

Abstract
The BH3-only members of the Bcl-2 protein family are essential for initiation of programmed cell death and stress-induced apoptosis. We have determined the expression pattern in mice of the BH3-only protein Bik, also called Blk or Nbk, and examined its physiological function by gene targeting. We found that Bik is expressed widely in the hematopoietic compartment and in endothelial cells of the venous but not arterial lineages. Nevertheless, its loss did not increase the numbers of such cells in mice or protect hematopoietic cells in vitro from apoptosis induced by cytokine withdrawal or diverse other cytotoxic stimuli. Moreover, whereas loss of the BH3-only protein Bim rescued mice lacking the prosurvival protein Bcl-2 from fatal polycystic kidney disease and lymphopenia, loss of Bik did not. These results indicate that any function of Bik in programmed cell death and stress-induced apoptosis must overlap that of other BH3-only proteins.
Programmed cell death (apoptosis), the orchestrated, genetically controlled removal of redundant, damaged, or potentially harmful cells from the body, is essential for correct tissue formation during development and subsequent maintenance of cellular homeostasis (1, 39). Apoptosis is governed by an evolutionarily conserved family of cell death regulators whose prototypic member, Bcl-2, was identified as a protein that promotes cell survival (9, 10). Bcl-2-related apoptosis inhibitors are found in mammals, nonmammalian vertebrates, invertebrates (e.g., Caenorhabditis elegans CED-9), and viruses (e.g., adenovirus E1B 19-kDa protein) and have three or four regions of homology, the so called Bcl-2 homology (BH) regions 1 to 4. The Bcl-2 family also contains two proapoptotic subgroups. Bax, Bak, Bok/Mtd, Bcl-GL, and Bfk have two or three BH regions, and at least Bax has extensive structural similarity to its prosurvival relatives (41). In contrast, mammalian Bik/Blk/Nbk, Bad, Hrk/DP5, Bid, Bim, Noxa, Puma, and Bmf, as well as C. elegans EGL-1, share with each other and the remainder of the Bcl-2 family only the short BH3 protein interaction domain and are therefore often called BH3-only proteins (19). Both subgroups of proapoptotic family members are essential for programmed cell death and stress-induced apoptosis. It appears that BH3-only proteins initiate cell death signalling (4, 19) whereas Bax/Bak-like proteins function further downstream (7, 25, 46).
Gene knockout studies have helped to determine the role of a number of Bcl-2 family members in apoptosis regulation (10). For example, mice lacking the proapoptotic BH3-only gene bim accumulate lymphoid, myeloid, and plasma cells (4). Their T-cell development is disrupted, and their lymphocytes are refractory to a range of apoptotic stimuli, including those mediating the deletion of autoreactive cells (5, 14). In contrast, mice lacking prosurvival Bcl-2 die early in life due to polycystic kidney disease (42) and their lymphocytes have an abnormally short life span and abnormal sensitivity to apoptotic stimuli (27, 42). The demonstration that the renal disease and lymphopenia in these mice were abolished by concomitant deficiency for Bim underlines the critical opposing roles of BH3-only and Bcl-2-like proteins for tissue homeostasis (3).
Determining the physiological roles of additional BH3-only proteins requires identification of the cell types in which each is expressed and establishment of the consequences of gene disruption. Here we report such studies for the mouse BH3-only gene originally designated blk (18). It is now evident that blk is the murine ortholog of human bik/nbk (6, 17) on the basis of their location in syntenic regions, gene organization, and nucleic as well as amino acid sequence homology (2, 11, 43). Consequently, we recommend that both the mouse and human gene be designated bik, as we will do below. Like other BH3-only proteins, Bik binds to prosurvival family members and kills cells when overexpressed (6, 17, 18). However, although Bik was one of the first BH3-only proteins to be discovered, little has been reported about its expression and regulation. We report here that mouse bik is widely expressed in the hematopoietic system and, like its human ortholog (22), can be induced in B cells following antigen receptor stimulation. Interestingly, we also found that bik is expressed in a subset of endothelial cells. We describe Bik-deficient mice and our efforts to determine whether Bik plays a nonredundant role in those cell types for their programmed death in vivo or for their responses to diverse cytotoxic stimuli in vitro.
MATERIALS AND METHODS
Generation of bik−/− and bik−/− bcl-2−/− mice.
The bik locus was characterized from four clones (010-k12, 216-a17, 131-h19 and 121-m7) isolated from an RPCI-23 female C57BL/6J mouse bacterial artificial chromosome library (Roswell Park Cancer Institutes, Roswell Park, N.Y.) screened using a probe encompassing the bik coding sequence. The targeting construct for bik was generated to include 2.8 kb of 5′ and 4.6 kb of 3′ homologous flanking sequence surrounding a nucleus localized Escherichia coli lacZ reporter gene [with its own poly(A) signal] fused in-frame with the bik initiator ATG and a loxP-flanked neomycin resistance cassette driven by the PGK promoter, replacing the first and second (BH3 region) coding exons of bik (see Fig. 1). The targeting construct was electroporated into C57BL/6 (Bruce4) embryonic stem (ES) cells. Targeting was confirmed by genomic Southern blotting using external 5′ and 3′ genomic probes and a Neo-specific probe to confirm single construct integration. Three correctly targeted ES lines were selected for blastocyst injection, and, of the resulting three independently derived mouse lines, two (denoted 373 and 375) were chosen for analysis and maintained on an inbred C57BL/6 background. The neomycin resistance cassette was deleted from one (mouse 375) by crossing to female C57BL/6 Cre deleter mice (36), to generate the del375 line. Neomycin cassette deletion was confirmed by PCR. All analysis reported was performed using del375 mice and verified where possible using 373 mice. Targeted bik mice of the 373, 375, and del375 lines were genotyped by PCR using a common 5′ oligonucleotide (5′-TCTGTAGCCACCGGTGCATAGTTCTG) in combination with a 3′ (antisense) targeted allele-specific oligonucleotide (5′-CGCCAGGGTTTTCCCAGTCACGAC) and a 3′ (antisense) wild-type (wt) allele-specific oligonucleotide (5′-TGACGTCTCTGGCCATAAGTCTCGC) in a three-oligonucleotide PCR. This reaction, designed to coamplify a wt, allele-specific product of 102 bp and a targeted allele-specific product of 162 bp, was performed using 25 cycles and an annealing temperature of 60°C.
FIG. 1.

bik expression in adult tissues and hematopoietic cell types. (A) Northern blot analysis of bik mRNA expression on poly(A)+-enriched RNA from indicated tissues of C57BL/6 mice, using a bik cDNA coding region probe. An ethidium bromide stain of 28S and 18S rRNA is included as a loading control. Muscle, skeletal muscle; salv. gland, salivary gland. (B) Northern blot analysis of bik mRNA expression on poly(A)+-enriched RNA from the indicated cell lines. (C) Semiquantitative RT-PCR analysis of bik mRNA expression in FACS-purified thymocyte subsets: pro-T3 (CD25+ CD44−), pro-T4 (CD25− CD44−), and large (cycling) and small (resting) pre-T (CD4+ CD8+). Analysis was performed on a titration of the cDNA template (neat, 1:5, and 1:25). Each amplicon was confirmed as that for bik by Southern blotting using a bik-specific internal oligonucleotide. Expression of hprt is included as a control for loading. (D) Semiquantitative RT-PCR analysis of bik mRNA expression in FACS-purified hematopoietic cell subsets: pro-B (B220+ CD43+ sIg−), pre-B (B220+ CD43− sIg−), naive B (B220+ sIgM+ sIgD−), mature B (B220+ sIgMlo sIgDhi), CD4+ CD8− T, CD4− CD8+ T, macrophages (Mac-1+ Gr-1−), granulocytes (Mac-1+ Gr-1+), or nucleated erythroid progenitors (Ter119+). B cells were activated with IgM cross-linking antibodies in the presence of cytokines. T cells were activated with PMA and ionomycin in the presence of cytokines. Analysis was performed as described in the legend to panel C.
The bcl-2+/− mice (29) were backcrossed for >10 generations to a C57BL/6 background as described previously (3). These mice were bred with bik−/− mice in successive intercrosses to generate doubly homozygous mutant mice.
Northern blotting, Southern blotting, and RT-PCR.
RNA for Northern blotting was extracted from tissues using guanidine isothiocyanate and phenol-chloroform and poly(A)+ mRNA enriched using oligo(dT) cellulose (Roche). RNA was size fractionated by electrophoresis on a denaturing 1% agarose-formaldehyde gel, and blots were hybridized to an α-32P-labeled bik coding region probe in Church buffer (8). Similarly, for Southern blotting, DNA (10 μg) was digested overnight and size separated by electrophoresis on a 0.7% agarose gel and the blots were probed with α-32P-labeled probes using Church buffer.
cDNA for reverse transcription-PCR (RT-PCR) expression analysis was prepared from fluorescence-activated cell sorter (FACS)-sorted hematopoietic cells, and PCR amplifications were performed as described previously (30). Oligonucleotides for bik were 5′-GCTTGGCAGGATCCATGTCGGAGGCGAGACTTATG (sense) and 5′-AACAAGCTTCCCTGCCCCAGCTGCACTTCACTG) (antisense), those for hprt were 5′-GCTGGTGAAAAGGACCTCT (sense) and 5′-CACAGGACTAGAACACCTGC (antisense), and those for gapdh were 5′ TGATGACATCAAGAAGGTGGTGAAG (sense) and 5′ TCCTTGGAGGCCATGTAGGCCAT (antisense). PCR products were subjected to Southern blotting with α-32P-end-labeled bik (5′-GTCAAGCTTCACTCAGGCGCCAGGAGTCAAGAC), hptr (5′-GGATACAGGCCAGACTTTGT), or a gapdh (5′-CCCGGCATCGAAGGTGGAAGAG) oligonucleotide in Church buffer.
Cell culture and viability assays.
All cells were cultured at 37°C in a humidified 10% CO2 incubator in high-glucose Dulbecco Modified Eagle medium supplemented with 10% heat-inactivated fetal calf serum, 50 μM 2-mercaptoethanol, and 10−6 M asparagine. FACS-sorted primary T and B lymphocytes were cultured at 1 × 105 to 4 × 105 cells/ml, and thymocytes were cultured at 2 × 106 cells/ml. FACS-sorted mature B cells (sIgMlo sIgDhi) (see below) were stimulated with 20 μg of goat anti-mouse immunoglobulin M (IgM) (μ chain) F(ab′)2 antibody fragments (Jackson ImmunoResearch) per ml in the presence of 100 U each of interleukin-2 (IL-2), IL-4, and IL-5 per ml at a starting concentration of 105 cells/ml. FACS-sorted T cells were stimulated with 2 ng of phorbol myristate acetate (PMA) (Sigma) per ml and 0.1 μg of ionomycin (Sigma) per ml plus 100 U of IL-2 per ml at 105 cells/ml. Cross-linked FasL was prepared by mixing FLAG-tagged human FasL (Alexis) with anti-FLAG M2 antibody (10 μg/ml) (Sigma). To assess viability, cell suspensions were incubated with 2 μg of propidium iodide (PI) (Sigma) per ml on ice for 5 min and analyzed on a FACS analyzer and viability was determined as the percentage of cells negative for PI.
Hematopoietic reconstitutions.
Fetal livers were dissected from Ly5.2+ E14.5 embryos and dispersed, and 2 × 106 cells were injected via the tail vein into irradiated (twice at 550 R) C57BL/6 Ly5.1 female recipient mice. Blood was taken 9 to 12 weeks postreconstitution and subjected to hematologic and FACS analysis. Host- and donor-derived cells were distinguished by staining for Ly5.1 and Ly5.2, respectively. Hematopoietic organ composition was determined by FACS 11 to 15 weeks postreconstitution. Donor-derived T and B lymphocytes (Ly5.2+) were isolated from recipient lymph nodes for viability assays by cell sorting (see below). All analysis was performed using recipients from multiple independent donors of each genotype.
Immunofluorescence staining and flow cytometric analysis.
FACS analysis and cell sorting were performed using RA3-6B2 anti-B220, S7 anti-CD43, 5.1 anti-IgM, 11-26C anti-IgD, YTA-321 anti-CD4, YTS-169 anti-CD8, A201.7 anti-Ly5.1, AL14A7 Ly5.2, 8C5 anti-Gr-1, and MI/70 anti-Mac-1 antibodies and Ter119 anti-erythroid surface marker conjugated to fluorescein isothiocyanate, Cy5, or biotin (Molecular Probes). R-Phycoerythrin-conjugated anti-B220 (Pharmingen) and anti-CD8 (Caltag) were also used Cell types were sorted as positive for the appropriate surface markers as described previously (30) using MoFlo (Cytomation) or Diva (Becton Dickinson) sorters. Pro-T and pre-T populations were sorted from thymus. Pro-B, pre-B, nucleated erythroid cells, and macrophages were sorted from bone marrow. Naive and mature B cells were sorted from spleen. T cells were sorted from lymph nodes, and granulocytes were isolated from peritoneal lavage fluid.
For hematopoietic analysis, single-cell suspensions were prepared from the thymus, lymph nodes (axial, branchial, mesenteric, and inguinal), bone marrow (two femurs), and spleen. Total viable white cells were then enumerated by trypan blue/hemocytometer counts. The composition of these organs was determined by FACS analysis as described previously (30) by using a FACScan apparatus (Becton Dickinson) or LSR analyser (Becton Dickinson). Hematological analysis was performed using an ADVIA hematology system (Bayer) on blood extracted via the retroorbital plexus.
For survival assays, wild-type and bik−/− lymphocytes were sorted from lymph nodes. T cells were sorted as CD4+ CD8− or CD4− CD8+ cells, whereas B cells were negatively sorted by collecting CD4− CD8− lymph node cells and shown to be ≥98% B220+.
Histology and β-galactosidase assay.
Tissues for frozen sections were dissected and fixed in 4 or 0.2% paraformaldehyde (pH 7.4) for 2 or 16 h, respectively. The tissues were infiltrated with sucrose before being transferred to OCT fixative (Sakura Finetechnical Co., Tokyo, Japan) and snap frozen. Blocks were sectioned at 10 μm on a Leica CM 3050 S cryostat (Leica).
Dual β-galactosidase/CD31 (PECAM-1) staining was performed on frozen sections from tissue fixed for 16 h with 0.2% paraformaldehyde. Sections were washed three times in phosphate buffered saline and then incubated with 0.03% H2O2 for 10 min. They were blocked with 10% normal goat serum (Vector Laboratories) before the addition of rat anti-mouse CD31− (7 μg/ml [Pharmingen], a kind gift from S. Stacker, Ludwig Institute, Melbourne, Australia) or isotype-matched rat anti-mouse B220 monoclonal antibody (RA3-6B2) for 2 h at room temperature. The sections were then incubated with biotin-conjugated mouse anti-rat Igκ light chain antibody (Mar-18) followed by ABC biotin-specific horseradish peroxidase reagent (Vector Laboratories). CD31 staining was then revealed by diaminobenzidine staining. The sections were rinsed in five changes of distilled H2O and, while still moist, immediately stained for β-galactosidase activity. β-Galactosidase activity was assayed on fixed frozen sections or whole tissue by incubation at 37°C for 16 h in an air incubator containing stain solution (5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 2 mM MgCl2, 0.02% Tween 20, 0.5 mg of 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside [X-Gal] per ml). Stained tissues were embedded in paraffin and sectioned at 10 μm. The sections were counterstained with nuclear fast red.
For hyaloid vessel analysis, eyes from postnatal day 16 (PN16) mice were fixed for 2 h in Bouin's fixative, embedded in paraffin, and sectioned longitudinally to reveal pupillary membrane (PM), tunica vasculosa lentis (TVL), and vasa hyaloidea propria (VHP). The sections were stained with hematoxylin and eosin. Vessel counts were conducted on 12 nonconsecutive sections from each eye, and the average number of vessels per section was calculated for each eye.
RESULTS
bik is expressed in many hematopoietic cell types.
Little has been reported about the expression patterns of mouse or human bik, especially in cells and tissues of the hematopoietic system. Consistent with the findings of Hegde et al. (18), Northern blot analysis of tissues from adult C57BL/6 mice showed bik mRNA to be prominent in the liver, lung, heart and kidneys, while faint signals were also detected in the spleen, skeletal muscle, and salivary gland (Fig. 1A). Northern blots also revealed bik mRNA in cell lines of B- and T-lymphoid and macrophage origin, indicating that Bik may be expressed in diverse hematopoietic cell types (Fig. 1B). That was confirmed by RT-PCR analysis of cDNA produced from sorted cell subsets. In the thymus, bik mRNA was present in cells from all stages of T-cell development (Fig. 1C), with the highest levels being found in immature cortical CD4+ CD8+ (pre-T) thymocytes, as well as in thymic stroma. Mature CD4+ CD8− and CD4− CD8+ T cells in the spleen also expressed bik (Fig. 1D), and its levels (relative to the hprt mRNA control) were not affected by activation of the cells following mitogenic stimulation (Fig. 1D). bik mRNA was also found in granulocytes and bone marrow-derived erythroid progenitors but was only faintly detected in macrophages (Fig. 1D). In the B lymphocyte lineage, pre-B, virgin B, and mature B cells all exhibited bik mRNA. In mature B cells, its expression was elevated by stimulation of the B-cell receptor (BCR) in the presence of cytokines (Fig. 1D). This observation is consistent with the reported elevation in Bik mRNA and protein levels in a human B-cell line following BCR stimulation (22).
Because the expression of several genes for BH3-only proteins is induced by cytotoxic insults (10, 35), we also investigated whether bik expression was induced in response to DNA damage or treatment with cytotoxic drugs. bik mRNA levels were examined by RT-PCR analysis on cDNA prepared from thymocytes exposed to dexamethasone, ionizing radiation, or the calcium ionophore ionomycin or thymocytes undergoing spontaneous death when cultured in the absence of cytokines. No change was discerniable in response to any of these stimuli (data not shown).
Generation of mice lacking Bik.
The bik gene was disrupted by replacing its first two coding exons (including that encoding the BH3 region) with a nucleus-localized E. coli β-galactosidase reporter gene and a loxP-flanked neomycin resistance cassette (Fig. 2A). Three independently derived C57BL/6 ES cell clones harboring the correctly integrated targeting construct were identified. From each of two of these clones, an independent line of mice having a heterozygous deletion of the bik locus was established and bred to homozygosity. Targeting was confirmed by performing DNA analysis with both the 5′ and 3′ external genomic probes (Fig. 2B) and by showing that no bik mRNA could be detected in the major Bik-expressing organs: the liver, lung, heart, and kidneys (Fig. 2C). One of these mouse lines was then bred to C57BL/6 Cre deleter females, and deletion of the neo cassette was confirmed by PCR. Results presented here are from mice with a neo deletion and were verified by using a second, independently derived knockout line.
FIG. 2.

Targeting of the bik locus. (A) Schematic diagram (to scale) of the bik targeting construct and the wt and targeted bik loci. Dashed lines demarcate regions of the wt locus used in the targeting construct. Locations of 5′ and 3′ external probes for genomic Southern blot analysis and expected fragment sizes from indicated restriction digests are shown. B, BamHI; RV, EcoRV; S, SpeI. (B) Confirmation with 5′ and 3′ genomic probes of the loss of both wt bik alleles in bik−/− mice. (C) Northern blot analysis of poly(A)+ RNA extracted from the indicated tissues of wt and bik knockout mice (littermates). The blot was probed with a cDNA probe containing the entire bik coding region. A gapdh loading control is shown.
Bik is not essential for normal development.
From intercrosses of heterozygotes, bik homozygous null mice were born at the expected Mendelian frequency (bik+/+, 44; bik+/−, 72; bik−/−, 38). Their general appearance, behavior and health up to at least 1 year of age were normal. The weights of both male and female mice, monitored up to 34 weeks of age, were normal, as were the appearance and weights of their major organs (liver, lungs, heart, thymus, spleen, and kidneys). Moreover, histologic analysis revealed no obvious pathology or abnormality. When mated, bik−/− males sired litters of normal size and Bik-deficient females produced and reared litters of normal size. Thus, Bik is dispensable for normal development and reproduction.
Loss of Bik does not affect hematopoiesis or cellular responses to apoptotic stimuli.
Given the widespread expression of Bik in the hematopoietic compartment (Fig. 1), we explored whether its loss disrupted development or homeostasis of this compartment by comparing bone marrow, thymus, and spleen from 5- to 11-week old bik−/− mice and their wt littermates. The total cellularity of these organs from bik−/− mice was normal (Fig. 3A to C). Moreover, their bone marrow contained normal numbers of pro-B and pre-B cells, immature and mature B cells, precursor and mature macrophages, granulocytes, and nucleated erythroid cells (Fig. 3A and data not shown). In the bik−/− thymus, the numbers of pro-T, pre-T, and mature single positive T cells were normal (Fig. 3B). Similarly, the bik−/− spleen contained normal numbers of mature CD4+ and CD8+ T lymphocytes, B cells, macrophages, granulocytes, and nucleated erythroid cells (Fig. 3C and data not shown). In bik−/− lymph nodes, the percentages of mature T and B lymphocytes were also unaffected by Bik loss (data not shown). Furthermore, analysis of peripheral blood from aged bik−/− mice and their wt littermates (30 to 46 weeks old) showed that the total numbers of lymphocytes, monocytes, neutrophils, eosinophils, and basophils were normal (Fig. 3D), as were the numbers of erythrocytes and platelets (data not shown). The relatively small absolute numbers of peripheral blood leukocytes observed (in both wt and bik−/− mice) are most probably due to the advanced age of the mice examined.
FIG. 3.

Cell type composition of hematopoietic organs from wt and bik−/− littermates. (A) The total number of bone marrow cells from 5- to 11-week-old wt (n = 6) and bik−/− (n = 8) mice was determined, and the numbers of pro- and pre-B cells (B220+ sIgM− sIgD−), immature and mature B cells (B220+ sIgM+ sIgD+), macrophages (Mac1+ Grl−) and granulocytes (Mac1+ Grl+) was determined by FACS analysis. (B) The total number of thymocytes from 5- to 11-week-old wt (n = 8) and bik−/− (n = 8) animals was determined, and the number of pro-T (CD4− CD8−), pre-T (CD4+ CD8+), and mature T (CD4+ CD8− or CD4− CD8+) cells was determined by FACS analysis. (C) The total number of splenocytes from 5- to 11-week-old wt (n = 8) and bik−/− (n = 8) animals was determined, and the number of naive and mature B cells (B220+ sIgM+ sIgD+), other B cells (B220+ sIgM− sIgD−), mature T cells (CD4+ CD8− or CD4− CD8+), macrophages (Mac1+ Grl−), and granulocytes (Mac1+ Grl+) was determined by FACS analysis. (D) Peripheral blood from 30- to 46-week-old wt (n = 6) and bik−/− (n = 9) littermates was examined, and the total number of lymphocytes and granulocytes (neutrophils, monocytes, eosinophils, and basophils) per milliliter was determined.
We next determined whether Bik deficiency affected the response of lymphocytes to various apoptotic stimuli. Its loss did not confer any protection on thymocytes undergoing spontaneous death in culture (“unstimulated” cultures) or apoptosis following exposure to dexamethasone, etoposide, PMA, ionomycin, or Fas ligand (FasL) (Fig. 4A). Mature T cells (CD4+ CD8− or CD4− CD8+) and B cells from lymph nodes of bik−/− mice were also normally sensitive to spontaneous death in culture and treatment with dexamethasone or etoposide (Fig. 4B and C). Since the levels of Bik increase in B cells in response to BCR stimulation (Fig. 1D) (22), mature B cells from bik−/− mice were exposed to IgM cross-linking antibodies. The Bik-deficient B cells died at the same rate as their wt counterparts (Fig. 4C). Mitogen-activated B cells are dependent on cytokines for continued survival and proliferation, and cytokine deprivation induces apoptosis by a mechanism regulated by the Bcl-2 family (26, 40). However, when starved of cytokines, mitogen-activated B cells from Bik-deficient mice underwent apoptosis at the same rate as wt cells did (data not shown).
FIG. 4.
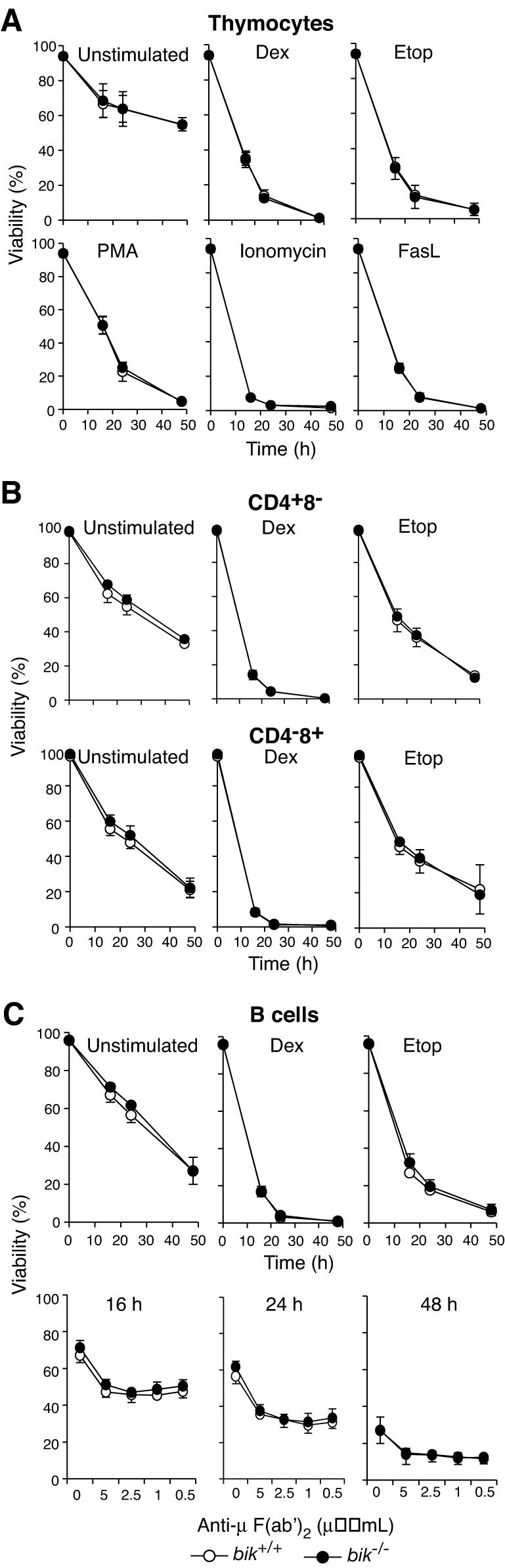
bik−/− lymphocyte susceptibility to apoptotic stimuli. (A) Thymocytes from wt (n = 4) and bik−/− (n = 4) littermates were harvested and cultured in the presence of dexamethasone (Dex, 10−7 M), etoposide (Etop, 1 μg/ml), PMA (2 ng/ml), ionomycin (1 μg/ml), or cross-linked FasL (100 ng/ml) or in the absence of exogenous stimuli (unstimulated) for the indicated times. (B) CD4+ or CD8+ T cells were sorted from lymph nodes of wt (n = 4) and bik−/− (n = 4) littermates and cultured in the presence of dexamethasone (10−7 M) or etoposide (1 μg/ml) or in the absence of exogenous stimuli (un-stimulated) for the indicated times. (C) Mature B cells were negatively sorted from lymph nodes of wt (n = 4) and bik−/− (n = 4) littermates and cultured in the presence of dexamethasone (10−7 M), etoposide (1 μg/ml), or anti-IgM cross-linking F(ab')2 fragments at the indicated concentrations or in the absence of exogenous stimuli (unstimulated) for the indicated times.
bik expression inferred from β-galactosidase reporter activity.
To identify the tissues in which Bik deficiency might cause abnormalities, bik expression was examined further by assessing the activity of the introduced β-galactosidase reporter in frozen tissue sections from bik+/− animals in which the neomycin resistance cassette had been deleted (Fig. 2). Consistent with results from Northern blot analysis of bik expression (Fig. 1), the liver, lungs, and kidneys were strongly positive for β-galactosidase activity, but no activity was detected in the brain (data not shown). We also found expression in the myocardium and, interestingly, the endocardium, as assessed by combined staining for β-galactosidase and the endothelial marker CD31 (PECAM-1) (Fig. 5A). Surprisingly, in the lymphoid organs, β-galactosidase activity was found only in vessels and not in the lymphoid areas, such as follicles in lymph nodes, cortex and medulla of the thymus, or periarteriolar sheath in the spleen (Fig. 5B to D and data not shown). This finding contrasts with the RT-PCR data described above (Fig. 1C and D), which revealed bik mRNA widely distributed in lymphocyte subsets. This discrepancy could simply mean that the RT-PCR assay is more sensitive, but the reporter activity might instead be silenced in these cells (see Discussion).
FIG. 5.

β-Galactosidase assay for bik expression. (A) bik expression in endocardium, confirmed by combined staining for β-galactosidase activity (blue) and immunohistochemical staining for CD31 (brown). (B) Whole-mount β-galactosidase staining of lymph nodes from bik+/− mice demonstrating bik expression in lymphoid vasculature. (C and D) The identity of β-galactosidase-positive cells in lymph nodes was determined as endothelium by morphology from thin sections of whole-mount stained nodes. (E and F) Thin sections from whole-mount β-galactosidase-stained mammary gland venule (E) and arteriole (F). Arrows indicate representative endothelial cells. (G) Combination of β-galactosidase staining and immunohistochemical staining for CD31 on frozen sections of blood vessels from the pancreas of bik+/− animals. (H to K) High-power images showing the morphology of the artery (H and I) and vein (J and K) and the distribution of β-galactosidase staining. Anti-B220 antibody staining (I and K) is included as an isotype-matched control for the anti-CD31 antibody (H and J). Arrows indicate representative endothelial cells. In no case was β-galactosidase staining observed in the above-mentioned cell types from wt animals.
While not associated with the lymphoid areas, β-galactosidase activity was prominent in tubular structures in the lymph nodes, thymus, and spleen. Immunostaining demonstrated that the positive cells also expressed CD31 (data not shown). In the lymph node, β-galactosidase activity was found in high endothelial venules as well as stellate endothelium in the marginal and medullary sinuses (Fig. 5B to D). Not all endothelial cells, however, were positive for β-galactosidase activity. In the mammary gland, activity was observed in endothelium from venules (Fig. 5E) but was undetectable in that from arterioles (Fig. 5F). Likewise, in the pancreas, endothelial cells from major veins but not those from major arteries were positive (Fig. 5G to K).
Loss of Bik does not prevent endothelial cell apoptosis in blood vessel regression.
Since Bik is expressed in endothelial cells, we sought to determine if the death of endothelial cells and vascular regression were impaired by the absence of Bik. To assess this, we studied hyaloid vessel regression in postnatal bik−/− and wt mice. Hyaloid vessels and PM, blood vessel networks that feed the developing eye, regress rapidly from postnatal (PN) day 8 and are generally undetectable by 4 weeks of age (20). Prior to vessel regression, the PM appears as a vascular mesh across the pupil, whereas the VHP resides in the vitreous body, leading from the hyaloid artery (HA) to the lens, where it diverges into the TVL capillaries that attach to the lens surface. Hyaloid vessels of bik+/− mice displayed β-galactosidase activity at PN7, indicating that bik was expressed in these vessels at the onset of their regression (Fig. 6A). Nevertheless, comparison of the eyes from PN16 bik−/− and wt mice revealed no difference in the numbers of TVL, VHP, or PM (Fig. 6B). Moreover, by 5 weeks, no hyaloid vessels remained in the eyes of bik−/− mice (data not shown). Hence, these vessels regress normally in the absence of Bik.
FIG. 6.

Hyaloid plexus blood vessel numbers in wt and bik−/− mice. (A) β-Galactosidase staining of an eye from a PN7 bik+/− pup indicates bik expression in hyaloid vessels. Shown is a lateral view of the lens with attached β-galactosidase-positive VHP. β-Galactosidase-positive PM is also evident on the anterior surface of the lens (top). No β-galactosidase activity was detected in hyaloid vessels of a control wt littermate. (B) Eyes of PN16 wt (n = 5) and bik−/− (n = 5) mice were sectioned longitudinally and counted for each hyaloid vessel type: TVL, VHP, and pupillary PM. Vessels were counted in 12 nonconsecutive sections taken at the level of the pupil. Each data point represents the average number of vessels observed per section per eye.
Absence of Bik fails to rescue the renal and hematopoietic defects in bcl-2−/− mice.
In another attempt to reveal a role for Bik in apoptosis regulation, we investigated the effect of Bik loss in mutant mice predisposed to excessive apoptosis by the concomitant loss of Bcl-2. Mice lacking Bcl-2 are runted, and all succumb to polycystic kidney disease between 3 and 6 weeks after birth on the C57BL/6 background (3, 42). They also have lower numbers of leukocytes and the lifespan of their mature B and T lymphocytes is greatly reduced (3, 27, 42). Loss of even a single allele of Bim, however, prevents the defects in bcl-2−/− mice (3). Since Bik is expressed in the hematopoietic system and kidneys (Fig. 1), we investigated whether loss of Bik could also rescue the hematopoietic and renal defects in bcl-2−/− mice.
Like bcl-2−/− mice, all bik−/− bcl-2−/− mice (n = 4) became runted and died between 2 and 4 weeks of age (Fig. 7A and B), and histologic examination confirmed that they had succumbed to polycystic kidney disease (Fig. 7C). An explanation emerged from an assessment of bik expression in the developing kidneys by a β-galactosidase assay performed at E14.5, the time at which pathological cell death occurs in bcl-2−/− kidneys (28). In contrast to adult kidneys, no β-galactosidase activity was found in the mesenchymal or tubule cells (Fig. 7D). The few, scattered β-galactosidase-positive cells in the E14.5 kidneys appeared to be endothelial cells. It was therefore unsurprising that Bik loss could not prevent the early postnatal death of bcl-2−/− mice.
FIG. 7.

Phenotype of bik−/− bcl-2−/− mice. (A) The survival rate of bik−/− bcl-2−/− mice (n = 4) and their bik−/− bcl-2+/− littermates (n = 12), monitored for a 10-week period and expressed as percent survival over time. (B) Comparison of the weight of bik-2−/− bcl-2−/− and bik−/− bcl-2+/− littermates at PN25. (C) Histological examination of kidneys from bik−/− bcl-2−/− and bcl-2−/− animals at the time of death, showing that they are clearly polycystic by comparison to an age-matched bik−/− control. bik−/− kidneys were indistinguishable from wt. (D) Expression of Bik in the embryonic kidney, assessed by measuring the β-galactosidase activity in a bik+/− E14.5 kidney frozen section.
We next explored whether Bik loss reversed any of the hematopoietic deficiencies caused by Bcl-2 loss. To preclude any effects of the renal abnormality on hematopoiesis, we compared the hematopoietic systems of lethally irradiated Ly5.1+ recipient mice reconstituted with fetal liver-derived hematopoietic stem cells (HSC) from Ly5.2+ E14.5 bik−/− bcl-2−/− or control embryos. HSC lacking Bik had normal reconstitution potential, since comparable numbers of leukocytes were found in the blood, bone marrow, thymus, spleen, or lymph nodes of mice reconstituted with wt or bik−/− HSC (Fig. 8 and data not shown). A number of cell populations were examined in these tissues. As expected, the absence of Bcl-2 produced marked deficiencies in most lymphoid populations, exceeding 90% for mature T and B cells in the periphery, and smaller but significant drops were seen with certain myeloid populations, e.g., monocytes in the spleen. In no cell population, however, did the concomitant absence of Bik increase the numbers of cells above that found in the absence of Bcl-2 alone (Fig. 8).
FIG. 8.

Composition of blk−/− bcl-2−/− reconstituted hematopoietic organs. (A) Total number of Ly5.2+ bone marrow cells from 11- to 15-week postreconstitution Ly5.1 recipients reconstituted with Ly5.2 wt (n = 3), bik−/− bcl-2+/+ (n = 5), bik+/+ bcl-2−/− (n = 3), and bik−/− bcl-2−/− (n = 5) fetal liver HSC. Numbers of Ly5.2+ pro- and pre-B cells (B220+ sIgM− sIgD−), immature B cells (B220+ sIgM+ sIgD−), recirculating mature B cells (B220+ sIgMlo sIgDhi), macrophages (Mac1+ Grl−), and granulocytes (Mac1+ Grl+) were then determined by FACS. (B) Total number of Ly5.2+ thymocytes and numbers of pro-T cells (CD4− CD8−), pre-T cells (CD4+ CD8+), and mature T cells (CD4+ CD8− and CD4− CD8+), determined by FACS. (C) Total number of Ly5.2+ splenocytes and numbers of naive B cells (sIgM+ sIgD−), mature B cells (sIgMlo sIgDhi), mature T cells (CD4+ CD8− and CD4− CD8+), macrophages (Mac1+ Grl−), and granulocytes (Mac1+ Gr1+), determined by FACS. (D) Peripheral blood from Ly5.1 recipient mice taken 9 to 12 weeks after reconstitution with Ly5.2 wt (n = 7), bik−/− bcl-2+/+ (n = 14), bik+/+ bcl-2−/− (n = 11), and bik−/− bcl-2−/− (n = 15) fetal liver HSC was examined, and the total number of Ly5.2+ lymphocytes and granulocytes (neutrophils, monocytes, eosinophils, and basophils) per milliliter was determined.
We next determined whether the sensitivity to apoptotic stimuli of mature lymphocytes lacking both Bik and Bcl-2 was lower than that of cells lacking only Bcl-2. Mature donor-derived (Ly5.2+) T and B lymphocytes purified from the lymph nodes of reconstituted animals were cultured in the absence of cytokines. Consistent with earlier reports (3, 27, 42), the viability of lymphocytes deficient in Bcl-2 was significantly reduced (Fig. 9). However, T or B lymphocytes lacking both Bik and Bcl-2 died as rapidly as those lacking Bcl-2 alone (Fig. 9A). Therefore, even in the sensitive context of lymphocytes lacking Bcl-2, the absence of Bik did not enhance survival.
FIG. 9.

Sensitivity of bik−/− bcl-2−/− lymphocytes to apoptotic stimuli. (A) bik−/− bcl-2−/− T lymphocytes were cultured in the absence of cytokines, and viability was assessed by measuring the PI uptake after 24 and 48 h. Lymphocytes were sorted from lymph nodes of Ly5.1 recipients reconstituted with Ly5.2 wt (n = 5), bik−/− bcl-2+/+ (n = 3), bik+/+ bcl-2−/− (n = 4), and bik−/− bcl-2−/− (n = 5) fetal liver HSC. (B) bik−/− bcl-2−/− B lymphocytes were cultured in the absence of cytokines, and viability assessed as described in the legend to panel A.
DISCUSSION
We found mRNA for bik in a wide range of murine hematopoietic cells, including granulocytes, macrophages, and developing as well as mature T and B lymphocytes (Fig. 1). Consistent with reports of bik in human B cells (22), bik expression increased in mouse B cells in response to BCR stimulation (Fig. 1D). Conceivably, the increased Bik expression in response to BCR stimulation could be responsible for, or at least contribute to, the death of B cells undergoing negative selection (23). Loss of Bik, however, conferred no protection on B cells against treatment with cross-linking anti-IgM antibodies (Fig. 4C). While this suggests that Bik on its own is not limiting for B-cell apoptosis following BCR stimulation, it does not exclude the possibility that Bik may function in a redundant fashion with other BH3-only proteins, such as Bim, which does play an essential role in BCR ligation-induced B-cell apoptosis (14).
Increasing the levels of BH3-only proteins such as Bik in response to activation may predispose B cells to apoptosis that occurs when growth factors become limiting, a process that is thought to be critical for the resolution of immune responses (26). This process can be mimicked in vitro by depriving activated B lymphocytes of their requisite cytokines. Although loss of Bik did not protect B-cell blasts against this death (data not shown), again it is possible that Bik may be operating in a redundant manner with other BH3-only proteins, such as Bim, which is required for B-cell apoptosis following cytokine withdrawal (4). The observation that overexpression of Bcl-2 affords B cells greater protection against cytokine withdrawal than does loss of Bim (4) suggests that other BH3-only proteins, such as Bik, do act in concert with Bim.
While Bim deficiency renders thymocytes highly resistant to ionomycin and spontaneous death in culture, it has only a minor effect on their response to glucocorticoids, DNA damage, or phorbol esters (4), death stimuli which can be countered by Bcl-2 overexpression (37, 38). Hence, Bim either is not required for apoptosis in response to these stimuli or acts in concert with other BH3-only proteins. However, Bik loss did not reduce the sensitivity of thymocytes to these or any other apoptotic stimuli tested (Fig. 4). Our results therefore show that Bik is not a major contributor to apoptosis in lymphoid cells in response to these stimuli but, again, that it might act in concert with other BH3-only proteins. Indeed, very recent work suggests that the BH3-only protein Puma plays an important role in these responses (21, 44).
Assessment of β-galactosidase reporter activity indicated that bik is expressed in the liver, kidneys, lungs, and heart but not the brain, consistent with the expression pattern of bik obtained by Northern blotting (Fig. 1) (18). This indicates that the β-galactosidase knock-in is a faithful reporter of Bik expression in many tissues. It is uncertain why β-galactosidase activity was not found in lymphocytes and other hematopoietic cells despite clear expression of bik mRNA in these cells. An equivalent discrepancy has been noted with a lacZ knock-in at the bim locus, which also presents very low or undetectable β-galactosidase activity in lymphoid areas of the lymph node, thymus, and spleen, even though RNA and protein analyses clearly indicate Bim expression in these cells (P. Bouillet, unpublished observation) (32), in which loss of Bim profoundly impairs apoptosis (4, 5). Silencing of lacZ transgenes in hematopoietic cells has been documented previously (12, 31), and hence silencing of the reporter gene might contribute to the lack of β-galactosidase activity in the lymphocytes in which it is located in the bik or bim locus. Another possibility is that a regulatory element needed for lymphoid expression of the locus resides within the region deleted in the replacement, such as in the deleted intron.
Bik is expressed in a restricted set of endothelial cells but is dispensable for vascular regression.
We found Bik to be expressed in endothelial cells in lymph node sinuses, high endothelial venules, heart (endocardium), and blood vessels (Fig. 5). Interestingly, expression in blood vessels was found in endothelial cells of veins but not arteries (Fig. 5). Discrete lineages of endothelial cells populate arteries and veins, and the lineages can be distinguished during embryogenesis from the earliest stages of vascular development, with arterial endothelial cells expressing EfnB2 and venous endothelial cells expressing EphB4 (45). Thus, inherent differences between venous and arterial endothelium may explain why Bik is confined to the former.
Since Bcl-2 overexpression inhibits endothelial cell apoptosis (13, 15, 16), Bik may contribute to apoptosis of endothelial cells during vessel regression or angiogenesis during development. To assess the impact of Bik loss on endothelial-cell apoptosis, we compared the regression of hyaloid vessels in the eyes of bik−/− and wt mice. These vessels service the developing lens and retina during embryogenesis but become redundant following birth and soon regress by apoptosis (20, 24). The PM and VHP become vestigial by 12 to 16 days of age. TVL and HA regression is slower, but both are largely undetectable by 1 month. Bik expression was detected in the TVL and PM, as assessed by β-galactosidase activity (Fig. 6A), but the PM, TVL, and VHP vessels did not persist in the eyes of adult bik−/− mice and seemed to be cleared at the normal rate (Fig. 6). Hence, if Bik regulates apoptosis in venous endothelial cells during either angiogenesis or vascular regression, it must do so in collaboration with other BH3-only proteins. By utilizing different BH3-only proteins for apoptosis induction, distinct populations of blood vessel endothelium might be eliminated in response to distinct developmental signals, thus fine-tuning the process of angiogenesis.
Loss of Bik does not rescue the defects caused by Bcl-2 deficiency.
The cellular life/death decision is considered determined by the balance of prosurvival and proapoptotic Bcl-2 family molecules (9). In several cell types, loss of Bcl-2 favors death but the balance can be restored by concomitant loss of its antagonist Bim (3). Would the loss of any BH3-only protein restore the balance and allow normal cell survival in Bcl-2-deficient mice? Since Bik, like Bim, is expressed in hematopoietic organs and the kidneys, tissues in which Bcl-2 deficiency has disastrous consequences (Fig. 1), loss of Bik might be expected to compensate for loss of Bcl-2. However, our analysis of mice lacking both Bcl-2 and Bik revealed that loss of Bik failed to rescue the hematopoietic and renal defects seen in mice lacking Bcl-2 (Fig. 7 to 9). The failure to rescue the renal defects can probably be ascribed to the absence of detectable bik expression in the relevant cell types in the developing kidneys at the time (around E14.5) when apoptosis occurs in the absence of Bcl-2 (28). The failure to rescue the hematopoietic defect was more surprising, but there are several potential explanations. First, since the activity of BH3-only proteins is regulated in multiple ways (10, 35), the Bik expressed in the hematopoietic system might be sequestered in an inactive state. If so, its absence might not restore the balance. Second, the presence of other BH3-only proteins (particularly Bim and Puma) may compensate, so that Bik loss alone is not sufficient to restore the balance. Third, Bik may play no role in apoptosis regulation at all but, instead, may perform another unidentified function in these cells. This possibility seems unlikely, however, given that at least in co-overexpression studies, Bik interacts with prosurvival family members and antagonizes their activity (6, 17, 18).
The explanation that we favor is that despite interacting with many prosurvival Bcl-2 family members when overexpressed, endogenous Bik may preferentially interact with only a few of these proteins under physiological conditions. Therefore, our observations in the bik−/− bcl-2−/− mice could be explained if Bik and Bcl-2 do not oppose each other under physiological conditions but are part of separate, parallel pathways. In lymphocytes, for example, where Bim (and certain other BH3-only proteins) antagonize Bcl-2, Bik may instead antagonize other prosurvival Bcl-2 homologues (e.g., Bcl-w) that play a less dominant survival role in those cells (33, 34). If so, although the absence of Bik does not affect the imbalance created by Bcl-2 loss in lymphocytes, it might still alter apoptosis in certain cells where Bcl-w is the vital guardian (34) or in cells also lacking another BH3-only protein. The assumption here that certain BH3-only “ligands” possess a degree of specificity for their prosurvival “receptors” would render control of apoptosis by the Bcl-2 family even more sophisticated. Proof of this hypothesis awaits careful affinity measurements of the interactions of BH3-only proteins with their prosurvival partners and further genetic studies. Indeed, Bik appears to favor Bcl-xL and Bcl-w binding over Bcl-2 in vitro (Chen et al., unpublished data).
Acknowledgments
We thank Kim Newton (Genentech, Inc., South San Francisco, Calif.) for providing cDNA templates for expression analysis; M. Cancilla (Exelixis, Inc., South San Francisco, Calif.) and L. Tai for assistance with BAC manipulation and blk locus mapping; A. Steptoe for ES cell manipulation; F. Battye, C. Tarlinton, V. Lapatis, and C. Clark for cell sorting; A. Naughton, C. Tilbrook, J. Morrow, N. Clark, and K. Birchall for animal husbandry and care; S. Mihajlovic and E. Tsui for histology; and S. Cory, D. Huang, A. Harris, D. Vaux, H. Puthalakath, and L. O'Reilly for insightful discussions.
This work was supported by fellowships and grants from the NHMRC (Canberra), the Dr Josef Steiner Cancer Research Foundation (Bern), the Leukemia and Lymphoma Society, and the NIH (CA80188). L.C. is a recipient of The Cancer Council Victoria Postdoctoral Cancer Research Fellowship.
REFERENCES
- 1.Adams, J. M. 2003. Ways of dying: multiple pathways to apoptosis. Genes Dev. 17:2481-2495. [DOI] [PubMed] [Google Scholar]
- 2.Amanna, I. J., K. Clise-Dwyer, F. E. Nashold, K. A. Hoag, and C. E. Hayes. 2001. Cutting edge: A/WySnJ transitional B cells overexpress the chromosome 15 proapoptotic Blk gene and succumb to premature apoptosis. J. Immunol. 167:6069-6072. [DOI] [PubMed] [Google Scholar]
- 3.Bouillet, P., S. Cory, L.-C. Zhang, A. Strasser, and J. M. Adams. 2001. Degenerative disorders caused by Bcl-2 deficiency are prevented by loss of its BH3-only antagonist Bim. Dev. Cell 1:645-653. [DOI] [PubMed] [Google Scholar]
- 4.Bouillet, P., D. Metcalf, D. C. S. Huang, D. M. Tarlinton, T. W. H. Kay, F. Köntgen, J. M. Adams, and A. Strasser. 1999. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 286:1735-1738. [DOI] [PubMed] [Google Scholar]
- 5.Bouillet, P., J. F. Purton, D. I. Godfrey, L.-C. Zhang, L. Coultas, H. Puthalakath, M. Pellegrini, S. Cory, J. M. Adams, and A. Strasser. 2002. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature 415:922-926. [DOI] [PubMed] [Google Scholar]
- 6.Boyd, J. M., G. J. Gallo, B. Elangovan, A. B. Houghton, S. Malstrom, B. J. Avery, R. G. Ebb, T. Subramanian, T. Chittenden, R. J. Lutz, and G. Chinnadurai. 1995. Bik, a novel death-inducing protein shares a distinct sequence motif with Bcl-2 family proteins and interacts with viral and cellular survival-promoting proteins. Oncogene 11:1921-1928. [PubMed] [Google Scholar]
- 7.Cheng, E. H., M. C. Wei, S. Weiler, R. A. Flavell, T. W. Mak, T. Lindsten, and S. J. Korsmeyer. 2001. BCL-2, BCL-xL sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 8:705-711. [DOI] [PubMed] [Google Scholar]
- 8.Church, G. M., and W. Gilbert. 1984. Genomic sequencing. Proc. Natl. Acad. Sci. USA 81:1991-1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cory, S., and J. M. Adams. 2002. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2:647-656. [DOI] [PubMed] [Google Scholar]
- 10.Cory, S., D. C. S. Huang, and J. M. Adams. The Bcl-2 family: role in cell survival and oncogenesis. Oncogene, in press. [DOI] [PubMed]
- 11.Coultas, L., D. C. S. Huang, J. M. Adams, and A. Strasser. 2002. Pro-apoptotic BH3-only Bcl-2 family members in vertebrate model organisms suitable for genetic experimentation. Cell Death Differ. 9:1163-1166. [DOI] [PubMed] [Google Scholar]
- 12.Cui, C., M. A. Wani, D. Wight, J. Kopchick, and P. J. Stambrook. 1994. Reporter genes in transgenic mice. Transgenic Res. 3:182-194. [DOI] [PubMed] [Google Scholar]
- 13.Dimmeler, S., and A. M. Zeiher. 2000. Endothelial cell apoptosis in angiogenesis and vessel regression. Circ. Res. 87:434-439. [DOI] [PubMed] [Google Scholar]
- 14.Enders, A., P. Bouillet, H. Puthalakath, Y. Xu, D. M. Tarlinton, and A. Strasser. 2003. Loss of the pro-apoptotic BH3-only Bcl-2 family member Bim inhibits BCR stimulation-induced apoptosis and deletion of autoreactive B cells. J. Exp. Med. 198:1119-1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fisher, S. A., B. L. Langille, and D. Srivastava. 2000. Apoptosis during cardiovascular development. Circ. Res. 87:856-864. [DOI] [PubMed] [Google Scholar]
- 16.Gerber, H. P., V. Dixit, and N. Ferrara. 1998. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J. Biol. Chem. 273:13313-13316. [DOI] [PubMed] [Google Scholar]
- 17.Han, J., P. Sabbatini, and E. White. 1996. Induction of apoptosis by human Nbk/Bik, a BH3-containing protein that interacts with E1B 19K. Mol. Cell. Biol. 16:5857-5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hegde, R., S. M. Srinivasula, M. Ahmad, T. Fernandes-Alnemri, and E. S. Alnemri. 1998. Blk, a BH3-containing mouse protein that interacts with Bcl-2 and Bcl-xL, is a potent death agonist. J. Biol. Chem. 273:7783-7786. [DOI] [PubMed] [Google Scholar]
- 19.Huang, D. C. S., and A. Strasser. 2000. BH3-only proteins—essential initiators of apoptotic cell death. Cell 103:839-842. [DOI] [PubMed] [Google Scholar]
- 20.Ito, M., and M. Yoshioka. 1999. Regression of the hyaloid vessels and pupillary membrane of the mouse. Anat. Embryol. (Berlin) 200:403-411. [DOI] [PubMed] [Google Scholar]
- 21.Jeffers, J. R., E. Parganas, Y. Lee, C. Yang, J. Wang, J. Brennan, K. H. MacLean, J. Han, T. Chittenden, J. N. Ihle, P. J. McKinnon, J. L. Cleveland, and G. P. Zambetti. 2003. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4:321-328. [DOI] [PubMed] [Google Scholar]
- 22.Jiang, A., and E. A. Clark. 2001. Involvement of Bik, a proapoptotic member of the Bcl-2 family, in surface IgM-mediated B cell apoptosis. J. Immunol. 166:6025-6033. [DOI] [PubMed] [Google Scholar]
- 23.Lam, K. P., and K. Rajewsky. 1998. Rapid elimination of mature autoreactive B cells demonstrated by Cre-induced change in B cell antigen receptor specificity in vivo. Proc. Natl. Acad. Sci. USA 95:13171-13175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lang, R. A., and J. M. Bishop. 1993. Macrophages are required for cell death and tissue remodeling in the developing mouse eye. Cell 74:453-462. [DOI] [PubMed] [Google Scholar]
- 25.Lindsten, T., A. J. Ross, A. King, W. Zong, J. C. Rathmell, H. A. Shiels, E. Ulrich, K. G. Waymire, P. Mahar, K. Frauwirth, Y. Chen, M. Wei, V. M. Eng, D. M. Adelman, M. C. Simon, A. Ma, J. A. Golden, G. Evan, S. J. Korsmeyer, G. R. MacGregor, and C. B. Thompson. 2000. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol. Cell 6:1389-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marsden, V., and A. Strasser. 2003. Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu. Rev. Immunol. 21:71-105. [DOI] [PubMed] [Google Scholar]
- 27.Matsuzaki, Y., K.-I. Nakayama, K. Nakayama, T. Tomita, M. Isoda, D. Y. Loh, and H. Nakauchi. 1997. Role of bcl-2 in the development of lymphoid cells from the hematopoietic stem cell. Blood 89:853-862. [PubMed] [Google Scholar]
- 28.Nagata, M., H. Nakauchi, K.-I. Nakayama, K. Nakayama, D. Loh, and T. Watanabe. 1996. Apoptosis during an early stage of nephrogenesis induces renal hypoplasia in bcl-2-deficient mice. Am. J. Pathol. 148:1601-1611. [PMC free article] [PubMed] [Google Scholar]
- 29.Nakayama, K., K.-I. Nakayama, I. Negishi, K. Kuida, H. Sawa, and D. Y. Loh. 1994. Targeted disruption of bcl-2αβ in mice: occurrence of gray hair, polycystic kidney disease, and lymphocytopenia. Proc. Natl. Acad. Sci. USA 91:3700-3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newton, K., A. W. Harris, and A. Strasser. 2000. FADD/MORT1 regulates the pre-TCR checkpoint and can function as a tumour suppressor. EMBO J. 19:931-941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogilvy, S., D. Metcalf, L. Gibson, M. L. Bath, A. W. Harris, and J. M. Adams. 1999. Promoter elements of vav drive transgene expression in vivo throughout the hematopoietic compartment. Blood 94:1855-1863. [PubMed] [Google Scholar]
- 32.O'Reilly, L. A., L. Cullen, J. Visvader, G. Lindeman, C. Print, M. L. Bath, D. C. S. Huang, and A. Strasser. 2000. The pro-apoptotic BH3-only protein Bim is expressed in hemopoietic, epithelial, neuronal and germ cells. Am. J. Pathol. 157:449-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Reilly, L. A., C. Print, G. Hausmann, K. Moriishi, S. Cory, D. C. S. Huang, and A. Strasser. 2001. Tissue expression and subcellular localization of the pro-survival molecule Bcl-w. Cell Death Differ. 8:486-494. [DOI] [PubMed] [Google Scholar]
- 34.Print, C. G., K. L. Loveland, L. Gibson, T. Meehan, A. Stylianou, N. Wreford, D. de Kretser, D. Metcalf, F. Köntgen, J. M. Adams, and S. Cory. 1998. Apoptosis regulator Bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc. Natl. Acad. Sci. USA 95:12424-12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puthalakath, H., and A. Strasser. 2002. Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 9:505-512. [DOI] [PubMed] [Google Scholar]
- 36.Schwenk, F., U. Baron, and K. Rajewsky. 1995. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 23:5080-5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sentman, C. L., J. R. Shutter, D. Hockenbery, O. Kanagawa, and S. J. Korsmeyer. 1991. bcl-2 inhibits multiple forms of apoptosis but not negative selection in thymocytes. Cell 67:879-888. [DOI] [PubMed] [Google Scholar]
- 38.Strasser, A., A. W. Harris, and S. Cory. 1991. Bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell 67:889-899. [DOI] [PubMed] [Google Scholar]
- 39.Strasser, A., L. O'Connor, and V. M. Dixit. 2000. Apoptosis signaling. Annu. Rev. Biochem. 69:217-245. [DOI] [PubMed] [Google Scholar]
- 40.Strasser, A., S. Whittingham, D. L. Vaux, M. L. Bath, J. M. Adams, S. Cory, and A. W. Harris. 1991. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc. Natl. Acad. Sci. USA 88:8661-8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki, M., R. J. Youle, and N. Tjandra. 2000. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 103:645-654. [DOI] [PubMed] [Google Scholar]
- 42.Veis, D. J., C. M. Sorenson, J. R. Shutter, and S. J. Korsmeyer. 1993. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 75:229-240. [DOI] [PubMed] [Google Scholar]
- 43.Verma, S., M. L. Budarf, B. S. Emanuel, and G. Chinnadurai. 2000. Structural analysis of the human pro-apoptotic gene Bik: chromosomal localization, genomic organization and localization of promoter sequences. Gene 254:157-162. [DOI] [PubMed] [Google Scholar]
- 44.Villunger, A., E. M. Michalak, L. Coultas, F. Müllauer, G. Böck, M. J. Ausserlechner, J. M. Adams, and A. Strasser. 2003. p53- and drug-induced apoptotic responses mediated by BH3-only proteins Puma and Noxa. Science 302:1036-1038. [DOI] [PubMed]
- 45.Wang, H. U., Z. F. Chen, and D. J. Anderson. 1998. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 93:741-753. [DOI] [PubMed] [Google Scholar]
- 46.Zong, W. X., T. Lindsten, A. J. Ross, G. R. MacGregor, and C. B. Thompson. 2001. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 15:1481-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]