

Abstract
Histone deacetylase inhibitors (HDACi) are a relatively new class of chemotherapy agents. Herein, we report a click-chemistry based approach to the synthesis of HDACi. Fourteen agents were synthesized from the combination of two alkyne and seven azido precursors. The inhibition of HDAC1 and HDAC8 was then determined by in vitro enzymatic assays, after which the cytotoxicity was evaluated in the NCI human cancer cell line screen. A lead compound 5g (NSC746457) was discovered that inhibited HDAC1 at an IC50 value of 104 ± 30 nM and proved quite potent in the cancer cell line screen with GI50 values ranging from 3.92 μM to 10 nM. Thus, this click HDACi design has provided a new chemical scaffold that has not only revealed a lead compound, but one which is easily amendable to further structural modifications given the modular nature of this approach.
Keywords: HDAC, histone, deacetylase, click chemistry, inhibitor, synthesis
Introduction
Posttranscriptional modifications of histones are important processes in epigenetics.1–3 The reversible acetylation of Lys residues,4–6 for example, can help control the remodeling status of chromatin via charge-charge interactions between negatively charged DNA and neutral or positively charged histones. The removal and addition of these acetyl groups is catalyzed by the enzymes histone deacetylase (HDAC) and histone acetyltransferase (HAT), respectively. In the past ten years, studies on this process have attracted an increasing amount of attention, due in part to the observations that aberrant hypoacetylation of histones frequently occurs in tumor cells, resulting in the silencing of specific (tumor suppressor) genes.7 Accordingly, clinical and biological studies have shown that inhibition of HDACs can selectively inhibit cancer cell growth.8 It is has also been revealed that HDACs have a panel of non-histone targets, many of which are involved in tumorigenesis.9–17 All of these features thus establish HDAC as an attractive target in cancer chemotherapy.18–21
Thus far, eighteen human HDAC subtypes have been identified and accordingly divided into four classes based upon their homology to yeast HDACs.20 Class I (yeast transcriptional regulator RPD3) and II (yeast Hda 1) HDACs require a zinc ion to mediate deacetylation and are therefore mechanistically different from the Class III (SIR2 yeast family) NAD+ dependent HDACs. While HDAC 11 possesses similar catalytic activity to Class I and II HDACs in terms of requiring a zinc ion, it actually serves as the sole member of Class IV due to low overall sequence similarity with Classes I and II.
In the case of the aforementioned Zn(II) dependant HDAC isozymes, the active site structure was first revealed by a homolog of the hyperthermophilic bacterium Aquifex aeolicus in 1999.22 In general, the active site is a tube-like pocket that accommodates the side chain of client Lys residues. Located at the bottom of this pocket is the Zn(II) cofactor, which acts as a Lewis acid to catalyze hydrolysis of the acetylated Lys side chain. The opening region (also called the cap region) of the active site consists of multiple loops and is thus highly malleable.
From this elucidation of the structure of the HDAC active site, progress on HDACi design has been greatly facilitated (Figure 1). In most cases, the reported inhibitor is composed of the following three moieties: a Zn(II) binding moiety, a spacer moiety and a “cap” moiety. Of the demonstrated Zn(II) binding moieties, the hydroxamic acid (HA) functionality is the most common. For example, the HA moiety exists in the naturally occurring compound TSA as well as in the clinically approved drug SAHA. In spite of the strong affinity of HA with Zn(II), however, the in vivo efficacy of HA-containing HDACi is impaired by the short clearance time of these agents.23–25 Therefore, non-HA HDACi design has been explored.26,27 These non-HA moieties include electrophilic ketones (e.g. 1 in Figure 1), O-aminoanilides (e.g. MS-275), thiols (or the pro-drug forms) (e.g. FK-228 and 2), and a variety of others. However, in terms of binding affinity, none are as strong as HA. As for the spacer moiety, proper length is the key factor in successful HDAC inhibition. For example, for SAHA and its analogs, six CH2 units are preferred, whereas chiral-center-&-HA-containing HDACi (e.g. CHAPs and 3) prefer five CH2 units between the HA and the chiral center.28–30 Similarly, chiral-center-&-thiol-containing HDACi (e.g. 2) prefer five CH2 units in the spacer moiety. In addition to these length requirements, introduction of an unsaturated moiety within the spacer region has also proved to be a valuable feature in enhancing potency (e.g. TSA, MS275, (S)-HDAC-42 and 4). Such value appears to arise from π-π stacking interactions between the unsaturated moiety and two conserved Phe residues that occur near the midpoint of the tube-like pocket (for HDAC8, they are Phe152 and Phe208) in the eleven Zn(II) dependant human HDACs.31 In regards to the “cap” moiety, a hydrophobic substituent, especially an aromatic substituent, is frequently incorporated. It should be noted that structure derivatization at the “cap” region is a key strategy in current HDACi design, since topological differences are observed in the corresponding “cap” regions of HDAC isozymes.
Figure 1.

Current HDACi.
Herein, we report a HDACi scaffold that is built upon a Huisgen 1,3-dipolar cycloaddition of alkyne and azido precursors (Figure 2). This reaction has been widely applied in chemical synthesis in recent years in efforts to develop new drugs.32–35 It is now recognized as the premier example of the click reaction due to the convenient availability and high stability of the alkyne and azide precursors, as well as the mild reaction conditions and nearly quantitative yields. In our design, the precursors corresponding to the “cap” moiety of the HDACi contain an azido group, whereas the zinc chelating functionality precursors contain an alkyne group. The “clicked” products have a triazole ring in the spacer moiety, which may enhance the HDAC binding affinity via π-π stacking interactions. We have accordingly named these HDACi as click HDACi. Currently, fourteen click HDACi (5a–f and 6a–f) have been prepared from two alkyne precursors (7 and 8) and seven azido-substituted precursors (9a–g) via this combinatorial approach. From these fourteen HDACi, a lead compound, 5g, was discovered which exhibited an in vitro potency comparable to that of SAHA which was clinically approved in 2007 as a chemotherapy agent.
Figure 2.

Combinatorial Design of HDACi viaClick Chemistry.
Results and Discussion
The seven azido precursors 9a–g (Scheme 1) were synthesized from the corresponding halide substrates 10a-g via treatment with 0.5 M sodium azide in DMSO (Scheme 1).36 All products, with the exceptions of 9c and 9d, were obtained after overnight stirring. In the case of 9e, however, the reaction had to be carried out at 0°C to provide satisfactory yields. For 9c and 9d, tetraethylammonium bromide was added to catalyze the reactions, which were subsequently complete after two days. Excluding 9e, all products could be used directly in the following steps without column purification.
Scheme 1.

Preparation of the Azido Precursors 9a–g.
The preparation of the alkyne precursor 8 began with the synthesis of the PMB-protected hydroxylamine 13 via established procedures (Scheme 2).37 Subsequent amidation of 13 with the commercially available acid 14 provided intermediate 8 in 61% yield.
Scheme 2.

Preparation of Alkyne Precursor 8.
In contrast, synthesis of the alkyne precursor 7 (Scheme 3) was initiated through conversion of the propiolic acid 15 to the trans-iodopropenoic acid 16 in 75% yield according to reported procedures.38 The synthesis of 18 from 16 utilized a known route, which was employed to prepare the corresponding (Z)-isomer.39 This route entailed initial protection of the acid 16 with MEMCl to furnish the ester 17 (86% yield). 18 was subsequently obtained in 87% yield via a Sonogashira reaction. Deprotection of 18 with HCl to afford 19 (quantitative) followed by treatment with oxalyl chloride and a catalytic amount of DMF yielded an acid chloride intermediate. This acid chloride was used directly in the next step in which DIPEA & 13 (2:1) were added within 1 min at −10 °C to give 7 in 84% yield. Such rapid addition of the amine 13 was necessary as slow addition dramatically increased the yield of the by-product 20.
Scheme 3.

Preparation of Alkyne Precursor 7.
Following preparation of these two libraries, a series of click reactions between alkyne precursors 7 and 8 and the seven azido precursors 9a–g were performed to afford intermediates 21a–g and 22a–g (Scheme 4). In this combinatorial chemistry step, the alkyne precursors were first desilylated with CsF (1 equiv.). Following workup, without purification, the crude desilylation intermediates were treated with 9a–g in the presence of a Cu(I) catalyst to give 21a–g and 22a–g. The purified products were then treated with TFA to yield the click HDACi 5a–g and 6a–g. These final target compounds were purified by C-18 reverse phase column chromatography due to the presence of an unidentified reddish impurity which appeared upon addition to normal phase silica gel.
Scheme 4.

Combinatorial Syntheses of Click HDACi.
In order to assess the biological activity of these fourteen compounds, they were first tested for inhibition of HDAC. Specifically, HDAC1 and HDAC8 were chosen for the preliminary screen. Selection of these two isozymes stemmed from the previously reported correlation between HDAC1 inhibition and cancer cell growth inhibition,40–44 as well as the existence of preferences among several HDAC inhibitors for HDAC1 or HDAC8.29,31,45,46 Accordingly, preliminary screening at a concentration of 0.50 μM (Table 1) was conducted. This screening revealed that one of the compounds, 5g, possessed an inhibitory potency towards HDAC1 that was quite similar to that of SAHA. In addition, several other inhibitors (5a-5f, 6a, 6b, 6g) possessed activity against HDAC1, albeit to a lesser extent than either SAHA or 5g. Of particular importance in this regard is 6g, given that it showed only 10 ± 1% inhibition at the concentration tested. This is notable due to the fact that it and 5g, which demonstrated 75 ± 5% inhibition, only differ in the length of the linker region. This suggests that a longer linker length considerably aids HDAC1 inhibition in this case.
Table 1.
Activity of all HDACi in the human HDAC1 and HDAC8 inhibition tests
| SAHA | 5a | 5b | 5c | 5d | 5e | 5f | 5g | 6a | 6b | 6c | 6d | 6e | 6f | 6g | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HDAC 1 | 72 ± 2 | 31 ± 4 | 10 ± 3 | 8 ± 2 | 13 ± 4 | 8 ± 2 | 15 ± 3 | 75 ± 5 | 18 ± 3 | 9 ± 1 | NS | NS | NS | NS | 10 ± 1 |
| HDAC 8 | NS | 12 ± 6 | NS | NS | NS | NS | NS | NS | 12 ± 2 | 12 ± 2 | NS | 26 ± 3 | NS | NS | 13 ± 2 |
The values given represent the percentage by which the HDAC activity was inhibited by a 0.50 μM HDACi treatment.
All of the assays were repeated at least three times.
When the inhibition capacity was less than 5%, NS (meaning no significant inhibition) was assigned.
While the aforementioned ten compounds showed inhibition against HDAC1, only five of the fourteen (5a, 6a, 6b, 6d, 6g) demonstrated any significant inhibition of HDAC8. Of these five, four also possessed activity against HDAC1 with 6d being the exception. Nonetheless, none of the inhibitors demonstrated as high of a degree of inhibition of HDAC8 as that seen with the inhibition of HDAC1 by SAHA and 5g.
Before better characterizing the activity of these compounds against HDAC, a list of the fourteen inhibitors was submitted to the National Cancer Institute (NCI) for acceptance into the human cancer cell line screening program which is provided at no cost. Of the fourteen, only one, 5g, ultimately advanced to the final five-dose screen. This selection for advancement stemmed from the results of the single-dose preliminary screen in which the percent inhibition of cell line growth was determined through comparison to untreated samples as described in detail in the Supplementary Information. Furthermore, in the five-dose screen (Figure 4), in which data for SAHA is included to serve as a standard, 5g proved to be active against each of the cancer cell lines in the screen, albeit to varying extents. The differences among the cell lines, however, were generally within or close to one order of magnitude. For example, the GI50 values obtained against the various leukemia cell lines all fell within the range of 0.249 – 2.99 μM. This same trend appeared among the other cancer types for which the ranges were as follows: non-small cell lung 0.199 – 2.26 μM, colon 0.347 – 2.07 μM, CNS 0.318 – 1.53 μM, melanoma 0.271 – 1.08 μM, ovarian 0.187 – 3.21 μM, renal 0.0100 – 2.38 μM, prostate 0.580 – 1.30 μM, and breast 0.180 – 3.92 μM. As can be seen, the lower end of the range for the renal cancer cell lines is noticeably lower than those of the other cancer types. Specifically, the RXF 393 cell line possessed a GI50 < 10 nM. This represents the lowest GI50 obtained across the 60 cell lines; however, this low value does not appear to be specific to this type of cancer, but rather cell line specific as the next most sensitive renal cell line (TK-10) possesses a GI50 value of 495 nM.
Figure 4.

Comparison of GI50 values from the NCI human cancer cell line screen for 5g and SAHA. Tested doses included 1×10−4, 1×10−5, 1×10−6, 1×10−7 and 1×10−8 M. a) Activity against leukemia and non-small cell lung cancer cell lines; b) Activity against colon, central nervous system (CNS) and prostate cancer cell lines; c) Activity against melanoma and ovarian cancer cell lines; d) Activity against renal and breast cancer cell lines
Given the results of the NCI screen and the fact that 5g did not show any significant inhibition of HDAC8 at the concentration used in the preliminary screen, a more detailed analysis of HDAC1 inhibition was performed. It was found that 5g possesses an IC50 value against HDAC1 (104 ± 30 nM) that is comparable to that found with SAHA (140 ± 65 nM). Thus, taken together, the results of the enzyme and cell-based assays provide support to the proposed correlation between the cytotoxicity of HDAC inhibitors and the ability of these agents to inhibit HDAC1. This notion is further supported by the NCI’s preliminary cell inhibition studies on the six candidates which were originally selected (Supplementary Information). Those compounds which demonstrated a high degree of HDAC1 inhibition (SAHA and 5g) also exhibited high levels of potency in the cell-based assay. In contrast, the weaker inhibitors of HDAC1 also generally exhibited reduced potency against cancer cells.
Conclusion
This paper highlights the utility of click chemistry in the synthesis of HDACi. Through this approach, a combinatorial strategy was employed which enabled generation of multiple HDACi. Such combinatorial approaches greatly enhance the efficiency of drug discovery as libraries of candidate molecules can be obtained rapidly. This fact was highlighted by the discovery of lead compound 5g, which was found to inhibit HDAC1 with an IC50 = 104 ± 30 nM while demonstrating no significant inhibition of HDAC8 in preliminary testing. This level of inhibition was found to be comparable to that exhibited by the clinically approved drug SAHA (140 ± 65 nM). Furthermore, 5g was also found to be active across all cell lines in the NCI human cancer cell line screen with GI50 values ranging from 3.92 μM to 10 nM.
In summary, we have developed a click chemistry approach to HDACi design which has not only led to a potent lead compound, but more importantly, one which is readily amendable to further derivatization. For example, functionalization of the aromatic ring, replacement of the aromatic ring with a heteroaromatic species, or introduction of a chiral center in the “cap” moiety can all occur quite readily by incorporating these features into the azide-containing fragment. This will allow modifications to occur prior to the combinatorial step, thus maintaining the modular nature of this approach. A large number of new candidate molecules can therefore be rapidly prepared via modification of the azide library.
Experimental Section
All solvents were dried with a solvent-purification system from Innovative Technology, Inc. All reagents were obtained from commercial sources and used without further purification. Analytical TLC was carried out on silica gel 60 F254 aluminum-backed plates (E. Merck). The 230–400 mesh size of the same absorbent was utilized for all chromatographic purifications. The 200–400 mesh silica gel 60 RP-18 (from EMD™) was utilized to purify the target click HDACi. 1H and 13C NMR spectra were recorded at the indicated field strengths. The high-resolution mass spectra were collected at The Ohio State University Campus Chemical Instrumentation Center. The purities of click HDACi were analyzed by HPLC (refer to the supporting document). The purity of the key target compound 5g is 98.3%, with the purities of the other thirteen target compounds all being higher than 96%. As for the HDAC1 and HDAC8 inhibition studies, the assay kits were obtained from Biomol International LP. All assays were repeated at least three times. Briefly, the assay was performed in two stages. In the first stage, HDAC8 solution (15 μL, 1 U totally), HDACi solution (10 μL) and HDAC8 substrate solution (25 μL) were added to the 96-well microplate. The reaction proceeded at 30 °C for 45 min. The second stage was initiated by the addition of 50 μL of Developer which stopped HDAC activity and produced the fluorescent signal (λex = 350 nm; λem = 450 nm). The cell line assays for the selected six target compounds, 5b (NSC746454), 5e (NSC746455), 5f (NSC746456), 5g (NSC746457), 6e (NSC746458) and 6g (NSC746459), were performed by the NIH.47 Cell lines tested consisted of the following: CCRF-CEM, HL-60(TB), K-562, MOLT-4, RPMI-8226 and SR (Leukemia); A549/ATCC, EKVX, HOP-62, HOP-92, NCI-H226, NCI-H23, NCI-H322M and NCI-H460 (Non-Small Cell Lung Cancer); COLO 205, HCT-116, HCT-15, HT29, KM12 and SW-620 (Colon Cancer); SF-628, SF-295, SF-539, SNB-19, SNB-75 and U251 (CNS Cancer); LOX IMVI, MALME-3M, M14, SK-MEL-28, SK-MEL-5, UACC-257 and UACC-62 (Melanoma); IGROV1, OVCAR-3, OVCAR-4, OVCAR-5, OVCAR-8 and SK-OV-3 (Ovarian Cancer); 786-0, A498, ACHN, CAKI-1, RXF 393, SN12C, TK-10 and UO-31 (Renal Cancer); PC-3 and DU-145 (Prostate Cancer); MCF7, NCI/ADR-RES, MDA-MB-231/ATCC, HS578T, MDA-MB-435, BT-549 and T-47D (Breast Cancer). Full experimental details are available free of charge via the Internet at http://dtp.nci.nih.gov/branches/btb/ivclsp.html.48, 49
General procedure for the preparation of Click HDACi 5a-g and 6a-g from 21a-g and 22a-g
Triisopropylsilane (3 mmol, 0.6 mL, 3 equiv), PMB protected substrate (21a–g or 22a–g, 1 mmol, 1.0 equiv) and TFA (2 mL) were added in sequence to a reaction vessel equipped containing DCM (40 mL). The mixture was stirred at room temperature and monitored by TLC. Upon completion, the reaction mixture was diluted with acetonitrile (100 mL) and then neutralized with DOWEX® MARATHON® WBA anion exchange resin (from Aldrich). The resin was washed additionally with acetonitrile (50 mL × 2). The combined organic solution was evaporated to provide a solid, which was purified to afford the final product.
(E)-3-(1-benzyl-1H-1,2,3-triazol-4-yl)-N-hydroxyacrylamide (5a)
The crude product was first dissolved with a minimal amount of 5% methanol in DCM, after which the pure compound was precipitated with hexanes (4× the original volume) as a slightly pink solid (28 mg 5a from 100 mg 21a, 42% yield). 1H NMR (500 MHz, CD3OD):δ 8.15 (s, 1H), 7.50 (d, J = 15.8 Hz, 1H), 7.42-7.28 (m, 5H), 6.62 (d, J = 15.8 Hz, 1H), 5.60 (s, 2H); 13C NMR (125 MHz, CD3OD):δ 165.8, 145.2, 136.7, 130.2, 129.8, 129.3, 125.7, 120.4, 55.1 (one peak less due to overlap ~130); HRMS (ESI) calcd for C12H12N4O2Na [M+Na]+ 267.0858, found 267.0860.
(E)-N-hydroxy-3-(1-phenethyl-1H-1,2,3-triazol-4-yl)acrylamide (5b)
The crude product was purified via C-18 reverse phase column chromatography (water: acetonitirile = 5: 1) to provide 5b as a white solid (65 mg 5b from 174 mg 21b, 55% yield). 1H NMR (500 MHz, DMF-d7):δ 8.32 (s, 1H), 7.48 (d, J = 15.7 Hz, 1H), 7.36-7.20 (m, 5H), 6.74 (d, J = 15.7 Hz, 1H), 4.73 (t, J = 7.3 Hz, 2H), 3.27 (t, J = 7.3 Hz, 2H); 13C NMR (125 MHz, CD3OD): δ 163.9, 144.4, 139.0, 129.9, 129.6, 128.5, 127.8, 125.5,120.6, 52.0, 37.1; HRMS (ESI) calcd for C13H14N4O2Na [M+Na]+ 281.1014, found 281.1017.
(E)-N-hydroxy-3-(1-(3-phenylpropyl)-1H-1,2,3-triazol-4-yl)acrylamide (5c)
The crude product was purified via C-18 reverse phase column chromatography (water: acetonitirile = 5: 1) to provide a pale colored solid (35 mg product 5c from 100 mg 21c, 50% yield). 1H NMR (500 MHz, CD3OD):δ 8.15 (s, 1H), 7.51 (d, J = 15.8 Hz, 1H), 7.30-7.24 (m, 2H), 7.21-7.15 (m, 3H), 6.62 (d, J = 15.8 Hz, 1H), 4.42 (t, J = 7.2 Hz, 2H), 2.64 (t, J = 7.5 Hz, 2H), 2.41 (tt, J1 = J2 = 7.4 Hz, 2H); 13C NMR (125 MHz, CD3OD):δ 165.9, 144.9, 142.0, 129.7, 129.6, 129.4, 127.4, 125.8,120.2, 51.0, 33.6, 33.0; HRMS (ESI) calcd for C14H16N4O2Na [M+Na]+ 295.1171, found 295.1168.
(E)-N-hydroxy-3-(1-(2-phenoxyethyl)-1H-1,2,3-triazol-4-yl)acrylamide (5d)
The crude product was purified via C-18 reverse phase column chromatography (water: acetonitirile = 6: 1) to provide a white solid (69 mg 5d from 163 mg 21d, 61% yield). 1H NMR (500 MHz, DMF-d7):δ 8.52 (s, 1H), 7.52 (d, J = 15.8 Hz, 1H), 7.38-7.24 (m, 2H), 7.07-6.92 (m, 3H), 6.79 (d, J = 15.7 Hz, 1H), 4.91 (t, J = 5.0 Hz, 2H), 4.51 (t, J = 5.0 Hz, 2H); 13C NMR (125 MHz, DMF-d7):δ 163.8, 159.3, 144.7, 130.6, 128.3, 126.0, 122,2, 120.8,115.7, 67.3 50.6; HRMS (ESI) calcd for C13H14N4O3Na [M+Na]+ 297.0964, found 297.0966.
(E)-N-hydroxy-3-(1-(2-oxo-2-phenylethyl)-1H-1,2,3-triazol-4-yl)acrylamide (5e)
The crude product was purified via C-18 reverse phase column chromatography (water acetonitirile = 6: 1) to provide a pale colored solid (22mg product 5e from 90 mg 21e, 35%). 1H NMR (500 MHz, DMF-d7):δ 11.19 (s, 1H), 8.48 (s, 1H), 8.17 (d, J = 7.4 Hz, 2H), 7.80 (t, J = 7.9 Hz, 1H), 7.67 (dd, J1 = J2 =7.8 Hz, 2H), 7.62 (d, J = 15.2, 1H), 6.85 (d, J = 15.7 Hz, 1H), 6.34 (s, 2H); 13C NMR (125 MHz, DMF-d7):δ 192.2, 143.6, 134.4, 134.4, 129.2, 128.3, 127.7, 126.4, 119.8, 56.1 (one peak less due to the overlap with solvent peak ~ δ 162); HRMS (ESI) calcd for C13H12N4O3Na [M+Na]+ 295.0807, found 295.0811.
(E)-N-hydroxy-3-(1-(2-oxo-2-(phenylamino)ethyl)-1H-1,2,3-triazol-4-yl)acrylamide (5f)
The crude product was purified via C-18 reverse phase column chromatography (water: acetonitrile: TFA = 6: 1: 0.03) to provide a pink solid (67 mg 5f from 187 mg 21f, 71% yield). 1H NMR (500 MHz, DMF-d7):δ 10.60 (s, 1H), 8.49 (s, 1H), 7.69 (d, J = 7.5 Hz, 2H), 7.57 (d, J = 15.7 Hz, 1H), 7.36 (apparent t, J = 7.4 Hz, 2H), 7.12 (t, J = 7.4 Hz, 1H), 6.81 (d, J = 15.7 Hz, 1H), 5.51 (s, 2H); 13C NMR (125 MHz, CD3OD):δ δ 165.4, 144.5, 140.0, 129.9, 128.5, 127.2, 124.9, 120.8, 120.4, 53.6; HRMS (ESI) calcd for C13H13N5O3Na [M+Na]+ 310.0916, found 310.0923.
(E)-3-(1-cinnamyl-1H-1,2,3-triazol-4-yl)-N-hydroxyacrylamide (5g)
The crude product was purified via C-18 reverse phase column chromatography (water: acetonitrile = 5: 1) to provide a white solid (96 mg 5g from 199 mg 21g, 70% yield). 1H NMR (500 MHz, DMF-d7):δ 10.2 (br s, 1H), 8.48 (s, 1H), 7.58-7.48 (m, 3H), 7.37 (apparent t, J = 7.4 Hz, 2H), 7.30 (t, J = 7.1 Hz, 1H), 6.82 (d, J = 16.0 Hz, 1H), 6.77 (d, J = 15.8 Hz, 1H), 6.60 (dt, J1 = 15.5 Hz, J2 = 6.2 Hz,1H), 5.29 (d, J = 6.1Hz, 2H); 13C NMR (125 MHz, DMF-d7):δ 163.9, 144.9, 137.2, , 129.7, 135.3, 129.2, 128.3, 127.8, 125.3, 124.5, 120.9, 52.8; HRMS (ESI) calcd for C14H14N4O2Na [M+Na]+ 293.1014, found 293.1020.
1-Benzyl-1H-[1,2,3]triazole-4-carboxylic acid hydroxamide (6a)
The crude product was purified via C-18 reverse phase column chromatography (water: acetonitirile = 6: 1) to provide a white solid (16 mg 6a from 53 mg 22a, 47% yield). 1H NMR (500 MHz, DMF-d7): δ 11.40-9.20 (bd s, 1H), 8.64 (s, 1H), 7.58-7.28 (m, 5H), 5.76 (s, 2H); 13C NMR (125 MHz, DMF-d7): δ 158.9, 143.1, 137.0, 129.9, 129.3, 129.1, 127.1, 54.4; HRMS (ESI) calcd for C10H10N4O2Na [M+Na]+ 241.0701, found 241.0697.
1-Phenethyl-1H-[1,2,3]triazole-4-carboxylic acid hydroxamide (6b)
The crude product was purified via C-18 reverse phase column chromatography (water: acetonitirile = 6: 1) to provide a white solid (21 mg 6b from 63 mg 22b, 51% yield). 1H NMR (500 MHz, DMF-d7): δ 11.21 (s, 1H), 9.30 (s, 1H), 8.48 (s, 1H), 7.40-7.20 (m, 5H), 4.78 (t, J = 7.3 Hz, 2H), 3.29 (t, J = 7.3 Hz, 2H); 13C NMR (125 MHz, DMF-d7): δ 158.3, 141.9, 138.0, 129.0, 128.7, 126.9, 126.2, 51.3, 36.2; HRMS (ESI) calcd for C11H12N4O2Na [M+Na]+ 255.0858, found 255.0862.
1-(3-Phenyl-propyl)-1H-[1,2,3]triazole-4-carboxylic acid hydroxamide (6c)
The crude product was purified via C-18 reverse phase column chromatography (water: acetonitirile = 6: 1) to provide a white solid (31 mg 6c from 63 mg 22c, 74% yield). 1H NMR (500 MHz, DMF-d7):δ 11.27 (s, 1H), 9.38 (s, 1H), 8.61 (s, 1H), 7.36-7.20 (m, 5H), 4.54 (t, J = 7.1 Hz, 2H), 2.66 (t, J = 7.7 Hz, 2H), 2.26 (tt, J1 = J2 = 7.1 Hz, 2H); 13C NMR (125 MHz, DMF-d7):δ 159.1, 142.8, 142.0, 129.4, 127.0, 126.9, 55.7, 50.5, 33.2, 32.7; HRMS (ESI) calcd for C12H14N4O2Na [M+Na]+ 269.1014, found 269.1014.
1-(2-Phenoxy-ethyl)-1H-[1,2,3]triazole-4-carboxylic acid hydroxamide (6d)
The crude product was purified via C-18 reverse phase column chromatography (water: acetonitirile = 6: 1) to provide a white solid (15 mg 6d from 40 mg 22d, 56% yield). 1H NMR (500 MHz, DMF-d7):δ 10.60-9.90 (bd s, 1H), 8.65 (s, 1H), 7.34-7.28 (m, 2H), 7.00-6.94 (m, 2H), 4.95 (t, J = 5.1 Hz, 2H), 4.53 (t, J = 5.1, 2H); 13C NMR (125 MHz, DMF-d7): δ 159.4, 159.1, 143.0, 130.6, 127.6, 122.2, 115.7, 67.3, 50.7; HRMS (ESI) calcd for C11H12N4O2Na [M+Na]+ 271.0807, found 271.0807.
1-(2-Oxo-2-phenyl-ethyl)-1H-[1,2,3]triazole-4-carboxylic acid hydroxyamide (6e)
The crude product was purified via C-18 reverse phase column chromatography (water: acetonitirile = 7: 1) to provide a white solid (56 mg 6e from 116 mg 22e, 72% yield). 1H NMR (500 MHz, DMF-d7):δ 11.40-9.20 (bd s, 2H), 8.62 (s, 1H), 8.16 (d, J = 7.3 Hz, 2H), 7.78 (t, J = 7.4 Hz, 1H), 7.65 (dd, J1 = J2 = 7.8 Hz, 2H), 6.38 (s, 2H); 13C NMR (125 MHz, DMF-d7):δ 192.9, 159.1, 143.0, 135.5, 135.3, 130.1, 129.3, 128.9, 57.3; HRMS (ESI) calcd for C11H10N4O3Na [M+Na]+ 269.0651, found 269.0629.
1-Phenylcarbamoylmethyl-1H-[1,2,3]triazole-4-carboxylic acid hydroxyamide (6f)
The crude product was purified via C-18 reverse phase column chromatography (water: acetonitirile = 7: 1) to provide a white solid (45 mg 6f from 120 mg 22f, 55% yield). 1H NMR (500 MHz, DMF-d7):δ 10.77 (s, 1H), 10.50-10.00 (bd s, 1H), 8.65 (s, 1H), 7.70 (d, J = 7.4 Hz, 2H), 7.35 (dd, J1 = J2 = 7.9 Hz, 2H), 7.11 (t, J = 7.4 Hz, 1H), 5.57 (s, 2H); 13C NMR (125 MHz, DMF-d7):δ 165.3, 159.1, 142.8, 140.1, 129.9, 128.8, 124.8, 120.4, 53.6; HRMS (ESI) calcd for C11H11N5O3Na [M+Na]+ 284.0760, found 284.0743.
1-(3-Phenyl-allyl)-1H-[1,2,3]triazole-4-carboxylic acid hydroxyamide (6g)
The crude product was purified via C-18 reverse phase column chromatography (water: acetonitirile = 5: 1) to provide a white solid (21 mg 6 from 62 mg 22 50% yield). 1H NMR (500 MHz, DMF-d7): δ 10.60-9.80 (bd s, 2H), 8.61 (s, 1H), 7.53 (d, J = 7.6 Hz, 2H), 7.38 (dd, J1 = J2 = 7.5 Hz, 2H), 7.31 (t, J = 7.3 Hz, 1H), 6.79 (d, J = 15.9, 1H), 6.62 (td, J1 = 15.7 Hz, J2 = 6.6 Hz, 1H), 5.33 (d, J = 6.5, 2H); 13C NMR (125 MHz, DMF-d7): δ 159.4, 159.1, 143.0, 130.6, 127.6, 122.2, 115.7, 67.3, 50.7; HRMS (ESI) calcd for C12H12N4O2Na [M+Na]+ 267.0858, found 267.0855.
(E)-N-(4-methoxybenzyloxy)-5-(trimethylsilyl)pent-2-en-4-ynamide (7)
The acid 19 (168 mg, 1.0 mmol, 1.0 equiv) was added to a reaction vessel containing DCM (3 mL). To this solution, oxalyl chloride (130 μL, 1.6 mmol, 1.6 equiv) and a trace amount of DMF were added. The mixture was stirred at room temperature for 1 hour. The solvent was then removed under vacuum to afford a yellow-reddish syrup. DCM (4 mL) was added, and the flask was cooled to 0°C (solution A). The amine 13 (285 mg 1.5 mmol, 1.5 equiv) and N,N′-diisopropyethylamine (DIPEA) (0.52 mL, 3.0 mmol, 3.0 equiv) were added to a second reaction vessel containing DCM (10 mL) (solution B). Solution B was cooled to 0°C, after which solution A was added within 1 min. The mixture was stirred for one half hour at 0°C. The reaction mixture was then poured onto ice cold 0.1 N HCl (20 mL), after which DCM (20 mL) was added. The organic layer was additionally washed with ice cold 0.1 N HCl (2 × 20 mL). The DCM solution was dried with Na2SO4, and the solvent was evaporated. The crude product was purified via flash column chromatography (hexanes: ethyl acetate, 87:13 to 83:17) to provide pure 7 as a white oil (253 mg, 84%). 1H NMR (500 MHz, CD3CN): δ 9.44 (bd s, 1H), 7.33 (d, J = 8.6 Hz, 2H), 6.92 (d, J = 8.6 Hz, 2H), 6.64 (d, J = 15.5 Hz, 1H), 6.17 (d, J = 14.2 Hz, 1H), 4.79 (s, 2H), 3.79 (s, 3H), 0.20 (s, 9H); 13C NMR (125 MHz, CD3CN):δ 163.0, 161.0, 132.3, 132.0, 128.7, 121.4, 114.8, 103.2, 102.8, 78.4, 55.9, – 0.33; HRMS (ESI) calcd for C16H21NO3SiNa [M+Na]+ 326.1188, found 326.1193.
(E)-N-(4-methoxybenzyloxy)-5-(trimethylsilyl)-N-((E)-5-(trimethylsilyl)pent-2-en-4-ynoyl)pent-2-en-4-ynamide (20)
Compound 20 was a by-product obtained during the preparation of 7. 1H NMR (500 MHz, CD3CN):δ 7.25 (d, J = 8.6 Hz, 2H), 6.90 (d, J = 8.7 Hz, 2H), 6.88 (d, J = 15.9 Hz, 1H), 6.48 (d, J = 16.0 Hz, 1H), 6.34 (d, J = 15.9 Hz, 1H), 6.12 (d, J = 16.0 Hz, 1H), 5.00 (s, 1H), 3.78 (s, 1H), 0.22 (s, 9H), 0.18 (s, 9H); 13C NMR (125 MHz, CD3CN):δ 161.3, 160.8, 149.0, 131.1, 131.0, 129.7, 129.6, 128.8, 116.4, 114.8, 108.2, 103.1, 103.1, 101.6, 77.8, 55.9, −0.30, −0.51; HRMS (ESI) calcd for C24H31NO4Si2Na [M+Na]+ 476.1689, found 476.1683.
N-(4-methoxybenzyloxy)-3-(trimethylsilyl)propiolamide (8)
Commercially available acid 14 (710 mg, 5.0 mmol, 1.0 equiv) was added to a reaction vessel containing DCM (10 mL). To this solution, oxalyl chloride (430 μL, 5.5 mmol, 1.1 equiv) and a trace amount of DMF were added. The mixture was stirred at room temperature for 2 hours. The solution was then placed in an ice bath, and most of the acidic gases (HCl & SO2) were removed under vacuum (~20 bar) to give a yellow solution. Freshly dried DCM (10 mL) was then added (solution A). The amine 13 (950 mg, 5.0 mmol, 1.0 equiv) and DIPEA (2.8 mL, 16.1 mmol, 3.2 equiv) were added to a second reaction vessel containing DCM (50 mL) (solution B). Solution B was cooled to 0 °C, after which solution A was added within 1 min. The mixture was then stirred for 30 min at 0 °C. The reaction mixture was poured onto ice cold 0.1 N HCl (200 mL), before DCM (200 mL) was also added. The organic layer was additionally washed with ice cold 0.1 N HCl twice (2 × 100 mL). The DCM solution was dried with Na2SO4, and the solvent was evaporated. The crude product was purified via flash column chromatography (hexanes: ethyl acetate, 85:15 to 80:20) to provide 8 as pale color oil (0.85 g, 61%). 1H NMR (500 MHz, CDCl3):δ 8.43 (bd s, 1H), 7.32 (d, J = 8.5 Hz, 2H), 6.89 (dd, J = 8.5 Hz, 2H), 4.85 (s, 2H), 3.80 (s, 3H), 0.20 (s, 9H); 13C NMR (125 MHz, CDCl3): δ 160.1, 151.0, 131.0, 126.9, 114.0, 95.0, 94.4, 78.2, 55.3, 0.8; HRMS (ESI) calcd for C14H19NO3SiNa [M+Na]+ 300.1032, found 300.1033.
General procedure to prepare the azido precursors 9a-g
An alkyl halide 10a–g (1.5 g, 1.0 equiv) was added to a reaction vessel containing a solution of NaN3 in DMSO (1.1 equiv, 0.5 M). The reaction was monitored by NMR analysis. Upon completion of the reaction, water (50 mL) was added, and the product was extracted with ether (3 × 50 mL). The combined organic layers were washed with water (2 × 50 mL) and brine (50 mL), and dried with magnesium sulfate. The organic solvent was removed to provide the azido compound. For 9c and 9d, a catalytic amount of tetraethylammonium bromide (0.05 mol%, 0.005 equiv) was added. The spectral data were in agreement with those reported.36, 50–53
(Azidomethyl)benzene (9a)
From 10a (1.5 g, 11.3 mmol, 1.0 equiv), 9a was obtained in 95% yield as a colorless liquid. 1H NMR (500 MHz, CDCl3):δ 7.75-7.40 (m, 5H), 4.35 (s, 2H).36
(2-Azidoethyl)benzene (9b)
From 10b (1.5 g, 10.2 mmol, 1.0 equiv), 9b was obtained in 93% yield as a colorless liquid. 1H NMR (500 MHz, CDCl3)δ 7.33 (t, J = 7.2 Hz, 2H), 7.26 (d, J = 7.1 Hz, 1H), 7.23 (d, J = 7.2 Hz, 2H), 3.51 (t, J = 7.3 Hz, 2H), 2.91 (t, J = 7.3 Hz, 2H).50,51
(3-Azidopropyl)benzene (9c)
From 10c (1.5 g, 9.30 mmol, 1.0 equiv), 9c was obtained in 90% yield as a colorless liquid. 1H NMR (500 MHz, CDCl3): δ 7.30 (t, J = 7.3 Hz, 2H), 7.24-7.16 (m, 3H), 3.29(t, J = 7.3 Hz, 2H), 2.71 (t, J = 7.6 Hz, 2H), 1.92 (tt, J1 = J2 = 7.2 Hz, 2H).50, 51
(2-Azidoethoxy)benzene (9d)
From 10d (1.5 g, 9.2 mmol, 1.0 equiv), 9d was obtained in 95% yield as a colorless liquid. 1H NMR (500 MHz, CDCl3)δ 7.34-7.28 (m, 2H), 7.02-6.97 (m, 1H), 6.95-6.89 (m, 2H), 4.16 (d, J = 5.0 Hz, 2H), 3.60 (d, J = 5.0 Hz, 2H); 13C NMR (125 MHz, CDCl3):δ 158.4, 129.8, 121.6, 114.8, 67.1, 50.4.
2-Azido-1-phenylethanone (9e)
From 10e (1.5 g, 8.0 mmol, 1.0 equiv), 9e was obtained in 75% yield after flash column chromatography (2% to 5% ether in hexanes) as a colorless liquid. 1H NMR (500 MHz, CDCl3): δ 7.91-7.88 (m, 2H), 7.65-7.60 (m, 1H), 7.53-7.48 (m, 2H), 4.56 (s, 2H).52
2-Azido-N-phenylacetamide (9f)
From 10f (1.5 g, 8.5 mmol, 1.0 equiv), 9f was obtained in 90% yield as a colorless liquid. 1H NMR (500 MHz, CDCl3):δ 8.00 (bd s, 1H), 7.54 (d, J = 8.6 Hz, 2H), 7.35 (dd, J1 = 8.4 Hz, J2 = 7.5 Hz, 2H), 7.16 (d, J = 8.6 Hz, 1H), 4.15 (s, 2H).53
2-Azido-N-phenylacetamide (9g)
From 10g (1.5 g, 9.4 mmol, 1.0 equiv), 9g was obtained in 87% yield as a colorless liquid. 1H NMR (500 MHz, DMSO-d6)δ 7.51-7.47 (m, 2H), 7.38-7.31 (m, 2H), 7.31-7.26 (m, 1H), 6.71 (d, J = 15.8 Hz, 1H), 6.39 (dt, J1 = 15.8 Hz, J2 = 6.6 Hz, 1H), 4.04 (d, J = 6.6 Hz, 2H).36
O-(4-methoxybenzyl)hydroxylammonium chloride (13)
N-hydroxyphthalimide 11 (4.9 g, 30 mmol, 1.0 equiv) & potassium carbonate (5.0 g, 36 mmol, 1.2 equiv) were added to a reaction vessel containing DMF (100 mL). To this solution, 1-(chloromethyl)-4-methoxybenzene (PMBCl) (4.7 mL, 33 mmol, 1.1 equiv) was added. The reaction continued overnight at room temperature. Water (500 mL) and benzene (500 mL) were then added. The aqueous layer was additionally extracted with benzene (2 × 100 mL). The combined organic layers were washed with sat. NaHCO3 solution (3 × 200 mL), and dried with magnesium sulfate. Crude 12 was obtained after removal of the solvent, and was directly employed in the next step. To this crude product in methanol (500 mL), hydrazine monohydrate was added (4.5 mL, 92.5 mmol, 3.1 equiv). The mixture was stirred overnight at room temperature. TLC analysis indicated the presence of a new compound. Upon completion of the reaction, the solution pH was adjusted to 2 via the addition of aqueous 2M HCl. The mixture was cooled in an ice bath for 1 hour, after which the white precipitate (by product) was removed. The solvent was removed, and the residue/syrup was partitioned between DCM (200 mL) & ammonium bicarbonate solution (pH = 8, 200 mL). The organic layer was dried with Na2SO4. Addition of 1M HCl in ether then caused the product 13 to precipitate as a white solid (4.3 g, 75.6% yield for two steps). Spectral data were in agreement with those reported.37 1H NMR (500 MHz, DMSO-d6): δ 11.10 (bd s, 3H), 7.35 (d, J = 8.6 Hz, 2H), 6.96 (d, J = 8.6 Hz, 2H), 4.97 (s, 2H), 3.76 (s, 3H).
(E)-3-Iodopropenoic acid (16)
Propiolic acid 15 (10 mL, 162.0 mmol, 1.0 equiv) was added over 1 min to a reaction vessel containing a solution of CuI (200 mg, 1.1 mmol, 0.007 equiv) and HI (40 mL, initially 57%, 303.2 mmol, 1.9 equiv). The reaction mixture was immediately immersed in a pre-heated oil bath (130°C) for 30 min. The reaction was then cooled to room temperature over a 15 min period. A white precipitate formed, after which the solution was kept at room temperature for another 15 min. The liquid was removed by filtration, and the solid was washed water (3 × 70 mL). Following a period of drying, the pure iodoacid 16 was obtained as white needles (23.9 g, 75%). 1H-NMR (500 MHz, CDCl3): δ 10.54-8.66 (bd s, 1H), 8.07 (d, J = 14.9 Hz, 1H), 6.88 (d, J = 14.9 Hz, 1H). Spectral data were in agreement with those reported.38
(2-Methoxyethoxy)methyl-(2E)-3-iodoprop-2-enoate (17)
K2CO3 (3.42 g, 24.8 mmol, 1.2 equiv) and 2-methoxyethoxymethyl chloride (MEMCl) (3.2 mL, 27.6 mmol, 1.3 equiv) were added to a reaction vessel containing a solution of E-iodopropenoic acid 16 (4.2 g, 21.2 mmol, 1.0 equiv) in DMF (20 mL). The mixture was stirred at ambient temperature for 1 hour, poured onto water (20 mL), and then extracted with ether (3 × 20 mL). The combined organic layers were washed with water, brine, dried over MgSO4, and concentrated. Flash column chromatograhphy (hexanes: ethyl acetate, 20:1 to 10:1) provided 5.2 g (86%) of 17 as a colorless oil: 1H NMR (500 MHz, C6D6): δ 7.62 (d, J = 14.8 Hz, 1H) 6.62 (d, J = 14.8 Hz, 1H), 5.13 (s, 2H), 3.49 (t, J = 4.7 Hz, 2H), 3.18 (t, J = 4.7 Hz, 2H), 3.04 (s, 3H); 13C NMR (125 MHz, C6D6):δ 163.1, 136.6, 100.2, 89.9, 71.8, 69.9, 58.7; HRMS (ESI) calcd for C7H11IO4Na [M+Na]+ 308.9600, found 308.9605.
(2-Methoxyethoxy)methyl-(2E)-5-(trimethylsilyl)pent-2-en-4-ynoate (18)
Pd(PPh3)2Cl2 (0.56 g, 0.80 mmol, 0.8 equiv) and CuI (0.31 g, 1.63 mmol, 0.1 equiv) were added to a reaction vessel containing a solution of the iodide 17 (4.65 g, 16.3 mmol, 1.0 equiv) in Et3N (60 mL). The solution was degassed with argon. Trimethylsilylacetylene (3.8 mL, 29.3 mmol, 1.8 equiv) was then added and the solution was stirred for 1 h. The mixture was poured onto sat. NH4Cl (200 mL) and extracted with ether (3 × 100 mL). The combined organic layers were washed with brine, dried over MgSO4, and concentrated. The thick oily residue was subjected to flash column chromatography (hexanes: ethyl acetate, 20:1 to 13:1) to yield 3.6 g (87%) of 18 as a pale oil: 1H NMR (500 MHz, CDCl3)δ 6.77 (d, J = 16.0 Hz, 1H), 6.23 (d, J = 16.0 Hz, 1H), 5.38 (s, 2H), 3.77(m, 2H), 3.53(m, 2H), 3.36 (s, 3H), 0.20 (s, 9H); 13C NMR (125 MHz, CDCl3)δ 165.1, 130.6, 126.0, 105.9, 101.1, 89.8, 71.5, 69.6, 59.1, −0.43; HRMS (ESI) calcd for C12H20O4SiNa [M+Na]+ 279.1029, found 279.1032.
(E)-5-(trimethylsilyl)pent-2-en-4-ynoic acid (19)
MEM-protected 18 (3.07 g, 12.0 mmol, 1.0 equiv) and 3N HCl (12 mL) were added to a reaction vessel containing THF (150 mL). The reaction was stirred at room temperature for 3 days, during which time it was monitored by TLC (5% MeOH in dichloromethane, Rf = 0.10). The solution was then concentrated and the residue partitioned between water (250 mL) and CH2Cl2 (100 mL). The water was further extracted with CH2Cl2 (2 × 100 mL). The combined organic layers were washed with brine, dried over MgSO4, and concentrate give 2.01 g (~100%) of the crude acid 19 as pale yellow solid. The crude product is pure enough for NMR analysis. 1H NMR (500 MHz, CDCl3):δ 0.20 (s, 9H), 6.22 (d, J = 15.9 Hz, 1H), 6.80 (d, J = 15.9 Hz, 1H), 12.5 ~ 11.0 (bd s, 1H); 13C NMR (125 MHz, CDCl3):δ −0.34, 101.1, 107.1, 127.6, 130.4, 171.4; HRMS (ESI) calcd for C8H12O2SiNa [M+Na]+ 191.0502, found 191.0504.
General procedure for the preparation of 21a-g
The alkyne precursor 7 (152 mg, 0.5 mmol, 1.0 equiv) was added to a reaction vessel containing THF (5 mL) and MeOH (5 mL). Cesium fluoride (91 mg, 0.6 mmol, 1.2 equiv) was then added, after which the reaction was monitored by TLC analysis. The desilylation intermediate was slightly more polar than the reactant. Upon disappearance of the reactant, the solvent was removed, followed by partitioning between DCM (50 mL) and brine (50 mL). A trace amount of acetic acid was added to neutralize the aqueous solution. The brine phase was additionally extracted with DCM (2 × 50 mL). The combined DCM solution was dried with Na2SO4, and the solvent was removed. The obtained white solid was dissolved with THF (10 mL). To this solution, the azido compound 9a–g (0.75 mmol, 1.5 equiv), catalyst CuIP(OEt)3 (7 mg, 0.02 mmol, 0.04 equiv) and one drop of DIPEA were added sequentially. The reaction continued overnight at room temperature, and was monitored by TLC analysis. Frequently, more and more white precipitate (product) formed as the reaction progressed. The solvent was removed, and the residue was partitioned between DCM (50 mL) and 1% CuSO4 aqueous solution (50 mL). The aqueous layer was additionally extracted with DCM (2 × 50 mL). The combined DCM layers were dried with Na2SO4. After removal of the solvent, the crude product was purified via flash column chromatography to afford the final product 21a–g.
(E)-3-(1-benzyl-1H-1,2,3-triazol-4-yl)-N-(4-methoxybenzyloxy) acrylamide (21a)
A white solid was obtained (87%, 158 mg 21a product from 0.5 mmol 7) via flash column chromatography (MeOH: DCM, from 1.2: 100 to 1.5: 100). 1H NMR (500 MHz, CD2Cl2 & CD3OD):δ 7.91 (s, 1H), 7.50 (d, J = 15.7 Hz, 1H), 7.42-7.28 (m, 7H), 6.90 (d, J = 8.4 Hz, 2H), 6.54 (d, J = 15.7 Hz, 1H), 5.56 (s, 2H), 4.84 (s, 2H), 3.79 (s, 3H); 13C NMR (125 MHz, CD2Cl2 & CD3OD):δ 164.8, 160.8, 144.5, 135.4, 131.6, 129.7, 129.4, 129.2, 128.7, 128.3, 125.0, 119.6, 114.4, 78.5, 55.6, 54.8; HRMS (ESI) calcd for C20H20N4O3Na [M+Na]+ 387.1433, found 387.1431.
(E)-N-(4-methoxybenzyloxy)-3-(1-phenethyl-1H-1,2,3-triazol-4-yl) acrylamide (21b)
A white solid was obtained (97% yield, 184 mg 21b from 0.5 mmol 7) via flash column chromatography (MeOH: DCM, from 0.8: 100 to 1.5: 100). 1H NMR (500 MHz, DMF-d7)δ 11.28 (s, 1H), 8.36 (s, 1H), 7.56 (d, J = 15.7 Hz, 1H), 7.40 (d, J = 8.5 Hz, 2H), 7.35-7.20 (m, 5H), 6.98 (d, J = 8.6 Hz, 2H), 6.72 (d, J = 15.5 Hz, 1H), 4.89 (s, 2H), 4.74 (t, J = 7.3 Hz, 2H), 3.83 (s, 3H), 3.27 (t, J = 7.3 Hz, 2H); 13C NMR (125 MHz, DMF-d7):δ 164.1, 161.0, 144.2, 138.9, 131.9, 129.9, 129.6, 129.6, 129.4, 127.7, 125.8, 120.2, 114.7, 78.1, 56.0, 52.0, 37.0; HRMS (ESI) calcd for C21H22N4O3Na [M+Na]+ 401.1590, found 401.1585.
(E)-N-(4-methoxybenzyloxy)-3-(1-(3-phenylpropyl)-1H-1,2,3-triazol-4-yl)acrylamide (21c)
A white solid was obtained (95% yield, 186 mg 21c from 0.5 mmol 7) via flash column chromatography (MeOH: DCM, from 0.8: 100 to 1.5: 100). 1H NMR (500 MHz, DMF-d7)δ 11.29 (s, 1H), 8.49 (s, 1H), 7.60 (d, J = 15.7 Hz, 1H), 7.40 (d, J = 8.5 Hz, 2H), 7.35-7.30 (m, 2H), 7.30-7.25 (m, 2H), 7.25-7.20 (m, 1H), 6.98 (d, J = 8.6 Hz, 2H), 6.75 (d, J = 15.5 Hz, 1H), 4.90 (s, 2H), 4.50 (t, J = 7.3 Hz, 2H), 3.83 (s, 3H), 2.67 (t, J = 7.3 Hz, 2H), 2.25 (t, J = 7.3 Hz, 2H); 13C NMR (125 MHz, DMF-d7): δ164.1, 161.0, 144.5, 142.2, 131.9, 129.7, 129.6, 129.5, 127.2, 125.8, 120.3, 114.8, 78.1, 56.0, 50.4, 33.3, 32.8 (one peak less due to overlap); HRMS (ESI) calcd for C22H24N4O3Na [M+Na]+ 415.1746, found 415.1745.
(E)-N-(4-methoxybenzyloxy)-3-(1-(2-phenoxyethyl)-1H-1,2,3-triazol-4-yl)acrylamide (21d)
A white solid was obtained (89 % yield, 176 mg 21d from 0.5 mmol 7) via flash column chromatography (MeOH: DCM, from 1.0: 100 to 2.0: 100). 1H NMR (500 MHz, DMF-d7):δ 11.26 (s, 1H), 8.55 (s, 1H), 7.61 (d, J = 15.7 Hz, 1H), 7.40 (d, J = 8.3 Hz, 2H), 7.36-7.27 (m, 2H), 7.06-6.90 (m, 5H), 6.77 (d, J = 14.8 Hz, 1H), 4.91 (t, J = 5.0 Hz, 2H), 4.89 (s, 2H), 4.51 (t, J = 5.1 Hz, 2H), 3.83 (s, 3H); 13C NMR (125 MHz, DMF-d7):δ 164.2, 161.0, 159.4, 144.5, 131.9, 130.6, 129.6, 129.5, 126.5, 122.3, 120.4, 115.7, 114.8, 78.1, 67.4, 56.0, 50.6; HRMS (ESI) calcd for C21H22N4O4Na [M+Na]+ 417.1539, found 417.1543.
(E)-N-(4-methoxybenzyloxy)-3-(1-(2-oxo-2-phenylethyl)-1H-1,2,3-triazol-4-yl)acrylamide (21e)
A pale yellow solid was obtained (38 % yield, 110 mg 21e from 0.73 mmol 7) via flash column chromatography (MeOH: DCM, from 1.5: 100 to 2: 100). 1H NMR (500 MHz, DMF-d7)δ 11.30 (s, 1H), 8.48 (s, 1H), 8.17 (d, J = 7.4 Hz, 2H), 7.78 (t, J = 7.4 Hz, 1H), 7.72-7.62 (m, 3H), 7.41 (d, J = 8.3 Hz, 2H), 6.99 (d, J = 8.5 Hz, 2H), 6.80 (d, J = 15.6 Hz, 1H), 6.35 (s, 2H), 4.91 (s, 2H), 3.83 (s, 3H); 13C NMR (125 MHz, DMF-d7):δ 192.3, 163.4, 160.2, 143.8, 134.8, 134.6, 131.1, 129.3, 128.9, 128.7, 128.6, 126.9, 119.7, 114.0, 77.3, 56.4, 55.3; HRMS (ESI) calcd for C21H20N4O4Na [M+Na]+ 415.1382, found 387.1378.
(E)-N-(4-methoxybenzyloxy)-3-(1-(2-oxo-2-(phenylamino)ethyl)-1H-1,2,3-triazol-4-yl)acrylamide (21f)
A white solid was obtained (92%, 187 mg product 21f from 0.5 mmol 7) via flash column chromatography (MeOH: DCM, from 2.5: 100 to 3.5: 100). 1H NMR (500 MHz, DMF-d7) δ 11.29 (s, 1H), 10.60 (s, 1H), 8.53 (s, 1H), 7.69 (d, J = 8.0 Hz, 2H), 7.65 (d, J = 15.7 Hz, 1H), 7.41 (d, J = 8.4 Hz, 2H), 7.36 (dd, J1 = 7.6 Hz, J2 = 8.4 Hz, 2H), 7.12 (t, J = 7.4 Hz, 1H), 6.98 (d, J = 8.6 Hz, 2H), 6.78 (d, J = 15.7 Hz, 1H), 5.52 (s, 2H), 4.90 (s, 2H), 3.83 (s, 3H); 13C NMR (125 MHz, DMF-d7) δ 164.6, 163.4, 160.2, 143.6, 139.3, 131.1, 129.2, 128.9, 128.7, 126.8, 124.1, 119.6, 114.0, 77.3, 55.2, 52.8 (one peak less due to the overlap); HRMS (ESI) calcd for C21H21N5O4Na [M+Na]+ 430.1491, found 430.1492.
(E)-3-(1-cinnamyl-1H-1,2,3-triazol-4-yl)-N-(4-methoxybenzyloxy)acrylamide (21g)
A white solid was obtained (86% yield, 216 mg product 21g from 150 mg 7) via flash column chromatography (MeOH: DCM, from 1.0: 100 to 2.0: 100). 1H NMR (500 MHz, DMF-d7):δ 11.29 (s, 1H), 8.52 (s, 1H), 7.62 (d, J = 15.7 Hz, 1H), 7.53 (d, J = 7.3 Hz, 2H), 7.42-7.36 (m, 4H), 7.31 (tt, J1 = 7.3 Hz, J2 = 2.0 Hz, 1H), 6.98 (d, J = 8.6 Hz, 2H), 6.78 (m, 2H), 6.61 (td, J1 = 15.8 Hz, J2 = 6.3 Hz, 1H), 5.30 (d, J = 6.1 Hz, 2H), 4.89 (s, 2H), 3.83 (s, 3H); 13C NMR (125 MHz, DMF-d7):δ 164.1, 161.0, 144.7, 137.3, 135.4, 131.9, 129.8, 129.7, 129.4, 129.3, 127.8, 125.7, 124.5, 120.4, 114.8, 78.1, 56.0, 52.8; HRMS (ESI) calcd for C22H22N4O3Na [M+Na]+ 413.1590, found 413.1581.
General procedure for the preparation of 22a-g
The alkyne precursor 8 (589 mg, 2.12 mmol, 1.0 equiv) was added to a reaction vessel containing THF (50 mL) and MeOH (50 mL). Cesium fluoride (322 mg, 2.12 mmol, 1.0 equiv) was then added, after which the reaction was monitored by TLC analysis. The desilylation intermediate was slightly more polar than 8. Upon the disappearance of 8, the solution was split into ten equal fractions. These fractions were used directly in the ensuing reactions as work-up led to degradation of the alkyne. To each fraction of the alkyne solution, the azido compound 9a–g (0.32 mmol, 1.5 equiv), catalyst CuIP(OEt)3 (10 mg, 0.028 mmol, 0.1 equiv) and one drop of DIPEA were added sequentially. The reaction was allowed to proceed to completion by stirring overnight at room temperature. The solvent was removed, and the residue was partitioned between DCM (50 mL) and 1% CuSO4 aqueous solution (50 mL). The aqueous phase was additionally extracted with DCM (2 × 50 mL). The combined organic fractions were then dried (Na2SO4). After removal of the solvent, the crude product was purified via flash column chromatography.
1-Benzyl-1H-[1,2,3]triazole-4-carboxylic acid (4-methoxy-benzyloxy)-amide (22a)
A white solid was obtained (83% yield, 60 mg 22a from 0.212 mmol 8) via flash column chromatography (MeOH: DCM, from 0.5: 100 to 0.8: 100). 1H NMR (500 MHz, DMF-d7): δ 11.76 (bd s, 1H), 8.72 (s, 1H), 7.60-7.25 (m, 7H), 6.96 (d, J = 7.3 Hz, 2H), 5.77 (s, 2H), 4.95 (s, 2H), 3.82 (s, 3H); 13C NMR (125 MHz, DMF-d7): δ 161.0, 159.1, 142.8, 137.1, 131.8, 130.0, 129.5, 129.3, 127.7, 114.7, 78.5, 56.0, 54.5; HRMS (ESI) calcd for C18H18N4O3Na [M+Na]+ 361.1277, found 361.1263.
1-Phenethyl-1H-[1,2,3]triazole-4-carboxylic acid (4-methoxy-benzyloxy)-amide (22b)
A white solid was obtained (95% yield, 63 mg 22b from 0.189 mmol 8) via flash column chromatography (MeOH: DCM, from 0.5: 100 to 0.75: 100). 1H NMR (500 MHz, DMF-d7):δ 11.69 (bd s, 1H), 8.56 (s, 1H), 7.42 (d, J = 8.6 Hz, 2H), 7.32-7.20 (m, 5H), 6.96 (d, J = 8.7 Hz, 2H), 4.93 (s, 2H), 4.77 (t, J = 7.3 Hz, 2H), 3.81 (s, 3H), 3.28 (t, J = 7.4 Hz, 2H); 13C NMR (125 MHz, DMF-d7):δ 161.0,142.4, 138.8, 131.8, 129.9, 129.6, 129.3, 127.8, 127.7, 114.8, 78.5, 56.0, 52.2, 37.0; HRMS (ESI) calcd for C19H20N4O3Na [M+Na]+ 375.1433, found 375.1425.
1-(3-Phenyl-propyl)-1H-[1,2,3]triazole-4-carboxylic acid (4-methoxy-benzyloxy)-amide (22c)
A white solid was obtained (67% yield, 53 mg 22c from 0.212 mmol 8) via flash column chromatography (MeOH: DCM, from 0.5: 100 to 0.6: 100). 1H NMR (500 MHz, DMF-d7): δ 11.74 (bd s, 1H), 8.69 (s, 1H), 7.45 (d, J = 8.4 Hz, 2H), 7.36-7.20 (m, 5H), 6.98 (d, J = 8.5 Hz, 2H), 4.97 (s, 2H), 4.54 (t, J = 7.0 Hz, 2H), 3.83 (s, 3H), 2.67 (t, J = 7.7 Hz, 2H), 2.27 (tt, J1 = J2 = 7.4 Hz, 2H); 13C NMR (125 MHz, DMF-d7):δ 161.0, 159.2, 142.6, 142.1, 131.8, 129.5, 129.3, 127.6, 127.2, 114.8, 78.5, 56.0, 50.7, 33.3, 32.8 (One less peak due to overlap); HRMS (ESI) calcd for C20H22N4O3Na [M+Na]+ 389.1590, found 389.1585.
1-(2-Phenoxy-ethyl)-1H-[1,2,3]triazole-4-carboxylic acid (4-methoxy-benzyloxy)-amide (22d)
A white solid was obtained (56 % yield, 44 mg 22d from 0.212 mmol 8) via flash column chromatography (MeOH: DCM, from 0.5: 100 to 1.0: 100). 1H NMR (500 MHz, DMF-d7):δ 11.81-11.72 (bd s, 1H), 8.73 (s, 1H), 7.44 (d, J = 8.6 Hz, 2H), 7.31 (t, J = 8.0 Hz, 2H), 7.01-6.95 (m, 5H), 4.98-4.94 (m, 4H), 4.54 (t, J = 5.2 Hz, 2H), 3.82 (s, 3H); 13C NMR (125 MHz, DMF-d7):δ 161.0, 159.4, 159.1, 142.6, 131.8, 130.7, 129.3, 128.2, 122.3, 115.7, 114.7, 78.5, 63.7, 56.0, 50.8; HRMS (ESI) calcd for C19H20N4O4Na [M+Na]+ 391.1382, found 391.1379.
1-(2-Oxo-2-phenyl-ethyl-1H-[1,2,3]triazole-4-carboxylic acid (4-methoxy-benzyloxy)-amide (22e)
A pale yellow solid was obtained (76 % yield, 74 mg 22e from 0.265 mmol 8) via flash column chromatography (MeOH: DCM, from 0.25: 100 to 1: 100). 1H NMR (500 MHz, DMF-d7):δ 11.83 (s, 1H), 8.69 (s, 1H), 8.17 (d, J = 7.6 Hz, 2H), 7.78 (t, J = 7.3 Hz, 1H), 7.65 (dd, J1 = J2 = 7.5 Hz, 2H), 7.46 (d, J = 8.2 Hz, 2H), 6.99 (d, J = 8.2 Hz, 2H), 6.39 (s, 2H), 4.99 (s, 2H), 3.84 (s, 3H); 13C NMR (125 MHz, DMF-d7):δ 192.9, 161.0, 159.2, 142.6, 135.6, 135.4, 131.9, 130.1, 129.5, 129.3, 114.8, 78.5, 57.4, 56.0 (One less peak due to overlap); HRMS (ESI) calcd for C19H18N4O4Na [M+Na]+ 389.1266, found 389.1218.
1-Phenylcarbamoylmethyl-1H-[1,2,3]triazole-4-carboxylic acid (4-methoxy-benzyloxy)-amide (22f)
The crude product was dissolved in a minimal amount of 5% MeOH in DCM. The pure compound was then precipitated via addition of hexane (2:1 = Hexane: Solution). Following centrifugation (5000 rpm, 5 min), the supernatant was removed, and the solid was sonicated in the presence of a 1% EDTA aqueous solution. The solution was then filtered to afford the pure compound (81% yield, 82 mg 22f from 0.265 mmol 8). 1H NMR (500 MHz, DMF-d7):δ 11.90-11.70 (bd s, 1H), 10.65 (s, 1H), 8.72 (s, 1H), 7.69 (d, J = 8.0 Hz, 2H), 7.45 (d, J = 8.5 Hz, 2H), 7.36 (dd, J1 = J2 = 7.8 Hz, 2H), 7.12 (t, J = 7.3 Hz, 1H), 6.98 (d, J = 8.5 Hz, 2H), 5.57 (s, 2H), 4.98 (s, 2H), 3.83 (s, 3H); 13C NMR (125 MHz, DMF-d7):δ 165.2, 161.0, 159.2, 142.4, 140.0, 131.8, 129.4, 129.3, 124.9, 120.4, 114.8, 78.5, 56.0, 53.7 (One less peak due to overlap); HRMS (ESI) calcd for C19H19N5O4Na [M+Na]+ 404.1335, found 404.1319.
1-(3-Phenyl-allyl)-1H-[1,2,3]triazole-4-carboxylic acid (4-methoxy-benzyloxy)-amide (22g)
A white solid was obtained (80% yield, 62 mg of 22g from 0.212 mmol 8) via flash column chromatography (MeOH: DCM, from 0.25: 100 to 0.5: 100). 1H NMR (500 MHz, DMF-d7):δ 11.76 (s, 1H), 8.69 (s, 1H), 7.54 (d, J = 7.4 Hz, 2H), 7.44 (d, J = 8.6 Hz, 2H), 7.38 (dd, J1 = J2 = 7.5 Hz, 2H), 7.31 (t, J = 7.3 Hz, 1H), 6.97 (d, J = 8.5 Hz, 2H), 6.80 (d, J = 15.9 Hz, 1H), 6.63 (td, J1 = 15.8 Hz, J2 = 6.6 Hz, 1H), 5.34 (d, J = 6.5, 2H), 4.96 (s, 2H), 3.82 (s, 3H); 13C NMR (125 MHz, DMF-d7):δ 161.0, 159.1, 142.7, 137.3, 135.6, 131.8, 129.8, 129.4, 129.3, 127.8, 127.5, 124.4, 114.7, 78.5, 56.0, 53.0; HRMS (ESI) calcd for C20H20N4O3Na [M+Na]+ 387.1433, found 387.1430.
Supplementary Material
Figure 3.
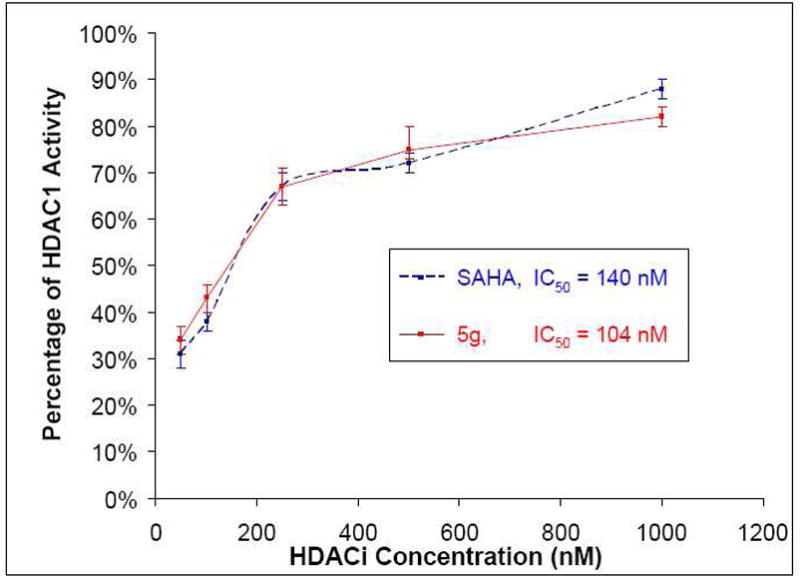
Comparison of HDAC1 inhibition by SAHA and 5g.
Acknowledgments
This project was supported by The Ohio State University (support to Peng George Wang). The Cell Line Studies were performed by the NIH.
Abbreviations
- HDAC
histone deacetylase
- HDACi
histone deacetylase inhibitor
- SAHA
Suberoylanilide hydroxamic acid
- TSA
trichostatin A
- HA
hydroxamic acid
Footnotes
Supporting Information Available: Proton and carbon NMR spectra of new compounds are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 3.Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nature Reviews Genetics. 2002;3:662–673. doi: 10.1038/nrg887. [DOI] [PubMed] [Google Scholar]
- 4.Trojer P, Reinberg D. Histone lysine demethylases and their impact on epigenetics. Cell. 2006;125:213–217. doi: 10.1016/j.cell.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 5.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nature Reviews Molecular Cell Biology. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolffe AP. Histone deacetylase: a regulator of transcription. Science. 1996;272:371–372. doi: 10.1126/science.272.5260.371. [DOI] [PubMed] [Google Scholar]
- 7.Lin RJ, Sternsdorf T, Tini M, Evans RM. Transcriptional regulation in acute promyelocytic leukemia. Oncogene. 2001;20:7204–7215. doi: 10.1038/sj.onc.1204853. [DOI] [PubMed] [Google Scholar]
- 8.Dokmanovic M, Marks PA. Prospects: Histone deacetylase inhibitors. Journal of Cellular Biochemistry. 2005;96:293–304. doi: 10.1002/jcb.20532. [DOI] [PubMed] [Google Scholar]
- 9.Drummond DC, Nobel CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. Clinical development of histone deacetylase inhibitors as anticancer agents. Annual Review of Pharmacology and Toxicology. 2005;45:495–528. doi: 10.1146/annurev.pharmtox.45.120403.095825. [DOI] [PubMed] [Google Scholar]
- 10.Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408:377–381. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- 11.Johnstone RW, Licht JD. Histone deacetylase inhibitors in cancer therapy: Is transcription the primary target? Cancer Cell. 2003;4:13–18. doi: 10.1016/s1535-6108(03)00165-x. [DOI] [PubMed] [Google Scholar]
- 12.Nusinzon I, Horvath CM. Interferon-stimulated transcription and innate antiviral immunity require deacetylase activity and histone deacetylase 1. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:14742–14747. doi: 10.1073/pnas.2433987100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan Z-l, Guan Y-j, Chatterjee D, Chin YE. Stat3 Dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269–273. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 14.Chen L-F, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kB action regulated by reversible acetylation. Science. 2001;307:269–273. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 15.Gu W, Roeder RG. Activation of p-53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 16.Martinez-Balbas MA, Bauer UM, Nielsen SJ, Brehm A, Kouzarides T. Regulation of E2F1 activity by acetylation. EMBO J. 2000;19:662–671. doi: 10.1093/emboj/19.4.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen HY, Lavu S, Bitterman KJ, Hekking B, Imahiyerobo TA, Miller C, Frye R, Ploegh H, Kessler BM, Sinclair DA. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Molecular Cell. 2004;13:627–638. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- 18.Pan L, Lu J, Huang B. HDAC inhibitors: a potential new category of anti-tumor agents. Cellular & Molecular Immunology. 2007;4:337–343. [PubMed] [Google Scholar]
- 19.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Molecular Cancer Research. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 20.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nature Reviews Drug Discovery. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 21.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nature Reviews Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 22.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 23.Elaut G, Laus G, Alexandre E, Richert L, Bachellier P, Tourwe D, Rogiers V, Vanhaecke T. A metabolic screening study of trichostatin A (TSA) and TSA-like histone deacetylase inhibitors in rat and human primary hepatocyte cultures. Journal of Pharmacology and Experimental Therapeutics. 2007;321:400–408. doi: 10.1124/jpet.106.116202. [DOI] [PubMed] [Google Scholar]
- 24.Sanderson L, Taylor GW, Aboagye EO, Alao JP, Latigo JR, Coombes RC, Vigushin DM. Plasma pharmacokinetics and metabolism of the histone deacetylase inhibitor trichostatin A after intraperitoneal administration of mice. Drug Metabolism and Disposition. 2004;32:1132–1138. doi: 10.1124/dmd.104.000638. [DOI] [PubMed] [Google Scholar]
- 25.Elaut G, Toeroek G, Papeleu P, Vanhaecke T, Laus G, Tourwe D, Rogiers V. Rat hepatocyte suspensions as a suitable in vitro model for studying the biotransformation of histone deacetylase inhibitors. Alternatives to Laboratory Animals. 2004;32:105–112. doi: 10.1177/026119290403201s16. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki T, Miyata N. Rational design of non-hydroxamate histone deacetylase inhibitors. Mini-Reviews in Medicianl Chemistry. 2006;6:515–526. doi: 10.2174/138955706776876186. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki T, Miyata N. Non-hydroxamate histone deacetylase inhibitors. Current Medicinal Chemistry. 2005;12:2867–2880. doi: 10.2174/092986705774454706. [DOI] [PubMed] [Google Scholar]
- 28.Furumai R, Komatsu Y, Nishino N, Khochbin S, Yoshida M, Horinouchi S. Potent histone deacetylase inhibitors built from trichosatin A and cyclic tetrapeptide antibiotics including trapoxin. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:87–92. doi: 10.1073/pnas.011405598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones P, Altamura S, Chakravarty PK, Cecchetti O, De Francesco R, Gallinari P, Ingenito R, Meinke PT, Petrocchi A, Rowley M, et al. A series of novel, potent and selective histone deacetylase inhibitors. Bioorganic & Medicinal Chemistry Letters. 2006;16:5948–5952. doi: 10.1016/j.bmcl.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 30.Kahnberg P, Lucke AJ, Glenn MP, Boyle GM, Tyndall JDA, Parsons PG, Fairlie DP. Design, synthesis, potency and cytoselectivity of anticancer agents derived by parallel synthesis from aminosuberic acid. Journal of Medicinal Chemistry. 2006;49:7611–7622. doi: 10.1021/jm050214x. [DOI] [PubMed] [Google Scholar]
- 31.Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:15064–15069. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angewandte Chemie, International Edition. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; (32) Moses JE, Moorhouse AD. The growing applications of click chemistry. Chemical Society Reviews. 2007;36:1249–1262. doi: 10.1039/b613014n. [DOI] [PubMed] [Google Scholar]
- 33.Roper S, Kolb HC. Click chemistry for drug discovery. Methods and Principles in Medicinal Chemistry. 2006;34:313–339. [Google Scholar]
- 34.Weber L. In vitro combinatorial chemistry to create drug candidates. Drug Discovery Today: Technologies. 2004;1:261–267. doi: 10.1016/j.ddtec.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 35.Kolb HC, Sharpless KB. The growing impact of click chemistry on drug discovery. Drug Discovery Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 36.Alvarez SG, Alvarez MT. A practical procedure for the synthesis of alkyl azides at ambient temperature in dimethyl sulfoxide in high purity and yield. Synthesis. 1997:413–414. [Google Scholar]
- 37.Ramsay SL, Freeman C, Grace PB, Redmond JW, MacLeod JK. Mild tagging procedures for the structural analysis of glycans. Carbohydrate Research. 2001;33:59–71. doi: 10.1016/s0008-6215(01)00115-x. [DOI] [PubMed] [Google Scholar]
- 38.Dixon DJ, Ley SV, Longbottom DA. Copper(I)-catalyzed preparation of (E)-3-iodoprop-2-enoic acid. Organic Syntheses. 2003;80:129–132. [Google Scholar]
- 39.Eichberg MJ, Dorta RL, Lamottke K, Vollhardt KPC. The formal total synthesis of (±)-strychnine via a cobalt-mediated [2+2+2] cycloaddition. Organic Letters. 2000;2:2479–2481. doi: 10.1021/ol006131m. [DOI] [PubMed] [Google Scholar]
- 40.Krusche CA, Wuelfing P, Kersting C, Vloet A, Boecker W, Kiesel L, Beier HM, Alfer J. Histone deacetylase-1 and -3 protein expression in human breast cancer: a tissue microarray analysis. Breast Cancer Research and Treatment. 2005;90:15–23. doi: 10.1007/s10549-004-1668-2. [DOI] [PubMed] [Google Scholar]
- 41.Park JH, Jung Y, Kim TY, Kim SG, Jong HS, Lee JW, Kim DK, Lee JS, Kim NK, Kim TY, Bang Y-J. Class I Histone Deacetylase-Selective Novel Synthetic Inhibitors Potently Inhibit Human Tumor Proliferation. Clinical Cancer Research. 2004;10:5271–5281. doi: 10.1158/1078-0432.CCR-03-0709. [DOI] [PubMed] [Google Scholar]
- 42.Halkidou K, Gaughan L, Cook S, Leung HY, Neal DE, Robson CN. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate. 2004;59:177–189. doi: 10.1002/pros.20022. [DOI] [PubMed] [Google Scholar]
- 43.Choi JH, Kwon HJ, Yoon BI, Kim JH, Han SU, Joo HJ, Kim D-Y. Expression profile of histone deacetylase 1 in gastric cancer tissues. Japanese Journal of Cancer Research. 2001;92:1300–1304. doi: 10.1111/j.1349-7006.2001.tb02153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Waltregny D, North B, Van Mellaert F, de Leval J, Verdin E, Castronovo V. Screening of histone deacetylases (HDAC) expression in human prostate cancer reveals distinct class I HDAC profiles between epithelial and stromal cells. European journal of histochemistry. 2004;48:273–290. [PubMed] [Google Scholar]
- 45.KrennHrubec K, Marshall BL, Hedglin M, Verdin E, Ulrich SM. Design and evaluation of ‘Linkerless’ hydroxamic acids as selective HDAC8 inhibitors. Bioorganic & Medicinal Chemistry Letters. 2007;17:2874–2878. doi: 10.1016/j.bmcl.2007.02.064. [DOI] [PubMed] [Google Scholar]
- 46.Hu E, Dul E, Sung C-M, Chen Z, Kirkpatrick R, Zhang GF, Johanson K, Liu R, Lago A, Hofmann G, et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. Journal of Pharmacology and Experimental Therapeutics. 2003;307:720–728. doi: 10.1124/jpet.103.055541. [DOI] [PubMed] [Google Scholar]
- 47.These NSC numbers were assigned by the NCI.
- 48.Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nature reviews Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 49.It should be noted that the NCI does not include an error analysis in their reports of compound activity in the human cancer cell line screen. However, they do perform an adequate number of determinations. Specifically, two microtiter plates are utilized for the testing of each cell line. Each drug is then tested at five concentrations along with a control. Furthermore, this screen is also well established at the NCI as it became operational in 1990 following a five year development period.
- 50.Katritzky AR, Liso G, Lunt E, Patel RC, Thind SS, Zia A. Heterocycles in organic synthesis. Part 42. Preparation of azides, phthalimides and sulfonamides from primary amines. Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry. 1980:849–850. [Google Scholar]
- 51.Benati L, Bencivenni G, Leardini R, Nanni D, Minozzi M, Spagnolo P, Scialpi R, Zanardi G. Reaction of azides with dichloroindium hydride: very mild production of amines and pyrrolidin-2-imines through possible indium-aminyl radicals. Organic Letters. 2006;8:2499–2502. doi: 10.1021/ol0606637. [DOI] [PubMed] [Google Scholar]
- 52.Mandel SM, Bauer JAK, Gudmundsdottir AD. Photolysis of α-azidoacetophenones: trapping of triplet alkyl nitrenes in solution. Organic Letters. 2001;3:523–526. doi: 10.1021/ol0068750. [DOI] [PubMed] [Google Scholar]
- 53.Srinivasan R, Uttamchandani M, Yao SQ. Rapid assembly and in situ screening of bidentate inhibitors of protein tyrosine phosphatases. Organic Letters. 2006;8:713–716. doi: 10.1021/ol052895w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.