

Abstract
This paper describes an enzymatic approach to obtain a thio-containing UDP-GlcNAc analog. We use an assay based on capture of the carbohydrate and analysis by mass spectrometry to quantitatively characterize the activity of this unnatural sugar donor in a LgtA-mediated glycosylation reaction.
Keywords: Enzymatic synthesis, UDP-GlcNAc, Glycosylation, SAMDI MS
1. Introduction
N-Acetylglucosamine (GlcNAc) is prevalent in nature and plays a significant role in several biological contexts. For example, alternating units of GlcNAc and N-acetylmuramic acid (MurNAc) form the peptidoglycan of bacterial cell walls.1–2 Typical N-linked glycoproteins are characterized by a glycosidic bond between a GlcNAc residue and an asparagine residue of the protein, and recent work has pointed to the importance of the O-GlcNAc modification—generated by the GlcNAcylation of serine and threonine residues of proteins—in modulating the functions and activities of proteins in the cell.3–4 The biosynthesis of GlcNAc-containing biomolecules normally employs glycosyltransferases of the Leloir pathway5 and uridine-5′-O-diphospho-GlcNAc (UDP-GlcNAc) as a glycosyl donor molecule.6 This donor is obtained from enzymatic pyrophosphorylation of GlcNAc-1-phosphate (GlcNAc-1-P), which is biosynthesized from either glucose or GlcNAc in a process comprising several steps including kinase-mediated 6-phosphorylation.7 We have previously reported that NahK (EC 2.7.1.162)8, which is an N-acetylhexosamine 1-kinase found in Bifidobacterium longum, could produce GlcNAc-1-P with high efficiency and is tolerant of a broad range of GlcNAc analogues.9–10 The resulting GlcNAc-1-P analogs could be further converted into the corresponding UDP-GlcNAc analogs in a reaction catalyzed by GlmU (EC 2.3.1.157 and EC 2.7.7.23)11, a GlcNAc-1-P uridyltransferase (pyrophosphorylase) from Escherichia coli.12 Since replacement of an oxygen atom with a sulfur in phosphate esters has been exploited frequently in mechanistic studies as well as in sugar nucleotides,13–16 here we reported the application of our two-enzyme system to synthesize GlcNAc-1-thiophosphate (1) using γ(S)ATP as an alternative phosphoryl donor and UDP(βS)-GlcNAc (2) (Scheme 1).17
Scheme 1.

Enzymatic synthesis of GlcNAc-1-thiophosphate (1) and UDP(βS)-GlcNAc (2).
2. Results and discussion
As shown in Scheme 1, we used NahK to transfer the γ-thiophosphate from a commercially available γ(S)ATP to construct GlcNAc-1-thiophosphate (1) in good yield (82% yield, >10 mg scale). We then used GlmU to catalyze a pyrophosphorylation reaction between GlcNAc-1-thiophosphate (1) and UTP to produce the corresponding UDP(βS)-GlcNAc (2) (43% yield, 9 mg of product obtained). A yeast inorganic pyrophosphatase (PPA) was added to the reaction to degrade the byproduct pyrophosphate (PPi) and increase the yield. We assigned the product as the β-thiophosphate product based on 31P NMR, which showed a downfield shift (~44 ppm) of the phosphorous atom due to its bonding to the sulfur atom, whereas the shift for the α-phosphorus atom was unchanged.13–14
We evaluated the ability of the Neisseria meningitidis 1,3-N-acetylglucosaminyltransferase (LgtA) to use the analog as a donor substrate in a glycosylation reaction.18 The LgtA-mediated glycosylation reactions were performed on self-assembled monolayers (SAMs) of alkanethiolates on gold that presented lactose acceptors at a density of 10% against a background of tri(ethylene glycol) groups and using UDP-GlcNAc or UDP(βS)-GlcNAc (2) as the donor. (Figure 1a). The glycol groups of the monolayer prevent non-specific interactions of the enzyme with the surface and maintain the activity of the immobilized carbohydrates.19 The assay was performed by applying a drop containing LgtA and either UDP-GlcNAc or UDP(βS)-GlcNAc (2) to the monolayer, allowing the mixture to react for a short time, and then rinsing the surface and analyzing the monolayer using matrix assisted laser desorption-ionization mass spectrometry (in a technique termed SAMDI MS) to reveal the masses of the substituted alkanethiols. Hence, this method can directly observe both the substrate and the product of the LgtA-mediated reaction, erasing the need for labels and also providing a measure of the yield for the glycosylation reaction. The assay is also notable because it requires only two microliters of sample and therefore minimizes the use of expensive reagents. Representative spectra for the glycosylation are shown in Figure 1b. The peaks at m/z 1835 and 1851 are the sodium and potassium adducts, respectively, of the lactose-terminated disulfide. After a 2 h reaction, the mass spectrum revealed new peaks at m/z 2038 and 2054 corresponding to the trisaccharide product. Hence, the analog described above is a substrate for LgtA. For the glycosyltransfer reaction shown in Figure 1a (using either natural UDP-GlcNAc or unnatural donor UDP(βS)-GlcNAc (2)), we performed a kinetic experiment by repeating the experiment for reaction times ranging up to 2 hours and show plots of the time course for both natural UDP-GlcNAc and UDP(βS)-GlcNAc in Figure 1c. It is apparent that the analog reacts much slower than the natural UDP-GlcNAc during the glycosyltransfer reaction. Additionally, when both donors were present in the same reaction, the reaction/time course was quite similar to that of natural UDP-GlcNAc alone, indicating the analog does not inhibit the activity of LgtA.
Figure 1.

To further understand the consequences of the sulfur substituted donor on enzyme activity, we first explored the influence of divalent ions on activity. Assays were performed in solution to avoid perturbations that may arise from presentation of the ligands at the surface. In this ‘pull-down’ format20, the reactions were performed in solution with an azido-modified lactose as the acceptor and either UDP-GlcNAc or UDP(βS)-GlcNAc (2) as the donor (Figure 2). The reactions were stopped by adding cold ethanol and then spotted onto a monolayer that presented a terminal alkyne group among tri(ethylene glycol) groups (For details see Experimental Section). In this way, a copper (I)-catalyzed click reaction between the azido sugars and the alkyne monolayer served to immobilize both the trisaccharide product and disaccharide acceptor, so that SAMDI analysis can then determine the extent of glycosylation (Figure 2). Some glycosyltranferases are known to require the presence of a divalent metal cation for enzymatic activity, and thus it is commonplace to assay the transferase. Different enzymes can utilize different cation metals in vivo, however which metal is actually used cannot be determined. Thus, we tested a panel of divalent metal cations to determine the optimal substrate for in vitro enzymatic activity. Table 1 summarizes the relative activities of different divalent ions for both of the donors. We find that the two donors share a similar metal ion preference for divalent manganese, magnesium and calcium over the other metal ions. Iron, molybdenum, palladium and cobalt salts result in diminished activity. The addition of EDTA totally abolished the activity of the enzyme. This similarity suggests that the divalent metal binds in comparable fashion to the pyrophosphate portion of both donors and the oxygen atoms, instead of the sulfur, probably make major contribution to the interaction.
Figure 2.
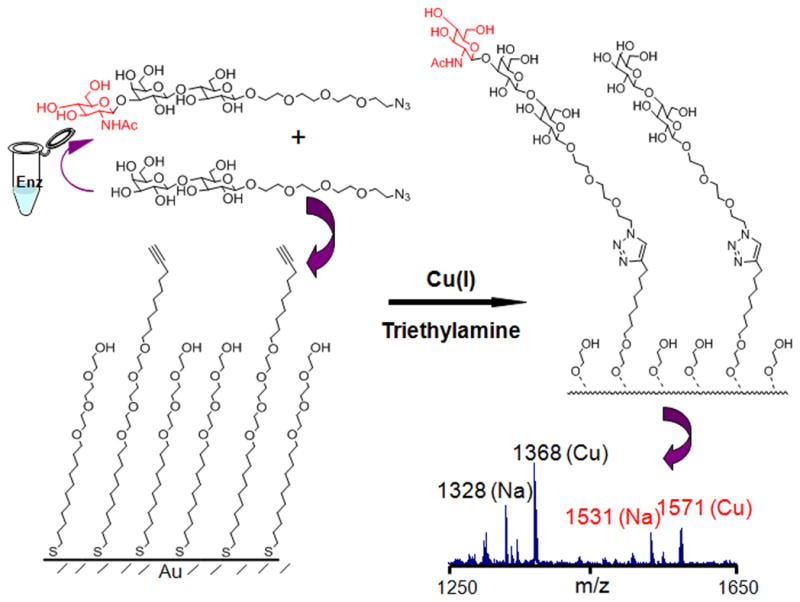
Glycosylation reactions were performed in solution using an azido modified substrate in order to determine the kinetic parameters for UDP-GlcNAc and the synthesized analog. Reactions were terminated at different time points and then applied to the monolayers to allow immobilization and SAMDI characterization of both the lactose substrate and trisaccharide product. The lower panel shows the spectrum from a 120 min reaction of UDP(βS)-GlcNAc. Black and red label the peaks of lactose and the trisaccharide products, respectively. Letters in parenthesis showed the different ion adducts appearing in the spectrum.
Table 1.
Effect of divalent ions and EDTA on the activity of UDP(βS)-GlcNAc and UDP-GlcNAc. Numbers in parenthesis are standard deviations of three parallel experiments.
| Cations (10 mM) | Relative Activity (%) UDP-GlcNAc |
Relative Activity (%) UDP(βS)-GlcNAc (2) |
|---|---|---|
| Mn2+ | 100 | 100 |
| Ca2+ | 115.9 (11.2) | 12.5 (1.33) |
| Mg2+ | 98.6 (7.9) | 92.6 (2.3) |
| Fe2+ | 51.2 (2.2) | 5.7 (0.98) |
| Mo2+ | 35.5 (3.62) | 21.7 (1.1) |
| Pd2+ | 21.5 (2.2) | 3.9 (0.55) |
| Co2+ | 8.0 (0.32) | 5.6 (1.28) |
| Cu2+ | <1 | <1 |
| Hg2+ | <1 | <1 |
| Ni2+ | <1 | <1 |
| Zn2+ | <1 | <1 |
| EDTA | <1 | <1 |
The kinetic parameters for enzyme-mediated transfer of the analog were also determined using the ‘pull-down’ format and SAMDI characterization. The enzyme assays were carried out by varying the concentration of one substrate in the presence of different fixed concentrations of the other. The initial velocities were plotted graphically in double reciprocal form to check the linearity of the relationship and to determine the patterns of the plots (Supplementary Information, Figure S1). The plots showed intersecting patterns and with the donor as the variable substrate, the plots intersect on the y axis. We therefore fit the data using Cleland’s equation for an ordered bi-bi model21: v0 = VAB/(KiAKb+KbA+KaB+AB). (v0: initial velocity; A and B: concentration of the variable substrate and fixed substrate, respectively; KiA: inhibition constant of A; Ka and Kb: apparent Michaelis-Menten constants) The kinetic parameters determined from a best-fit of the data are summarized in Table 2. Kinetic parameters for UDP-GlcNAc are consistent with the previously reported values.18 The Km for the analog, by contrast, is about 150-fold greater and the Vmax is substantially decreased. This decrease in activity likely owes to the lower electronegativity of the sulfur relative to the oxygen atom, which makes the uridine-5′-O-β-thiodiphosphate a poorer leaving group in the glycosyltransfer reaction. It is also possible that larger sulfur atom can result in a less favorable interaction between the anomeric center of the donor and the hydroxyl group of the acceptor in the transition state mimicking the oxo-carbenium ion of the SN2 glycosyltransfer reaction.
Table 2.
Summary of kinetic data for LgtA catalyzed reaction using azido modified lactose and UDP(βS)-GlcNAc and UDP-GlcNAc as substrates. a: azido modified lactose; b: donor. The number is reported as the average of three parallel experiments. The number in parenthesis shows the standard error.
| Ka,Apparant (mM) | Kia (mM) | Kb (mM) | Vmax (mM min−1) | [E0] (mg mL−1) | |
|---|---|---|---|---|---|
| UDP(βS)-GlcNAc (2) | 6.08 (0.93) | 15.0 (2.75) | 51.5 (4.3) | 0.211 (0.194) | 1.63 |
| UDP-GlcNAc | 5.84 (0.71) | 14.8 (1.18) | 0.40 (0.22) | 0.456 (0.131) | 0. 204 |
3. Conclusion
Replacement of an oxygen atom with a sulfur in phosphate esters has been exploited frequently in mechanistic studies.15–16 For example, uridine-5′-O-(2-thiodiphosphoglucose) and uridine-5′-O-(2-thiodiphosphoglucuronic acid) were enzymatically synthesized and their properties towards related enzymes were explored.13,22 Here we provide another example where the milligram scale synthesis of sulfur containing UDP-GlcNAc analog was easily achieved using a two-enzyme pathway. Furthermore, our work took advantage of the combination of self-assembled monolayers and MALDI MS to assay for glycosyltransferase activities. This assay is convenient, label-free and also requires very little material to achieve both quantitative and qualitative measurements.23–24 This work will benefit the investigation of more unnatural sugar donors which have chemical or biological significance in the future.
4. Experimental
4.1 General
Adenosine 5′-[γ-thio]triphosphate tetralithium salt (γ(S)ATP), N-acetyl-D-glucosamine hydrochloride were purchased from Sigma-Aldrich. Unless indicated, all commercial reagents were used as received without further purification. Analytical TLC was carried out on silica gel 60 F254 aluminum-backed plates (E. Merck). The 200–400 mesh size of the same absorbent was utilized for all chromatographic purifications. Unless noted, all compounds isolated by chromatography were sufficiently pure by 1H NMR analysis for use in subsequent reactions. Direct On-Chip MALDI-TOF mass spectrometry is performed on Applied Biosystems™ 4800 plus MALDI TOF/TOF mass spectrometer.
4.2 NMR and HRMS data for GlcNAc-1-thiophosphate (1)
1H NMR (500 MHz, D2O) δ 5.57 (dd, J = 10.0, 3.2 Hz, 1H), 4.01–4.05 (m, 1H), 3.97–3.99 (m, 1H), 3.94 (dd, J = 12.2, 2.2 Hz, 1H), 3.80–3.87 (m, 2H), 3.54 (app t, J = 9.3 Hz, 1H), 2.11 (s, 3H); 13C NMR (125 MHz, D2O) δ 174.8, 92.9 (d), 72.4, 71.5, 70.0, 60.7, 54.1 (d), 22.3; 31P NMR (162 MHz, D2O) δ 43.3; HRMS (ESI) calcd for C8H15NO8PS (M - H)− 316.0261, found 316.0267 m/z.
4.3 NMR and HRMS data for UDP(βS)-GlcNAc (2)
1H NMR (400 MHz, D2O) δ 8.03 (d, J = 8.2 Hz, 1H), 6.01–6.04 (m, 2H), 5.72 (dd, J = 9.8, 3.3 Hz, 1H), 4.41–4.47 (m, 2H), 4.25–4.34 (m, 3H), 3.98–4.04 (m, 2H), 3.82–3.92 (m, 3H), 3.61 (app t, J = 9.8 Hz, 1H), 2.10 (s, 3H); 13C NMR (100 MHz, D2O) δ 174.8, 166.3, 151.9, 141.8, 102.8, 94.7 (d), 88.3, 83.4 (d), 73.8, 73.2, 70.9, 69.9, 69.5, 65.1 (d), 60.2, 53.7 (d), 22.1; 31P NMR (162 MHz, D2O) δ 42.6 (d, J = 29.4 Hz), −12.1 (d, J = 29.3 Hz); HRMS (ESI) calcd for C17H26N3O16P2S (M - H)− 622.0514, found 622.0531 m/z.
4.4 Preparation of self-assembled monolayers on gold coated slides
The gold substrate was prepared as previously reported.24 Briefly, glass coverslips were cleaned by sonication for 30 min first in deionized ultrafiltered (DIUF) water and then in ethanol and dried under a stream of nitrogen. Titanium (5 nm) and gold (50 nm) were evaporated onto the glass coverslips using an electron beam evaporator (Thermionics) at a rate of 0.05–0.10 nm s−1 and at a pressure of 1.0 × 10−6 Torr. The azido modified lactose and alkyne-terminated alkanethiol (as shown in Figure 2) were prepared as previously reported.25–26 Monolayers were prepared as described previously.26 Briefly, gold-patterned slides were immersed in an ethanolic solution of alkyne-terminated alkanethiol (or lactose-terminated disulfide) and tri(ethylene glycol)-terminated alkanethiol (or disulfide) in a ratio of 1:9 for 12 h at room temperature (total concentration of alkanethiol or disulfide: 1 mM). The substrates were washed with ethanol and dried under nitrogen.
4.5 Enzyme assays
The enzyme buffer used in both the on-chip and pull-down assay was Tris-HCl (100 mM, pH 7.5) with MnCl2 or other divalent ions (10 mM). For the on-chip assay, 2 μL reaction cocktail, which contains the enzyme buffer, LgtA (0.816 mg mL−1) and one of the donors (2 mM), was applied to the lactose-presenting monolayer on the gold-patterned slide. Reactions were carried out for times ranging from 5 to 120 min for the reaction progress plots and stopped by adding 1 μL ethanol to the corresponding gold chip and quickly removing the mixture by pipetting. At the end of the last reaction, the slide was rinsed with water, ethanol and dried under nitrogen. For the in-solution assay, the reactions for each donor were performed under the same conditions except for higher LgtA concentration (1.63 mg mL−1) for UDP(βS)-GlcNAc. The reactions for measuring relative activities of different divalent ions were stopped at 10 min for each metal. The reactions for kinetics measurements were carried out for times ranging from 2 min to 30 min for UDP-GlcNAc and 2 min to 60 min for UDP(βS)-GlcNAc with intervals of 2 or 3 minutes for the former and 5 min for the latter and stopped by adding cold ethanol mixed with 1 mM EDTA. At the end of the last reaction, a volume of 1 μL of the reaction mixture from each time point was transferred onto individual gold chips of the same sizes modified with the alkyne-terminated monolayer. An aqueous solution (1 μL per chip) containing copper bromide (2 mM) and triethylamine (0.5 mM) was applied to each circle and the reactions were incubated at room temperature for 30 min. The slide was then rinsed with water, ethanol and dried under nitrogen. For SAMDI measurement, monolayers were treated with matrix solution (2,4,6-trihydroxyacetophenone, 5 mg mL−1 in acetone). The slide was allowed to dry under atmospheric pressure and loaded in the MALDI TOF mass spectrometer. A 355 nm Nd:YAG laser was used as the desorption/ionization source with an accelerating voltage of 20 kV and extraction delay time of 50 ns. All spectra were acquired using positive reflector mode, and were Gaussian-smoothed and baseline corrected. For quantification, the extent of glycosylation (R) was determined from the relative peak intensity for product (Ip) and lactose substrate (Is) on SAMDI spectra using R = Ip/(Ip+Is). Previous report shows that this peak area ratio represents the actual ratio in solution in these concentration ranges.27 For determination of kinetics parameters, the donor substrate concentrations range from from 10 mM to 66.7 mM and the acceptor ranges from 5 mM to 30 mM. Double reciprocal plots of initial velocities (primary plots) are plotted in Supporting Figure 1 (see supporting information). The kinetics parameters are obtained from the secondary plots (Supporting Figure 2) derived from the primary plots (see supplementary information).
Supplementary Material
Acknowledgments
P.G.W. acknowledges NIH (R01 AI083754, R01 HD061935 and R01 GM085267) for financial support.
Footnotes
Supplementary data (NMR spectra for compound 1 and 2 and supplementary figures) associated with this article can be found, in the online version, at doi:10.1016/j.carres..
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goffin C, Ghuysen JM. Microbiol Mol Biol Rev. 2002;66:702–738. doi: 10.1128/MMBR.66.4.702-738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hesek D, Lee M, Morio K, Mobashery S. J Org Chem. 2004;69:2137–2146. doi: 10.1021/jo035583k. [DOI] [PubMed] [Google Scholar]
- 3.Rexach JE, Clark PM, Hsieh-Wilson LC. Nat Chem Biol. 2008;4:97–106. doi: 10.1038/nchembio.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zachara NE, Hart GW. Chem Rev. 2002;102:431–438. doi: 10.1021/cr000406u. [DOI] [PubMed] [Google Scholar]
- 5.Leloir LF. Science. 1971;172:1299–1303. doi: 10.1126/science.172.3990.1299. [DOI] [PubMed] [Google Scholar]
- 6.Kornfeld R, Kornfeld S. Annu Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 7.Barreteau H, Kovac A, Boniface A, Sova M, Gobec S, Blanot D. FEMS Microbiol Rev. 2008;32:168–207. doi: 10.1111/j.1574-6976.2008.00104.x. [DOI] [PubMed] [Google Scholar]
- 8.Nishimoto M, Kitaoka M. Appl Environ Microbiol. 2007;73:6444–6449. doi: 10.1128/AEM.01425-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai L, Guan W, Wang W, Zhao W, Kitaoka M, Shen J, O’Neil C, Wang PG. Bioorg Med Chem Lett. 2009;19:5433–5435. doi: 10.1016/j.bmcl.2009.07.104. [DOI] [PubMed] [Google Scholar]
- 10.Cai L, Guan W, Kitaoka M, Shen J, Xia C, Chen W, Wang PG. Chem Commun. 2009:2944–2946. doi: 10.1039/b904853g. [DOI] [PubMed] [Google Scholar]
- 11.Olsen LR, Roderick SL. Biochemistry. 2001;40:1913–1921. doi: 10.1021/bi002503n. [DOI] [PubMed] [Google Scholar]
- 12.Guan W, Cai L, Fang J, Wu B, George Wang P. Chem Commun. 2009:6976–6978. doi: 10.1039/b917573c. [DOI] [PubMed] [Google Scholar]
- 13.Singh AN, Newborn JS, Raushel FM. Bioorg Chem. 1988;16:206–214. [Google Scholar]
- 14.Kowalska J, Lewdorowicz M, Darzynkiewicz E, Jemielity J. Tetrahedron Lett. 2007;48:5475–5479. [Google Scholar]
- 15.Eckstein F. J Am Chem Soc. 1970;92:4718–4723. doi: 10.1021/ja00718a039. [DOI] [PubMed] [Google Scholar]
- 16.Frey PA. Tetrahedron. 1982;38:1541–1567. [Google Scholar]
- 17.Zhao G, Guan W, Cai L, Wang PG. Nat Protoc. 2010;5:636–646. doi: 10.1038/nprot.2010.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blixt O, van Die I, Norberg T, van den Eijnden DH. Glycobiology. 1999;9:1061–1071. doi: 10.1093/glycob/9.10.1061. [DOI] [PubMed] [Google Scholar]
- 19.Su J, Mrksich M. Angew Chem Int Ed. 2002;41:4715–4718. doi: 10.1002/anie.200290026. [DOI] [PubMed] [Google Scholar]
- 20.Min DH, Yeo WS, Mrksich M. Anal Chem. 2004;76:3923–3929. doi: 10.1021/ac049816z. [DOI] [PubMed] [Google Scholar]
- 21.Morrison JF, Ebner KE. J Biol Chem. 1971;246:3977–3984. [PubMed] [Google Scholar]
- 22.Klinger MM, McCarthy DJ. Bioorg Med Chem Lett. 1992;2:197–200. [Google Scholar]
- 23.Mrksich M. ACS Nano. 2008;2:7–18. doi: 10.1021/nn7004156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ban L, Mrksich M. Angew Chem Int Ed. 2008;47:3396–3399. doi: 10.1002/anie.200704998. [DOI] [PubMed] [Google Scholar]
- 25.Blixt O, Vasiliu D, Allin K, Jacobsen N, Warnock D, Razi N, Paulson JC, Bernatchez S, Gilbert M, Wakarchuk W. Carbohydr Res. 2005;340:1963–1972. doi: 10.1016/j.carres.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Thiara PS, Mrksich M. Langmuir. 2007;23:11826–11835. doi: 10.1021/la701638d. [DOI] [PubMed] [Google Scholar]
- 27.Guan W, Ban L, Cai L, Li L, Chen W, Liu X, Mrksich M, Wang PG. Bioorg Med Chem Lett. 2011;21 doi: 10.1016/j.bmcl.2011.1004.1100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.