doubled solutions exist near this latter bifurcation, and the limit cycle can collide with these solutions, producing abrupt changes in the interval between successive spikes. Bursting solutions tend not to repeat over many bursts, and thus appear to be chaotic (not analyzed in detail).
Including small levels of Gaussian white noise in simulations causes burst characteristics to fluctuate and yields more long-lived STOs, consistent with the reported propensity of injured axons to exhibit STO phenomena.
Inhomogeneous CLS is sufficient to explain a rich variety of axon excitability behaviors. Combinations of CLS-injured Nav channels, all exhibiting realistic m3 activation behavior, yield flattened gNa(V) curves similar to those of other models that, of necessity, include at least one gNa with m1 activation.
Nav-CLS and pathophysiological Nav leak
Regions of membrane with mild Nav-CLS have a “subthreshold” persistent gNa (a Nav-leak conductance) due to gwindow operating abnormally close to the cell's normal resting Vm; when only a small fraction of Nav channels are affected, the cell's gwindow plot has a shoulder as in Figure 4C; such plots strongly resemble coupling-induced subthreshold persistent conductance of pacemaker neurons [56]. Like that physiological pacemaker, our injury model includes no slow mode (or non-inactivating) gNa. Calculations [3] show, in any case, that at voltages near typical AP thresholds, left-shifted Iwindow would dominate over equivalently left-shifted non-inactivating INa. In much of this study, we mimicked “smeared” extents of bilayer damage via Gaussian distributions of Nav-CLS, reflecting the notion that nodal axolemmal injury is unlikely to exhibit a unique intensity. Smear, though expected in situ [2], [8], is not actually required for neuropathic activity. Use of assorted smears confirmed that CLS-fuzziness in no way abrogates dysfunctionality and it demonstrated the robustness of Nav-CLS as a model for leak. Nav-CLS injury thus provides a minimalist explanation for multiple spontaneous modes of firing in which APs, bursts and subthreshold oscillations (STOs) occur in various patternings. In severe cases, it also predicts depolarizing block [11]. We used bistability diagrams and various bifurcation analyses to help clarify the mechanistic underpinnings of the Nav-CLS-induced dynamics.
A mildly injured excitable cell cannot survive for hours or days (the time course, e.g., of diffuse axonal injury) without operational Na/K pumps. In electrical models of axon injury, pumps do not only maintain ion gradients, they also contribute to slowly varying excitability patterns of the damaged-but-viable axons [11], [29], [30]. Pursuing this further here, we found the following: mild Nav-CLS induces STOs through a gradual reduction of firing threshold mediated by slow EIon dynamics whose characteristic times depend on pump-mediated fluctuations of extra- and intracellular ion levels. Threshold voltages for firing are not fixed but depend on EIon. EIon fluctuations induced by Nav-CLS produced, in a bistable fashion, both STOs and bursts of full-sized APs. With mild Nav-CLS injuries, adding Gaussian white noise to the current-balance equation induced erratic bursting, or, where injury was already causing sustained bursting, noise influenced the rates and durations of the ectopic signals. These effects of noise are consistent with those seen in a recent computational study of neuropathic firing in a hyper-excitable system with more ion channel species than used here, but without pumps [57]. In the following we discuss and provide more context for the above results.
Problematic rhythmicity patterns associated with mild axonal injury
Trauma-induced [58] and ischemia-induced [59] Nav leak triggers Ca2+-excitotoxicity, the proximate cause, in diffuse axonal injury and oxygen glucose deprivation, of axon demise. A mechanistic understanding of early stages of Nav channel dysfunction, when salvage is possible [60], is needed. Although pathological TTX-sensitive Na+ leaks are attributed to increased “persistent INa” (e.g. [61], [62]) or slow-inactivating INa [63] it is not known how membrane damage elicits chronic Nav-leaks. A molecular-level understanding is lacking as to what constitutes leaky native Nav channels. A common Nav-leak culprit in diverse sick excitable cell conditions is, we propose, Nav-CLS [3]. For recombinant Nav1.6, this phenomenon occurs as an immediate consequence of mechanical membrane damage [8], [11]. Any fraction of Iwindow (an attribute of fast gNa) that has become left-shifted acts as a leak [3], [8] that could trigger positive feedback excitation.
In previous modeling [11], we established that mild axonal Nav-CLS would effectively dissipate [Na+] gradients, bringing on excitotoxicity. Total nodal gNa was modeled by an intact fraction plus a second fraction with a discrete level of CLS. Various “Nav-CLS injuries” were imposed, Vm (t) was plotted as new steady-states were attained, then excitability was probed with current injection. Mild Nav-CLS caused spontaneous APs (tonic or damped trains). With Na/K pump activity included, it caused AP bursts whose specifics varied with compartmental volumes. From temporal patterns of [Na+] gradient rundown, it was evident that Nav-CLS type injuries could cause axon demise by overwhelming an axon's capacity for ion homeostasis. Injured nodes, moreover, generated pernicious ectopic signals and impaired transmission fidelity during saltatory propagation. Pathological activity patterns, plotted as a function of the extent (in mV) of Nav-CLS, and of the fraction (0–1) of affected nodal Nav channels, fell into several regimes.
What can we infer about responses to stimulating inputs when there is Nav-CLS injury? Effects of stimulation, a multi-faceted issue, was addressed for nodes with fixed reversal potentials in a preliminary study [11] involving two Nav populations, one intact and one with a specified CLS injury. Spontaneous firing occurs over a range of damage, then beyond a critical threshold level (LSc) the system is quiescent. Stimulating with constant current yields a larger maximal firing rate but, due to depolarizing block, quiescence occurs with less damage (LS<LSc). Effects of stimulation are currently under study for bursting nodes (model with pumps as in Figure 5). Because bursting is a globally attracting behaviour, a single spike as a form of input may transiently change a bursting cycle but not its steady state. For the regime we considered, bifurcation analysis reveals that a single spike cannot “trigger” bursting behavior.
Nav-CLS and neuropathic pain
Though ectopic signals in vivo are generated at both axons and somata [64], peripheral neuropathic (as distinct from nociceptive) pain mechanisms are mostly studied via ectopic activity in damaged somata such as those of primary mechanoreceptor afferents [62], [65], [66]. A closely-related issue of relevance here is that of low threshold afferents, dubbed “algoneurons” [67]. These are non-pain sensory neurons that can serve double functions, signalling pain only when peripherally hypersensitized due to trauma, inflammation and the like. Spontaneous activity patterns described here are like those of both damaged mechano-afferents neurons and hypersensitized algoneurons. This is a post hoc assessment; we monitored behaviors across parameter space, but did not adjust parameters to mimic particular neuron activities. Clearly, it would be interesting to determine if “hypersensitization” in algoneurons corresponds to Nav-CLS in damaged peripheral endings.
Mechanisms for generating STOs
Emerging from that “injury”-modified Hodgkin-Huxley axon (two gNa sub-populations and the Na/K pump) was a critical feature of the electrical dysfunction of injured excitable membranes, abnormal STO phenomena [11]. We lacked, however, a dynamical understanding of the phenomenon and its robustness; given the prevalence of STOs in neuropathic firing (plus physiological correlates) this represented a significant gap. Typically, STOs and injury have been modeled with non-inactivating and/or slow-gating gNa. Our model - Nav-CLS - includes only a fast-gating gNa to which a simple biophysically-justified (and biophysically-explicable) modification has been applied at varying intensities. Here, using smeared-intensity versions of Nav-CLS to model injury, we generated diverse STO phenomena.
The “spared but impaired axons” of injured tissue are targets for Nav antagonists (see [21]) and such mildly-injured axons were our focus here. We examined sustained spontaneous (ectopic) behaviors, extending our analysis first to three gNa sub-populations (i.e., for otherwise identical gNa mechanisms, the Nav population was given three extents of Nav-CLS), then to various Gaussian-distributed (“smeared”) Nav-CLS populations. Realistically untidy, but conceptually simple, all these Nav-CLS injury variants exhibited the key components of the injury repertoire. STOs arise with (Figure 8D) or without smear. Along with the basic spontaneous activities (APs and bursts), injured nodes spontaneously produced sustained STOs (Figure 3) and STOs as parts of bursts consisting of clusters of spikes and STOs (Figure 4).
Pump/CLS-leak: a slow “component” of an oscillatory system
Na/K pump turnover rates are sensitive to substrate concentrations, membrane potential [68] and lipid packing [39], [40]. Quiescent (“at rest”) axons maintain a steady basal pump activity that counteracts all system leaks. In our model Ipump is always substantially smaller than INa and IK (∼22-fold in Figure 4A) and pump rates oscillate significantly more slowly than APs (e.g., Figures 8C,D, ∼2 orders of magnitude slower). In our simulations of injury, small amplitude slowly oscillating Ipump plays an essential role in mediating spontaneous Vm excursions (Figures 3, 4 and 9). Larger magnitudes for Imaxpump (see Figure 3D and Figure 9) accelerate the efflux/influx of Na/K ions. This was associated with dynamical changes, from RP to STOs to bursts or tonic APs. Figure 3D in particular is a bifurcation diagram; it characterizes the transition from quiescence to tonic firing as Imaxpump increases. This shows clearly the increase in the amplitude of voltage excursions Vm and the decrease in firing rate. Figure 9 shows the effect of Imaxpump in the bursting regime. As Imaxpump increases, we go from quiescence to bursting. All characteristics of the burst summarized in Figure 4 shorten with this increase.
In the injured nodes, therefore, a reduced Imaxpump (due, say, to ATP depletion and/or to a more fluidized pump-embedding bilayer) would modify ectopic excitation patterns. In line with this, in endogenously bursting snail neurons, inhibiting the pump shortens burst periods and increases spike frequency [69], though unlike injured nodes, pacemaker neurons have multiple slow processes that confer resilience on specific oscillatory patterns. Dynamic transitions of Ipump are seen in a number of rhythmically active cells including sinoatrial node cells [44] and trigeminal motoneurons [70].
Insofar as Na/K pumps show greater ATPase activity in membranes with greater lipid packing order [39], [40], membrane injury that caused Nav-CLS should simultaneously impair pump activity. To better understand ectopia arising from injured nodes or axon initial segments, it would be helpful to know if Na/K pump inhibitors (e.g. ouabain, but also generic membrane fluidizers [43], [71]) reduce ectopic burst firing in mildly damaged axons. In this regard, we note that peripherally-applied ethanol (shown recently to inhibit neuronal Na/K–ATPase [72]) induces a “feeling no pain” condition when injected locally to relieve peripheral trigeminal neuralgia [73].
Bifurcation analysis: Nav-CLS/pump STOs help trigger spikes by decreasing firing threshold
Although neuropathic STOs trigger APs in many neurons [58], [74], STOs are not principally a neuropathic phenomenon. In the CNS [75], STOs are a filtering mechanism by which neurons decode oscillatory inputs from other neurons in a frequency dependent manner [76]. Disparate underlying mechanisms for STOs have been noted, including Ca channel Iwindow [77] and HCN-channel based Ih [75], and, of particular interest here, a fast gNa-based [56] subthreshold persistent INa [78]). Our mild Nav-CLS-based Iwindow resembles that mechanism, but generates STOs and AP bursts only in conjunction with the electrogenic pump. STOs arising spontaneously at nodes are, by definition, pathologic, and given the dominance of nodal conductances by gNa (specifically, Nav1.6 channels) would likely be simpler in nature than the STOs elicited (by current stimulation) in multi-Nav isoform sensory neuron somata [21] or in double-Nav nociceptive terminals [23].
STOs simulated here involve a small-amplitude rhythmic mechanism that develops with mild Nav-CLS Iwindow (Figures 3 and 4) operating near what would normally be the RP level. Bifurcation analysis of this system enables us to propose that slow dynamics of the firing threshold are responsible for the timing of spikes. In a system where the EIon vary, the firing threshold varies too, gradually decreasing in the interval between successive STOs (Figure 9). This is broadly consistent with experimental findings of a decreased spike rheobase in injured dorsal root ganglion neurons [79] and, more general terms, seems consistent with the injury-induced hypersensitization of algoneurons [67]. Models involving fixed EIon cannot exhibit spontaneous oscillations of firing threshold, but in vivo, and especially in fine processes (e.g., Nav-rich nociceptive endings), fluctuating [Ion]s and pump currents are expected once channels activate.
Leak/pump counterplay and the oscillating threshold: implications for healthy membranes
Analyses of the electrical excursion diversity seen in excitable cells gradually going through their death throes have, historically, been fruitful in providing sketches of the behavioral diversity of healthy excitable cells. For example, the Hodgkin [80] classification of various spiking behaviors in isolated crayfish axons and the Morris-Lecar [81] analysis of giant barnacle myocyte voltage oscillations provide the springboard for analytical descriptions of oscillatory types in mammalian interneurons [82]. Here we simulated sick cell behaviors in a context where large fluxes into small volumes made it crucial to include the contributions of the electrogenic Na/K pump. From this emerged an STO mechanism that triggers bursts of spikes in a manner not, to our knowledge, previously recognized, i.e., the oscillating threshold. Since threshold occurs at Vm values where fluxes unbalance, threshold varies with EIon variations. An ultra-simple biological exemplar of this point is a gCa-based biphasic AP (Figure 3c of [83]) that shows different thresholds for its depolarizing and hyperpolarizing excursions and that is modeled simply by letting [Ca2+]intracellular accumulate with gCa activation (Figure 3d and Equation (8) of [81]).
Here we explicitly considered injury-induced Nav-CLS in a setting where Nav1.6 would be the exclusive Nav isoform. But Nav-CLS may have resonances for healthy excitable membranes with two or more Nav isoforms with different voltage midpoints, such as seen in axon initial segments [33] and nociceptive nerve endings [23]. Moreover, even with a single isoform, non-homogeneous physiological modulation (e.g., from inhomogeneous lipid packing density) could yield CLS-like situations that might affect tuning (e.g,. in the cochlea [83], [84]). As in vivo lipid imaging becomes refined, it may be possible to detect lipid-packing changes before and after injurious stimuli at nodes or axon initial segments. This is important given that the activities of voltage-gated channels and the Na/K pump are both modulated by lipid packing [37], [39]. The kinetic phenotypes of the membrane proteins simulated here are, of course, constrained by amino acid sequence, but they also vary according to the bilayer mechanical milieu. Our work here suggests how, in the face of bilayer injury, the sensitivity of Nav channels to bilayer mechanics can be perilous. By the same token, it may spell opportunity, with developmentally controlled CLS-type kinetic modulations spawning behavioral amplifications beyond what would initially be expected from a limited set of membrane proteins.
Methods
Node of Ranvier with variable amounts of membrane damage
Axonal voltage excursions are modeled at an individual node of Ranvier with the Hodgkin-Huxley model [85] using the values listed in Ochab-Marcinek et al [86]. We start with the basic equation for membrane potential (Vm):
| (1) |
where C is the nodal membrane capacitance. The total INa for all Nav channels and the potassium current IK are given as:
| (2) |
| (3) |
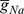 and
and 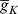 are the maximal conductances of the Nav and Kv channels, respectively; ENa and EK are the sodium and potassium reversal potentials, respectively; and n4 gives the steady-state open probability of potassium channels. By the HH formulation, gating variables m, h, and n evolve according to:
are the maximal conductances of the Nav and Kv channels, respectively; ENa and EK are the sodium and potassium reversal potentials, respectively; and n4 gives the steady-state open probability of potassium channels. By the HH formulation, gating variables m, h, and n evolve according to:
| (4) |
| (5) |
| (6) |
The forward (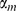 and
and 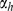 ) and backward rate functions (
) and backward rate functions (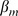 and
and 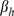 ) describe the first-order transitions between the deactivated and activated states of activation (m) and inactivation (h) processes in Eqs (4–5). They are determined by the membrane potential Vm:
) describe the first-order transitions between the deactivated and activated states of activation (m) and inactivation (h) processes in Eqs (4–5). They are determined by the membrane potential Vm:
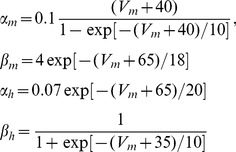 |
(7) |
The rate functions αn and βn of potassium gating variable n in Eq (6) also depend on Vm:
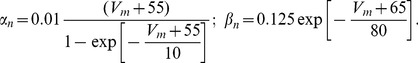 |
(8) |
Experimental findings for recombinant INa from nodal type Nav1.6 channels [8] show that, with mechanical membrane damage (“injury”), transient INa at a given Vm irreversibly accelerates because the activation and steady state inactivation (availability) processes undergo irreversible hyperpolarizing (“left”)-shifts. Consistent with this, the steady-state product of activation and availability (m3 h(Vm)t→∞), also called the window conductance, also left-shifts. This injury response we term “coupled left-shift” (CLS). This CLS can be easily modeled within the above HH model by shifting the voltage dependences of the activation (m) and inactivation (h) functions by LS, i.e replacing Vm by (Vm+LS) in Eqs (7). In other words whatever used to be observed at a given Vm in any function of m and h is now observed at (Vm−LS). This CLS is the basis of our model for axon damage. In patch clamp experiments the activation/inactivation time course data are explained with a single LS value for each degree of injury [8].
However, axon injury is spatially non-homogeneous [2] and is therefore likely to result in spatially inhomogeneous CLS. To account for this possibility we introduce fractions fi of Nav channels in damaged membrane, with left-shift intensity LSi (in millivolts). A Gaussian distribution of LSi seems a realistic model of axon injury, hence we explore various Gaussians. But discrete populations with a small number of LS values are more amenable to a thorough analysis, such as the bifurcation analysis done on a three population system exhibiting bursting dynamics (see Figure 5).
Specifically for a population of left-shifted Nav 1.6 channels {fi, LSi}i = 1,N, for each fi of intensity LSi, independent activation variables mi (i = 1,2,…N) and inactivation variables hi (i = 1,2,…N) are introduced. The total INa for all Nav channels is now given as:
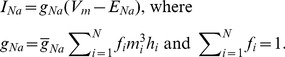 |
(9) |
Three classes of Nav-CLS are considered here: (a) two gNa populations: one intact, one left-shifted by a specified fixed amount (as in [11]); (b) three populations: one intact, two with discrete LS values; and (c) assorted versions of LS of all or specified fractions of the total Nav population, but with fi(LS) following Gaussian distributions to mimic the expectation that nodal injury would produce “smeared” (as opposed to discrete) extents of “Nav-CLS”. At the node of Ranvier, gNa dominates over gK, but previously [11] we briefly discussed the effects of also applying a LS to the gating of the Kv channels.
The following features are also included in the model. Leakage currents [29], INaleak and IKleak, account for the background permeabilities needed to bring the basal system to a quiescent steady-state Vm with an active pump present, along with the usual unspecific leakage current Ileak. These leak currents take the form:
| (9) |
where conductances gNaleak, gKleak and gleak are constants. Parameter values are summarized in Table 1.
With an active Na/K pump, the model system has dynamic intracellular and extracellular Na+ and K+ concentrations, denoted by [Na+]i, [Na+]o, [K+]i, and [K+]o. The Na/K pump balances the accumulation and depletion of ions in the intra- and extracellular space by exchanging 3 internal Na+ for 2 external K+ ions. Thus [K+]o and [Na+]i determine the pump amplitude [29], [87]:
| (10) |
where 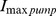 is the maximal current generated by the pump,
is the maximal current generated by the pump, 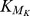 and
and 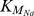 are Michaelis-Menten kinetic constants, and the Na+ and K+ currents flowing through the pump are INapump = 3Ipump and IKpump = −2Ipump. Ion fluxes across the node of Ranvier membrane cause the ion concentration changes:
are Michaelis-Menten kinetic constants, and the Na+ and K+ currents flowing through the pump are INapump = 3Ipump and IKpump = −2Ipump. Ion fluxes across the node of Ranvier membrane cause the ion concentration changes:
| (11) |
| (12) |
where F is the Faraday constant, A is the surface area of the nodal membrane, and 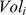 (or
(or 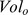 ) is the intracellular (or extracellular) volume of the node of Ranvier under study (we take
) is the intracellular (or extracellular) volume of the node of Ranvier under study (we take 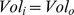 for simplicity). Ion concentration dynamics alter the reversal potentials EIon in Eqs (2–3) by the Nernst equations:
for simplicity). Ion concentration dynamics alter the reversal potentials EIon in Eqs (2–3) by the Nernst equations:
| (13) |
Model equations are solved using a 4-th order Runge-Kutta scheme with 10−3 ms as time step in MATLAB and bifurcation diagrams were plotted using XPPAUT.
Acknowledgments
We thank PA Boucher for sharing code and useful discussions.
Funding Statement
The authors acknowledge funding from NSERC (Canada) (AL and BJ) as well as from a CIHR training grant in Neurophysics (AL). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Lorincz A, Nusser Z (2010) Molecular identity of dendritic voltage-gated sodium channels. Science 328: 906–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maxwell WL (1996) Histopathological changes at central nodes of Ranvier after stretch injury. Microsc Res Tech 34: 522–535. [DOI] [PubMed] [Google Scholar]
- 3. Morris CE, Boucher PA, Joos B (2012) Left-shifted Nav channels in injured bilayer: primary targets for neuroprotective Nav antagonists? Frontiers Pharmacol 3: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morris CE (2011) Why are so many ion channels mechanosensitive? In: Sperelakis N, editor. Cell Physiology Source Book, 4th ed. Amsterdam: Elsevier. pp 493–505.
- 5. Zhang Y, Gao F, Popov VL, Wen JW, Hamill OP (2000) Mechanically gated channel activity in cytoskeleton-deficient plasma membrane blebs and vesicles from Xenopus oocytes. J Physiol 523: 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sheetz MP, Sable JE, Döbereiner HG (2006) Continuous membrane-cytoskeleton adhesion requires continuous accommodation to lipid and cytoskeleton dynamics. Annu Rev Biophys Biomol Struct 35: 417–34. [DOI] [PubMed] [Google Scholar]
- 7. Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH (2001) Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci 21: 1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang JA, Lin W, Morris T, Banderali U, Juranka PF, et al. (2009) Membrane trauma and Na+ leak from Nav1.6 channels. Am J of Physiol 297: C823–34. [DOI] [PubMed] [Google Scholar]
- 9. Morris CE, Juranka PF (2007) Nav channel mechanosensitivity: activation and inactivation accelerate reversibly with stretch. Biophys J 93: 822–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Banderali U, Juranka PF, Clark RB, Giles WR, Morris CE (2010) Impaired stretch modulation in potentially lethal cardiac sodium channel mutants. Channels (Austin) 4: 12–21. [DOI] [PubMed] [Google Scholar]
- 11. Boucher PA, Joós B, Morris CE (2012) Coupled left-shift of Nav channels: modeling Na+ loading and dysfunctional excitability of damaged axons. J Comput Neurosci 32 E-pub ahead of print. [DOI] [PubMed] [Google Scholar]
- 12. Tabarean IV, Juranka P, Morris CE (1999) Membrane stretch affects gating modes of a skeletal muscle sodium channel. Biophys J 77: 758–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shcherbatko A, Ono F, Mandel G, Brehm P (1999) Voltage-dependent sodium channel function is regulated through membrane mechanics. Biophys J 77: 1945–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beyder A, Rae JL, Bernard C, Strege PR, Sachs F, et al. (2010) Mechanosensitivity of Nav1.5, a voltage-sensitive sodium channel. J Physiol 588 Pt 24:4969–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun GC, Werkman TR, Wadman WJ (2006) Kinetic changes and modulation by carbamazepine on voltage-gated sodium channels in rat CA1 neurons after epilepsy. Acta Pharmacol Sin 27: 1537–1546. [DOI] [PubMed] [Google Scholar]
- 16. Yang JJ, Yang Z, Zhang T (2010) Action potential changes associated with impairment of functional properties of sodium channels in hippocampal neurons induced by melamine. Toxicol Lett 198: 171–6. [DOI] [PubMed] [Google Scholar]
- 17. Novak KR, Nardelli P, Cope TC, Filatov G, Glass JD, et al. (2009) Inactivation of sodium channels underlies reversible neuropathy during critical illness in rats. J Clin Invest 119: 1150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rusinova R, Herold KF, Sanford RL, Greathouse DV, Hemmings HC Jr, et al. (2011) Thiazolidinedione insulin sensitizers alter lipid bilayer properties and voltage-dependent sodium channel function: implications for drug discovery. J Gen Physiol 138: 249–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Filatov GN, Pinter MJ, Rich MM (2009) Role of Ca2+ in injury-induced changes in sodium current in rat skeletal muscle. Am J Physiol Cell Physiol 297: C352–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Song XS, Huang ZJ, Song XJ (2009) Thiamine suppresses thermal hyperalgesia, inhibits hyperexcitability, and lessens alterations of sodium currents in injured, dorsal root ganglion neurons in rats. Anesthesiology 110: 387–400. [DOI] [PubMed] [Google Scholar]
- 21. Lenkey N, Karoly R, Epresi N, Vizi E, Mike A (2011) Binding of sodium channel inhibitors to hyperpolarized and depolarized conformations of the channel. Neuropharmacology 60: 191–200. [DOI] [PubMed] [Google Scholar]
- 22. Kovalsky Y, Amir R, Devor M (2009) Simulation in sensory neurons reveals a key role for delayed Na current in subthreshold oscillations and ectopic discharge: implications for neuropathic pain. J Neurophysiol 102: 1430–1442. [DOI] [PubMed] [Google Scholar]
- 23. Choi JS, Waxman SG (2011) Physiological interactions between Na(v)1.7 and Na(v)1.8 sodium channels: a computer simulation study. J Neurophysiol 106: 3173–3184. [DOI] [PubMed] [Google Scholar]
- 24. Rotstein HG, Oppermann T, White JA, Kopell N (2006) The dynamic structure underlying subthreshold oscillatory activity and the onset of spikes in a model of medial entorhinal cortex stellate cells. J Comput Neurosci 21: 271–292. [DOI] [PubMed] [Google Scholar]
- 25.Izhikevich EM (2007) Dynamical systems in neuroscience: The geometry of excitability and bursting. Cambridge, Massachusetts: The MIT Press. 441p.
- 26. Prescott SA, Sejnowski TJ, De Koninck Y (2006) Reduction of anion reversal potential subverts the inhibitory control of firing rate in spinal lamina I neurons: towards a biophysical basis for neuropathic pain. Mol Pain 2: 32–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Coggan JS, Prescott SA, Bartol TM, Sejnowski TJ (2010) Imbalance of ionic conductances contributes to diverse symptoms of demyelination. Proc Natl Acad Sci U S A 107: 20602–20609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Coggan JS, Ocker G, Sejnowski TJ, Prescott SA (2011) Explaining pathological changes in axonal excitability through dynamical analysis of conductance-based models. J Neural Eng 8: 065002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Käger H, Wadman W, Somjen G (2000) Simulated seizures and spreading depression in a neuron model incorporating interstitial space and ion concentrations. J Neurophysiol 84: 495–512. [DOI] [PubMed] [Google Scholar]
- 30. Cressman JR Jr, Ullah G, Ziburkus J, Schiff SJ, Barreto E (2009) The influence of sodium and potassium dynamics on excitability, seizures, and the stability of persistent states: I. Single neuron dynamics. J Comput Neurosci 26: 1 59–170 Erratum in (2011) 30: 781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barreto E, Cressman JR (2011) Ion concentration dynamics as a mechanism for neuronal bursting. J Biol Phys 37: 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Milton RL, Caldwell JH (1990) Na current in membrane blebs: implications for channel mobility and patch clamp recording. J Neurosci 10: 885–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hu W, Tian C, Li T, Yang M, Hou H, et al. (2009) Distinct contributions of Nav1.6 and Nav1.2 in action potential initiation and backpropagation. Nat Neuro 12: 996–1002. [DOI] [PubMed] [Google Scholar]
- 34. Roza C, Castillejo S, Lopez-García JA (2011) Accumulation of Kv7.2 channels in putative ectopic transduction zones of mice nerve-end neuromas. Mol Pain 7: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schmidt D, MacKinnon R (2008) Voltage-dependent K+ channel gating and voltage sensor toxin sensitivity depend on the mechanical state of the lipid membrane. Proc Natl Acad Sci U S A 105: 19276–19281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Finol-Urdaneta RK, McArthur JR, Juranka PF, French RJ, Morris CE (2010) Modulation of KvAP unitary conductance and gating by 1-alkanols and other surface active agents. Biophys J 98: 762–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Morris CE (2011) Voltage-gated channel mechanosensitivity: fact or friction? Front Physiol 2: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dobrota D, Matejovicova M, Kurella EG, Boldyrev AA (1999) Na/K-ATPase under oxidative stress: molecular mechanisms of injury. Cell Mol Neurobiol 19: 141–149. [DOI] [PubMed] [Google Scholar]
- 39. Wu BJ, Else PL, Storlien LH, Hulbert AJ (2001) Molecular activity of Na+/K+-ATPase from different sources is related to the packing of membrane lipids. J Exp Biology 204: 4271–4280. [DOI] [PubMed] [Google Scholar]
- 40. Turner N, Hulbert AJ, Else PL (2005) Sodium pump molecular activity and membrane lipid composition in two disparate ectotherms, and comparison with endotherms. J Comp Physiol B 175: 77–85. [DOI] [PubMed] [Google Scholar]
- 41. Johnson SW, Seutin V, North RA (1992) Burst firing in dopamine neurons induced by N-methyl-D-aspartate: role of electrogenic sodium pump. Science 258: 665–667. [DOI] [PubMed] [Google Scholar]
- 42. Darbon P, Tscherter A, Yvon C, Streit J (2003) Role of the electrogenic Na/K pump in disinhibition-induced bursting in cultured spinal networks. J Neurophysiol 90: 3119–3129. [DOI] [PubMed] [Google Scholar]
- 43. Giraud F, Claret M, Bruckdorfer KR, Chailley B (1981) The effects of membrane lipid order and cholesterol on the internal and external cationic sites of the Na+-K+ pump in erythrocytes. Biochim Biophys Acta 647: 249–258. [DOI] [PubMed] [Google Scholar]
- 44. Sakai R, Hagiwara N, Matsuda N, Kassanuki H, Hosoda S (1996) Sodium-potassium pump current in rabbit sino-atrial node cells. J Physiol 490: 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ha J, Dobretsov M, Kurten RC, Grant DF, Stimers JR (2002) Effect of linoleic acid metabolites on Na(+)/K(+) pump current in N20.1 oligodendrocytes: role of membrane fluidity. Toxicol Appl Pharmacol 182: 76–83. [DOI] [PubMed] [Google Scholar]
- 46. Longtin A, Hinzer K (1996) Encoding with bursting, subthreshold oscillations, and noise in mammalian cold receptors. Neural Comput 8: 215–255. [DOI] [PubMed] [Google Scholar]
- 47. Longtin A (1997) Autonomous stochastic resonance in bursting neurons. Phys Rev E 55: 868–876. [Google Scholar]
- 48. Muratov CB, Vanden-Eijnden E (2008) Noise-induced mixed-mode oscillations in a relaxation oscillator near the onset of a limit cycle. Chaos 18: 015111–015120. [DOI] [PubMed] [Google Scholar]
- 49. Paydarfar D, Forger DB, Clay JR (2006) Noisy inputs and the induction of on-off switching behavior in a neuronal pacemaker. J Neurophysiol 96: 3338–3348. [DOI] [PubMed] [Google Scholar]
- 50. Neiman AB, Yakusheva TA, Russell DF (2007) Noise-Induced Transition to Bursting in Responses of Paddle fish Electroreceptor Afferents. J Neurophysiol 98: 2795–2806. [DOI] [PubMed] [Google Scholar]
- 51. White JA, Klink R, Alonso A, Kay ARA (1998) Noise from voltage-gated ion channels may influence neuronal dynamics in the entorhinal cortex. J Neurophysiol 80: 262–269. [DOI] [PubMed] [Google Scholar]
- 52. Kuske R, Baer SM (2002) Asymptotic analysis of noise sensitivity in a neuronal burster. Bull Math Bio 64: 447–481. [DOI] [PubMed] [Google Scholar]
- 53. Wang YY, Wen ZH, Duan JH, Zhu JL, Wang WT, et al. (2011) Noise enhances subthreshold oscillations in injured primary sensory neurons. Neurosignals 19: 54–62. [DOI] [PubMed] [Google Scholar]
- 54. Yu N, Kuske R, Li YX (2006) Stochastic phase dynamics: Multiscale behavior and coherence measures. Phys Rev E 73: 056205. [DOI] [PubMed] [Google Scholar]
- 55. Lundbaek JA, Birn P, Hansen AJ, Søgaard R, Nielsen C, et al. (2004) Regulation of sodium channel function by bilayer elasticity: the importance of hydrophobic coupling. Effects of micelle-forming amphiphiles and cholesterol. J Gen Physiol 123: 599–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Taddese A, Bean BP (2002) Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron 33: 587–600. [DOI] [PubMed] [Google Scholar]
- 57. Rho YA, Prescott SA (2012) Identification of molecular pathologies sufficient to cause neuropathic excitability in primary somatosensory afferents using dynamical systems theory. PLoS Comput Biol 8: e1002524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yuen TJ, Browne KD, Iwata A, Smith DH (2009) Sodium channelopathy induced by mild axonal trauma worsens outcome after a repeat injury. J Neurosci Res 87: 3620–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schafer DS, Jha S, Liu F, Akella T, McCullough LD, et al. (2009) Disruption of the axon initial segment cytoskeleton is a new mechanism for neuronal injury. J Neurosci 29: 13242–13254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Douglas HA, Callaway JK, Sword J, Kirov SA, Andrew RD (2011) Potent inhibition of anoxic depolarization by the sodium channel blocker dibucaine. J Neurophysiol 105: 1482–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Misawa S, Sakurai K, Shibuya K, Isose S, Kanai K, et al. (2009) Neuropathic pain is associated with increased nodal persistent Na(+) currents in human diabetic neuropathy. J Peripher Nerv Syst 14: 279–284. [DOI] [PubMed] [Google Scholar]
- 62. Xie RG, Zheng DW, Xing JL, Zhang XJ, Song Y, et al. (2011) Blockade of persistent sodium currents contributes to the riluzole-induced inhibition of spontaneous activity and oscillations in injured DRG neurons. PLoS One 6: e18681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hildebrand ME, Smith PL, Bladen C, Eduljee C, Xie JY, et al. (2011) A novel slow-inactivation-specific ion channel modulator attenuates neuropathic pain. Pain 152: 833–843. [DOI] [PubMed] [Google Scholar]
- 64. Ma C, LaMotte RH (2007) Multiple sites for generation of ectopic spontaneous activity in neurons of the chronically compressed dorsal root ganglion. J Neurosci 27: 14059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ma C, Greenquist KW, Lamotte RH (2006) Inflammatory mediators enhance the excitability of chronically compressed dorsal root ganglion neurons. J Neurophysiol 95: 2098–2107. [DOI] [PubMed] [Google Scholar]
- 66. Devor M (2009) Ectopic discharge in Abeta afferents as a source of neuropathic pain. Exp Brain Res 196: 115–128. [DOI] [PubMed] [Google Scholar]
- 67. Fried K, Sessle BJ, Devor M (2011) The paradox of pain from tooth pulp: low-threshold “algoneurons”? Pain 152: 2685–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rakowski RF, Gadsby DC, De Weer P (1989) Stoichiometry and voltage dependence of the sodium pump in voltage clamped, internally dialyzed squid giant axon. J Gen Physiol 93: 903941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tobin AE, Calabrese RL (2005) Myomodulin increases Ih and inhibits the Na/K pump to modulate bursting in leech heart interneurons. J Neurophysiol 94: 3938–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. del Negro CA, Hsiao D-F, Chandler SH (1999) Outward currents influencing bursting dynamics in guinea pigtrigeminal motoneurons. J Neurophysiol 81: 1478–1485. [DOI] [PubMed] [Google Scholar]
- 71. Muddana HS, Chiang HH, Butler PJ (2012) Tuning membrane phase separation using nonlipid amphiphiles. Biophys J 102: 489–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Botta P, de Souza FM, Sangrey T, De Schutter E, Valenzuela CF (2010) Alcohol excites cerebellar Golgi cells by inhibiting the Na+/K+ ATPase. Neuropsychopharmacology 35: 1984–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shah SA, Khan MN, Shah SF, Ghafoor A, Khattak A (2011) Is peripheral alcohol injection of value in the treatment of trigeminal neuralgia? An analysis of 100 cases. Int J Oral Maxillofac Surg 40: 388–392. [DOI] [PubMed] [Google Scholar]
- 74. Amir R, Michaelis M, Devor M (2002) Burst discharge in primary sensory neurons: triggered by subthreshold oscillations, maintained by depolarizing afterpotentials. J Neurosci 22: 1187–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yoshida M, Giocomo LM, Boardman I, Hasselmo ME (2011) Frequency of subthreshold oscillations at different membrane potential voltages in neurons at different anatomical positions on the dorsoventral axis in the rat medial entorhinal cortex. J Neurosci 31: 12683–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nadasdy Z (2010) Binding by asynchrony: the neuronal phase code. Front Neurosci 4: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Choi S, Yu E, Kim D, Urbano FJ, Makarenko V, et al. (2010) Subthreshold membrane potential oscillations in inferior olive neurons are dynamically regulated by P/Q- and T-type calcium channels: a study in mutant mice. J Physiol 588: 3031–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Koizumi H, Smith JC (2008) Persistent Na+ and K+-dominated leak currents contribute to respiratory rhythm generation in the pre-Bötzinger complex in vitro. J Neurosci 28: 1773–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Xing JL, Hu SJ, Long KP (2001) Subthreshold membrane potential oscillations of type A neurons in injured DRG. Brain Res 901: 128–136. [DOI] [PubMed] [Google Scholar]
- 80. Hodgkin AL (1948) The local electric changes associated with repetitive action in a non-medullated axon. J Physiol 107: 165–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Morris C, Lecar H (1981) Voltage oscillations in the barnacle giant muscle fiber. Biophys J 35: 193–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Prescott SA, De Koninck Y, Sejnowski TJ (2008) Biophysical basis for three distinct dynamical mechanisms of action potential initiation. PLoS Comput Biol 4: e1000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ricci AJ, Crawford AC, Fettiplace R (2003) Tonotopic variation in the conductance of the hair cell mechanotransducer channel. Neuron 40: 983–990. [DOI] [PubMed] [Google Scholar]
- 84. Levic S, Yamoah EN (2011) Plasticity in membrane cholesterol contributes toward electrical maturation of hearing. J Biol Chem 286: 5768–5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hodgkin AL, Huxley AF (1952) A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117: 500–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ochab-Marcinek A, Schmid G, Goychuk O, Hänggi P (2009) Noise-assisted spike propagation in myelinated neurons. Phys Rev 79: 011904. [DOI] [PubMed] [Google Scholar]
- 87.Läuger P (1991) Electrogenic ion pumps, Distinguished lecture series of the society of general physiologists. Sunderland, Massachusetts: Sinauer Associates. 313p.