

Abstract
Reliable evidence supports the role of sleep in learning and memory processes. In rodents, sleep deprivation (SD) negatively affects consolidation of hippocampus-dependent memories. As memory is integral to post-traumatic stress symptoms, the effects of post-exposure SD on various aspect of the response to stress in a controlled, prospective animal model of post-traumatic stress disorder (PTSD) were evaluated. Rats were deprived of sleep for 6 h throughout the first resting phase after predator scent stress exposure. Behaviors in the elevated plus-maze and acoustic startle response tests were assessed 7 days later, and served for classification into behavioral response groups. Freezing response to a trauma reminder was assessed on day 8. Urine samples were collected daily for corticosterone levels, and heart rate (HR) was also measured. Finally, the impact of manipulating the hypothalamus–pituitary–adrenal axis and adrenergic activity before SD was assessed. Mifepristone (MIFE) and epinephrine (EPI) were administered systemically 10-min post-stress exposure and behavioral responses and response to trauma reminder were measured on days 7–8. Hippocampal expression of glucocorticoid receptors (GRs) and morphological assessment of arborization and dendritic spines were subsequently evaluated. Post-exposure SD effectively ameliorated long-term, stress-induced, PTSD-like behavioral disruptions, reduced trauma reminder freezing responses, and decreased hippocampal expression of GR compared with exposed-untreated controls. Although urine corticosterone levels were significantly elevated 1 h after SD and the HR was attenuated, antagonizing GRs with MIFE or stimulation of adrenergic activity with EPI effectively abolished the effect of SD. MIFE- and EPI-treated animals clearly demonstrated significantly lower total dendritic length, fewer branches and lower spine density along dentate gyrus dendrites with increased levels of GR expression 8 days after exposure, as compared with exposed-SD animals. Intentional prevention of sleep in the early aftermath of stress exposure may well be beneficial in attenuating traumatic stress-related sequelae. Post-exposure SD may disrupt the consolidation of aversive or fearful memories by facilitating correctly timed interactions between glucocorticoid and adrenergic systems.
Keywords: post-traumatic stress disorder (PTSD), animal model, sleep deprivation, corticosterone, epinephrine, mifepristone
INTRODUCTION
Intrusive memories that are chronically active, disruptive, and exceptionally vivid, and which can be factual, emotional, or somatosensory, are fundamental characteristics of post-traumatic stress disorder (PTSD) (American Psychiatric Association, 2004). The memories of a traumatic event, together with the emotions at the time of the event, shape symptoms such as intrusive thoughts, physiological hyperarousal, and avoidance of traumatic reminders.
It is well established that following initial encoding, memory remains temporarily vulnerable to disruption (McGaugh, 2000) until it is consolidated. Emerging evidence suggests that pharmacological secondary prevention (ie, intervening after a traumatic event to forestall development of PTSD) can alter brain mechanisms regulating the formation, storage, and retrieval of different type of traumatic memories. A diverse group of compounds including corticosteroids (Aerni et al, 2004; de Quervain et al, 1998, 2000; Schelling, 2002; Schelling et al, 1999, 2001, 2003, 2004), beta-adrenergic antagonists (propranolol) (Pitman et al, 2002; Vaiva et al, 2003), and opiate analgesics (morphine) (Bryant et al, 2009) have been shown to reduce hormonally enhanced memories and fear conditioning and have potentially therapeutic effects on the clinical course of subsequent PTSD symptoms.
Ample evidence indicates that sleep participates in the consolidation of recent memory traces (Born et al, 2006; Maquet, 2001; Stickgold, 2005; Walker and Stickgold, 2006). Sleep following learning, independent of time of day, is known to enhance the consolidation of newly acquired memory traces (Gais and Born, 2004; Maquet, 2000, 2001; Peigneux et al, 2001; Wagner et al, 2006) through an active reorganization of representations, whereas acute sleep deprivation (SD) may disrupt this process and impair retrieval functions (Hagewoud et al, 2010). Wagner et al (2006) reported that brief periods of sleep immediately following learning cause preservation of emotional memories over 4 years. We therefore hypothesized that interfering with memory consolidation processes by SD immediately after traumatic experience will reduce post-traumatic stress symptoms and incidence.
In this study, the effects of post-exposure SD on behavioral responses to predator scent stress (PSS) were evaluated in a controlled, prospective animal model of PTSD. In this model, populations of exposed rodents are classified according to the degree of their individual behavioral response using standardized ‘cut-off behavioral criteria' (CBC), creating three distinct groups: ‘extreme behavioral response' (EBR) and ‘minimal behavioral response' (MBR) at the extremes, and a middle group of ‘partial behavioral responders' (PBR) (Cohen and Zohar, 2004; Cohen et al, 2003, 2004, 2005, 2012). The relative prevalence rates of individuals displaying the different degrees of disrupted behavior provide an indication of the potential efficacy of the ‘treatment' under study.
The aim was to perform a controlled, prospective trial to examine the effect of 6 h of SD after PSS and to investigate possible mechanisms for these effects. For this purpose physiological, molecular and morphological parameters were assessed in animals classified according to their behavioral responses in the elevated plus-maze (EPM) and acoustic startle response (ASR) paradigms 7 days post-exposure, and trauma-cue-triggered freezing responses were assessed on day 8. Prevalence rates for EBR, MBR, and PBR individuals among animals treated with SD after exposure were calculated from these data and compared with untreated controls and unexposed controls. The effects on memory were assessed by the response to the neutral reminder of the trauma (the trauma-cue) on day 8. Urine samples were collected daily for corticosterone levels; radio-telemetrically collected heart rate (HR) was also recorded. In light of an association between corticosterone levels and behavioral response patterns, the effect of preventing the elevation of corticosterone or stimulating adrenergic activation on behavior was assessed. Mifepristone (MIFE), a glucocorticoid receptor (GR) antagonist, and epinephrine (EPI) were administered systemically 10-min post-PSS exposure (50 min before SD) and behavioral responses were measured on day 7 and trauma reminder on day 8. The expression of GR was evaluated in the hippocampus and dendritic arborization and spine density in Golgi-impregnated neurons in dentate gyrus (DG) granule cells were assessed.
MATERIALS AND METHODS
Animals
Adult male Sprague–Dawley rats weighing 180–230 g were habituated to housing conditions for at least 7 days, housed four per cage in a vivarium with stable temperature and a reversed 12-h light–dark cycle (lights off: 1900 hours), with unlimited access to food and water. All testing was performed during the dark phase in dim red light conditions.
Experimental Design
Three experiments were conducted. In the first (N=66), the behavioral effects of SD for 6 h, started 1 h after PSS exposure, were evaluated with the EPM and the ASR tests on day 7. One day later, animals were exposed to a trauma-cue (unsoiled cat litter) for 10 min and freezing response was assessed. Urine samples were collected daily for corticosterone levels. The rats were killed within 24 h (8 days post-exposure) and their brains collected for measurement of GR in the hippocampus and for dendritic profile of Golgi-stained granule cells in the DG. We hypothesized that interfering with memory consolidation processes by SD immediately after traumatic experience will reduce post-traumatic stress symptoms and incidence. We expected that SD immediately after stress exposure would be manifested in a distinctive activation pattern of the hypothalamus–pituitary–adrenal (HPA)-axis and the autonomic nervous system. In parallel, we hypothesized that SD immediately after exposure would result in increased synaptic plasticity, synaptic strength, and dendritic complexity, with a concomitant attenuation of behavioral stress responses (less prevalence of PTSD-like response). In the second experiment (N=22), radio-telemetrically collected HR from stress-exposed animals ‘treated' with SD was recorded. The last experiment (N=30) assessed the effects of systemic MIFE or EPI administrated 10 min after PSS (50 min before SD) on behavioral, molecular, and morphological responses to the intervention. We hypothesized that antagonizing glucocorticoid and/or stimulating adrenergic activity would effectively abolish the effect of SD.
Predator Scent Stress
The test animals were placed on well-soiled cat litter for 10 min (in use by the cat for 2 days). Control sham-exposed animals were exposed to unused litter for the same amount of time (Cohen and Zohar, 2004; Cohen et al, 2003, 2004, 2005).
SD was performed for 6 h using gentle handling (Moldovan et al, 2010; Tobler and Jaggi, 1987). Although prolonged SD has been found to induce a neuroendocrine stress reaction, this method, that is, gentle handling, demonstrated lower stressful, and anxiety responses as compared with other methods (Kopp et al, 2006; Longordo et al, 2009). This mild form of SD was based on the spontaneous exploratory behavior of rats, whereas constraining and directly manipulating the animals was avoided. To maximize sleep pressure, SD was performed in the first half of the light period, during the inactive phase of the animals, (between 1000 and 1600 hours), when the rats showed their main resting period and lowest level of motor activity in a 24-h cycle. The procedure of gentle handling consisted of continuous monitoring and keeping the rats awake with minimal disturbance, using novel objects, tapping on, and moving their cages and, if necessary, using tactile stimulation by brush (without direct handling of the animal) when the rats showed behavioral signs of sleepiness (immobility without any gross body, head, or whisker movements) (Moldovan et al, 2010). Notably, mice exposed to this SD procedure showed unaltered levels of the stress hormone corticosterone compared with control undisturbed animals (Kopp et al, 2006; Longordo et al, 2009).
Behavioral responses were assessed in the EPM and the ASR paradigms as described previously (Cohen et al, 2003, 2006a, 2006b).
In the EPM, behaviors assessed were: time spent in open and closed arms and on the central platform; number of open and closed arm entries; and total exploration. ‘Anxiety index', an index that integrates the EPM behavioral measures, was calculated as follows:
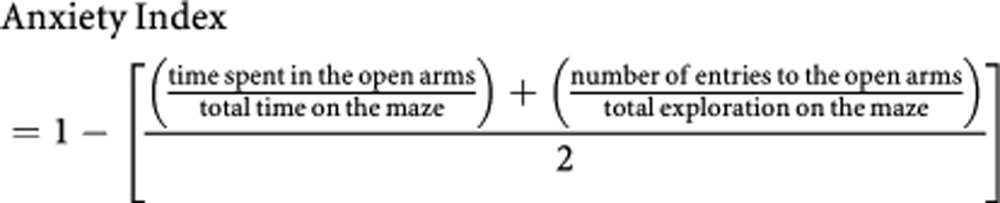 |
Anxiety index values range from 0 to 1, where an increase in the index expresses increased anxiety-like behavior.
Behavioral assessment in the ASR consisted of: mean startle amplitude (averaged over all 30 trials) and percentage of startle habituation to repeated presentation of the acoustic pulse. Percent habituation—the percent change between the response to the first block of sound stimuli and the last—was calculated as follows:
 |
Contextual Freezing Measurement
The situational reminder consisted of placing the animals on fresh, unused cat litter for 10 min (ie, identical to sham exposure), which acts to mimic the context of the initial exposure experience. Behavior was recorded using an overhead video camera and scored for immobility (freezing) using the recorded images (Cohen et al, 2011c, 2012). Freezing behavior during trauma-cue exposure was defined as an absence of all movement (except for respiration) (Ma et al, 2010). Total cumulative freezing time (total seconds spent freezing during each assessment period) was measured and calculated as a percentage of total time.
The CBC Model
The model was originally motivated by the fact that the clinical diagnosis of PTSD is made only if an individual exhibits a certain number of symptoms of sufficient severity from each of three quite well-defined symptom-clusters over a specific period of time. The criteria are based on the EMP and ASR paradigms taken together and clearly define the two opposing extremes of individual responses. The one extreme describes animals whose exploration of the open areas of the EPM is nil throughout the test and whose startle response is maximal and does not undergo any habituation throughout the ASR test. This EBR is considered to parallel PTSD-like responses—extreme and unabating maximal stress. At the other extreme are animals whose behavior is virtually unaffected by the stressor. They are termed MBR and likened to individuals who clearly have no PTSD-like response to the stressor. The remainder are poorly understood and termed PBR (by default) (Cohen and Zohar, 2004; Cohen et al, 2003, 2004, 2005). The procedure is detailed in Figure 1.
Figure 1.

The cut-off behavioral criteria (CBC) algorithm: in order to approximate the approach to understanding animal behavioral models more closely to contemporary clinical conceptions of PTSD, we use an approach that enables the classification of study animals into groups according to degree of response to the stressor, that is, the degree to which individual behavior is altered or disrupted. In order to achieve this, behavioral criteria were defined and then complemented by the definition of cut-off criteria reflecting severity of response; this parallels inclusion and exclusion criteria applied in clinical research. The procedure requires the following steps: (a) verification of global effect: the data must demonstrate that the stressor had a significant effect on the overall behavior of exposed vs unexposed populations at the time of assessment. (b) Application of the CBC's to the data: in order to maximize the resolution and minimize false positives, extreme responses to both elevated plus-maze (EPM) and acoustic startle response (ASR) paradigms, performed in sequence, were required for ‘inclusion' into the EBR group, whereas a negligible degree of response to both was required for inclusion in the MBR group.
Immunofluorescence
All animals were euthanized 24 h after the last behavioral tests (between 1400 and 1430 hours). Animals were deeply anesthetized (ketamine and xylazine mixture) and perfused transcardially with cold 0.9% physiological saline followed by 4% paraformaldehyde (Sigma-Aldrich, Israel) in 0.1 M phosphate buffer (pH 7.4). Brains were quickly removed, postfixed in the same fixative for 12 h at 4 °C, and were cryoprotected overnight in 30% sucrose in 0.1 M phosphate buffer at 4 °C. Brains were frozen on dry ice and stored at −80 °C. Serial coronal sections (10 μm) at the level of the dorsal hippocampus were collected from each animal, using a cryostat (Leica CM 1850) and mounted on coated slides. Immunofluorescence was assessed as previously described (Cohen et al, 2011b).
Sliced sections were air dried and incubated for 2 min in frozen methanol and 4 min in 4% PFA. After washing the section with PBS/0.01% tween 20 (PBS/T), the sections were incubated for 60 min in a blocking solution (normal goat or horse serum in PBS) and then overnight at 4 °C with the primary antibodies against GR (1 : 250 each; Santa Cruz Biotechnology). The sections were washed three times in PBS/T, and incubated with either DyLight-488 labeled goat-anti-rabbit IgG or Dylight-594 goat anti-mouse IgG (1 : 250; KPL, MD, UDA) in PBS containing 2% normal goat or horse serum for 2 h. Sections were washed with PBS/T, and mounted with mounting medium (Vectrastain Vector laboratories). Control sections were incubated without any primary antibodies to check for any nonspecific binding of the secondary antibodies.
Quantification
A computer-assisted image analysis system (Leica Application Suite V3.6, Leica, Germany) was used for quantitative analysis of the immunostaining and a 50 × objective lens was employed to assess the number of GR-positive cells in the hippocampus, divided into three (counted separately) areas: CA1 subfield, CA3 subfield, and DG. The regions of interest were outlined and computer-aided estimation was used (ImageJ analysis NIH) to calculate the number of GR cells in the pyramidal layer of CA1 and CA3, and in the granular layer of DG. Six representative sections of the hippocampus were chosen (between Bregma −2.30 and Bregma −3.60) from each animal, from each group (Paxinos and Watson, 2005). The sections were analyzed by two observers blinded to the treatment protocol. Standard technique was used to estimate the number of GR cell profiles per unit area for each investigated hippocampal structure.
Golgi–Cox Staining
All animals were euthanized 24 h after the last behavioral tests (between 1400 and 1430 hours). Animals were deeply anesthetized and perfused intracardially with 0.9% saline. The brains were immediately dissected and processed. Tissue was prepared by using the rapid Golgi kit (FD Neuro-technologies) according to the manufacturer's instructions. Golgi–Cox staining was assessed as previously described (Zohar et al, 2011).
The hippocampal DG was chosen as a target in this study for four reasons: (1) the DG is a unique structure in that it is one of the few telencephalic brain areas that reliably produces new neurons well into adulthood (Redila and Christie, 2006). (2) The DG is also highly sensitive to stress (Kavushansky et al, 2006). (3) DG granule cells have a critical role in the function of the entorhinal-hippocampal circuitry in health and disease. (4) DG granule cells are situated to regulate the flow of information into the hippocampus.
In order to obtain accurate measurements of dendritic parameters, strict criteria were adopted for the selection of the filled neurons before quantitative analysis: (1) only well-impregnated neurons were chosen for the histological analysis. (2) Granule cells were included in this analysis only if the cell body and primary dendrites were clearly stained and easily distinguishable from those of neighboring cell bodies and their dendrites. (3) Granule cells were sampled from the suprapyramidal blades of the DG, in both the right and left sides of the brain. (4) Granule cells from the inner granule zone (IGZ) were included in this analysis (because the dendritic morphology of hippocampal DG cells varies with their position in the granule cell layer (GCL) (Green and Juraska, 1985)). A cell was classified as belonging to the IGZ if the entire soma was positioned in the inner half of the GCL. Granule cells whose soma was intersected by the midline of the GCL, in the outer granule zone, or in the subgranular zone were not included in any analysis.
We performed an analysis to characterize the extent that dendrites branched out from both somal and dendritic sites. Primary dendrites were defined as direct extensions from the soma of at least 10 μm in length. Only DGs with at least one primary dendrite >10 μm in total length were analyzed. When a primary dendrite bifurcated at a branch point, the dendrites extending from that branch point were classified as secondary dendrites. We extended this analysis to include tertiary (3), quaternary (4), quinary (5), and senary (6) order dendrites. This procedure provides an additional measure of the pattern of dendritic arborization, allowing a more comprehensive analysis of differences in the branch patterns of the dendrites themselves. We also performed a Sholl-analysis (Sholl, 1956). A series of concentric rings, spaced 25 μm apart, was placed over the neuron and centered on the cell body, and the number of dendrite crossings as a function of distance was recorded.
Analysis of Neuronal Morphology
All slides were coded and the analysis was performed with the experimenter blinded as to the origin of the slides. Dendritic morphology was observed by epifluorescent microscopy (Leica). A 0.5 μm interval z-series was captured throughout the extent of the dendritic arbor of the DG with a CCD camera (Leica) controlled by LAS software.
Telemetric Transmitter Implantation
Animals in this study were monitored telemetrically from baseline throughout 9 h following stress exposure, allowing sampling of HR without the presence of a human. Wireless radiofrequency transmitters (Data Sciences International (DSI), St Paul, MN; model TA10ETA-F20) were implanted for continuous electrocardiographic (ECG) recordings, under aseptic conditions, during the light period, using procedures described previously (Cohen et al, 2011a).
Rats were anesthetized with ketamine (60–80 mg/kg, intraperitoneally) and xylazine (5–10 mg/kg, intraperitoneally) and transmitters were implanted intraperitoneally. Briefly, the body of the transmitter was placed into the abdominal cavity, and the two electrodes (wire loops) were fixed to the dorsal surface of the xiphoid process and in the anterior mediastinum close to the right atrium. The leads were directed rostrally (subcutaneously) and anchored in place with permanent sutures (DII positioning). Rats were prophylactically injected with penicillin (Natrium-penicillin G, 40 000 IU/kg body wt sc; Hanford's United States Veterinary Products) 10–15 min before incision, and were given codeine (1 mg/100 ml) in their drinking water for 3 days post-procedure. Following the surgical procedures, all animals were housed for 5 days in custom-designed divided cages to permit adequate healing of suture wounds (Grippo et al, 2007) and then were returned to the home cages to recover for an additional 5–7 days.
Radio Telemetric Recordings
ECG signals were recorded with a radiotelemetry receiver (DSI; sampling rate 5 kHz, 12-bit precision digitizing). The radiotelemetry receiver was controlled by the vendor software (Dataquest ART, Version 4.1 Acquisition software; DSI).
Quantification of Telemetric Variables
The data were evaluated for HR. HR conditioning data were collected for three consecutive days. All ECG parameters were evaluated using continuous data that were not confounded by movement artifact. The pulse-modulated signal at the output of the receiver was simultaneously routed to IBM-compatible personal computers containing the LABPRO data-acquisition system (Data Sciences), which was used only for monitoring, storage, and visual inspection of ECG waves. HR was evaluated using software developed in the Israeli Naval Medical Institute, following the recommendations in Fahlm and Sornmo (1984). Evaluation is based on a peak detector with several adaptive time and amplitude thresholds incorporated into the decision rule. The software allows user interaction in editing the detection results, displayed graphically as a time series of R-R intervals. Other than verification that all detected peaks (as marked on the original trace) indeed belonged to QRS complexes and that no complexes were missed, all ectopies and pathological beats were retained in the analysis series. The final edited output of the software was a filed list of M consecutive R-R intervals. The list was then converted into a point array in an N-dimensional space, the axes being either successive interval durations or absolute values of duration differentials.
Corticosterone Sampling
Urine samples were collected daily for corticosterone levels by gently removing each rat to metabolic cages for 30 min. Animals were placed in these cages from their home and once the procedure was complete the animal was returned to its home cage. Rats were allowed to acclimate to the metabolic cage for 7 days before urine collection (Brennan et al, 2000). These are regular cages with grooves along the floor, allowing urine collection in suspended calibrated cylinders. All samples were immediately frozen (−80 °C) after collection. Samples were taken before PSS, immediately after PSS exposure, after the SD procedure, and then daily through day 7 (between 1230 and 1300 hours). CORT was measured with a DSL-10-81000 ELISA kit according to the instructions of the manufacturer (Diagnostic Systems Laboratories, Webster, TX) by a person blind to experimental procedures. The sensitivity of the corticosterone assay is 12.5 μg/l. Within-assay variation is <10% and between-assay variation is <14% at 100 μg/l. All samples were measured in duplicate.
Injections
MIFE (Sigma-Aldrich) in a dose of 7.5 mg (approximately 30 mg/kg) was dissolved in 0.5 ml propylene glycol vehicle. EPI (Sigma-Aldrich) in a dose of 0.1 mg/kg was injected subcutaneously. Drugs were prepared fresh before use. Drug doses were chosen on the basis of previous studies (Pitman et al, 2011; Weinberger et al, 1984).
Statistical Analyses
For the behavioral and molecular results, the statistical analyses were performed using two-way analysis of variance (ANOVA) (three-way for Sholl-analysis). For urine corticosterone levels and HR, the statistical analyses were performed using repeated-measure (RM)ANOVA. Post-hoc Bonferroni test examined differences between individual groups. The prevalence of affected rats as a function of rat group was tested using cross-tabulation and nonparametric χ2 tests. In all cases, p<0.05 was considered statistically significant.
Ethical Approval
All procedures were carried out under strict compliance with ethical principles and guidelines of the NIH Guide for the Care and Use of Laboratory Animals. All treatment and testing procedures were approved by the Animal Care Committee of Ben-Gurion University of the Negev, Israel. All efforts were made to minimize pain, stress, and the number of animals used.
RESULTS
Post-Exposure SD-Attenuated Behavioral Stress Responses
As shown in Table 1, two-way ANOVA revealed significant exposure, treatment, and exposure–treatment interaction effects in terms of time spent in open arms (F(1, 62)=7.1, p<0.01, F(1, 62)=50.3, p<0.0001, and F(1, 62)=7.8, p<0.007, respectively), number of entries to the open arms (F(1, 62)=7.5, p<0.0085, F(1, 62)=18.3, p<0.0001, and F(1, 62)=12.4, p<0.0009, respectively) and anxiety index (F(1, 62)=11.5, p<0.0015, F(1, 62)=37.4, p<0.0001, and F(1, 62)=12.0, p<0.001, respectively). In terms of total activity, two-way ANOVA revealed significant effects of treatment and exposure–treatment interaction (F(1, 62)=19.3, p<0.0001 and F(1, 62)=5.6, p<0.025, respectively). No effects were observed for exposure. Bonferroni test confirmed that the exposed-untreated group exhibited a significant decrease in overall time spent in the open arms and in open arm entries and a significantly increased anxiety index compared with the unexposed-untreated group (Bonferroni test: p<0.0003, p<0.008, and p<0.0001, respectively) and to exposed rats treated with SD (Bonferroni test: time open: p<0.0001 for all). Moreover, SD increased total activity both in the unexposed and exposed groups (p<0.0025 and p<0.0001, respectively).
Table 1. Effect of Post-Exposure SD on Behavioral Stress Responses.

Startle response
Two-way ANOVA revealed a significant effect for exposure (F(1, 62)=28.3, p<0.0001), a treatment effect (F(1, 62)=4.45, p<0.04) and an exposure–treatment interaction effect (F(1, 62)=5.7, p<0.025). Bonferroni test confirmed that the exposed-untreated group showed a significantly increased mean startle amplitude compared with unexposed controls (p<0.0001). The post-exposure SD group (PSS-SD) exhibited significantly decreased mean startle amplitude compared with the exposed-untreated group (p<0.002).
Startle habituation
Two-way ANOVA revealed a significant effect for exposure (F(1, 62)=41.2, p<0.0001) and an exposure–treatment interaction effect (F(1, 62)=27.7, p<0.0001). Bonferroni test confirmed that the exposed SD group exhibited significantly increased startle habituation compared with the exposed-untreated group (p<0.0001).
Freezing response to trauma-cue exposure
The trauma-cue elicited significant exposure–treatment interaction (F(1, 37)=112.1 p<0.0001). No effect was observed for exposure or treatment. Bonferroni post-hoc tests confirmed that exposed-untreated rats displayed significantly more immobility than unexposed-untreated control and PSS-SD rats (p<0.0001 for both groups). No differences were found between unexposed groups, or between PSS-SD and unexposed groups (Table 1).
Relative prevalence rates according to CBC classification
There were significant differences in the prevalence rates of individuals displaying EBR among groups (Pearson χ2=17.6, df=3, p<0.00055) (Figure 2a). The prevalence of EBR animals among exposed-untreated animals was 33.3% of the total population (N=6/18) and differed significantly from the unexposed-untreated animals, and PSS-SD groups (χ2=6.1, p<0.015; χ2=6.5, p<0.0015, respectively), in which there were no EBR individuals.
Figure 2.

Effect of post-exposure SD on relative prevalence rates according to CBC classification: top line: the behavioral procedure used for the unexposed and PSS-exposed rats. Vertical arrow represents total SD procedure. Post-exposure SD reduced the prevalence of PTSD-like behavioral responses (EBR) (a) relative to the exposed-untreated group and concomitantly increased the prevalence of minimal behavioral responders (b). No differences were observed in the prevalence of PBRs (c). CON, unexposed control; EBR, extreme behavioral response; MBR, minimal behavioral response; PBR, partial behavioral response; PSS, predator scent stress; SD, sleep deprivation.
Significant differences were found in the prevalence of MBR among groups (Pearson χ2=18.4, df=3, p<0.0004) (Figure 2b). MBR prevalence among the CON-SD and PSS-SD animals was 68.75 and 64.7% of the total population (N=11/16 and N=11/17, respectively) and differed significantly from unexposed-untreated animals (χ2=7.4, p<0.007; χ2=8.4, p<0.004, respectively), and from exposed-untreated animals (χ2=11.9, p<0.0007; χ2=10.76, p<0.0015, respectively). No significant differences were found between SD groups.
There were significant differences in the prevalence rates of individuals displaying PBR among groups (Pearson χ2=9.3, df=3, p<0.03) (Figure 2c). The prevalence of PBR individuals in the unexposed-untreated group was 80.0% of the total population (N=12/15) and not significantly different from the exposed-untreated animals (55.5%) (N=10/18). However, prevalence among the CON-SD and PSS-SD animals was 31.2 and 35.2% of the total population (N=5/15 and N=6/17, respectively) and differed significantly from unexposed-untreated animals (χ2=7.4, p<0.007; χ2=6.47, p<0.015, respectively). No significant differences were found between SD groups or between SD groups and the exposed-untreated group.
Post-Exposure SD Changed Urine Corticosterone Levels
RM-ANOVA revealed a significant effect for time (F(4, 16)=6.6, p<0.004) and a time–group interaction effect (F(12, 16)=3.5, p<0.015) (Figure 3a). No effect was observed for groups. Bonferroni test confirmed no significant differences among groups at baseline. Six hours of SD significantly increased urine corticosterone levels in the exposed and unexposed groups as compared with the unexposed-untreated control group (p<0.03 and p<0.0025, respectively). At day 6 post-exposure, the exposed-untreated group exhibited significantly elevated urine corticosterone as compared with the unexposed-untreated group (p<0.0025). Moreover, the SD groups (exposed and unexposed) exhibited significantly lower urine corticosterone as compared with the unexposed-untreated control group (p<0.035). Figure 3b depicts the area under the curves (AUCs) and demonstrates that the exposed-untreated group displayed a significantly higher total urine corticosterone level as compared with all other groups (p<0.05).
Figure 3.

Effect of post-exposure SD on urine corticosterone levels: post-exposure SD, 1 h after the end of the real or sham stress, caused urine corticosterone levels (a) to increase in a rapid spike, followed by a rapid decline within the next 12 h, in both the stress-exposed and -unexposed groups. (b) The area under the curve. All data represent group mean±SEM. CON, unexposed control; PSS, predator scent stress; SD, sleep deprivation.
Post-Exposure SD Decreased HR
RM-ANOVA revealed a significant effect for groups (F(1, 20)=6.4, p<0.025) and a group–time interaction effect (F(1, 20)=6.9, p<0.02) (Figure 4a). No effect was observed for time. At baseline, there were no significant differences in HR between the groups. Seven hours post-exposure (1 h after SD), the exposed-untreated animals exhibited significantly elevated mean HR, as compared with their baseline (p<0.035) and to the post-exposure, SD-treated animals (p<0.001).
Figure 4.

Effect of post-exposure SD on HR: (a) HR (bpm) in exposed-untreated rats (N=12) and exposed rats treated with SD (N=10). (b) HR profile in telemetry-instrumented rats. Post-exposure SD abolished the PSS-induced tachycardia. All data represent group mean±SEM. bpm, beat per minute; CON, unexposed control; HR, heart rate; PSS, predator scent stress; SD, sleep deprivation.
Post-Exposure SD Changed Expression of Hippocampal GR-Immunoreactive Cells
In the hippocampal subregions CA1 (Figure 5a) and DG (Figure 5b), two-way ANOVA revealed a significant effect for exposure (F(1, 26)=7.8, p<0.01 and F(1, 26)=8.9, p<0.006, respectively) and an exposure–treatment interaction effect (F(1, 26)=3.6, p<0.05 and F(1, 26)=7.8, p<0.01, respectively). No effects were observed for treatment. Bonferroni test confirmed that post-exposure SD significantly decreased expression of GR-IR cells in the CA1 and DG hippocampus subregions 7 days later, compared with exposed-untreated animals (p<0.00035, p<0.0015, respectively).
Figure 5.

Effect of post-exposure SD on GR immunoreactivity in the hippocampus: top line: the behavioral procedure used for the unexposed and PSS-exposed rats. Vertical arrow represents total SD procedure. The quantitative analysis of GR immunostaining in the hippocampus subregions CA1 (a), and DG (b) of unexposed-untreated rats (N=8), unexposed rats treated with SD (N=8), exposed-untreated rats (N=8) and exposed rats treated with SD (N=6). On the right are representative photographs of GR immunoreactivity for each area. Photographs were acquired at × 20 (scale bar, 100 μm) and × 40 magnification (scale bar, 50 μm). The cells in red were GR positive. All data represent group mean±SEM. CA1, cornu ammonis 1; CON, unexposed control; DG, dentate gyrus; PSS, predator scent stress; SD, sleep deprivation. The color reproduction of this figure is available on the Neuropyschopharmacology journal online.
Post-Exposure SD Increased Cytostructure of DG Granule Cells
Total dendritic length
Two-way ANOVA revealed a significant effect for treatment (F(1, 28)=12.3, p<0.002). No effects were observed for exposure or exposure–treatment interaction. Bonferroni test confirmed that exposure significantly decreased the total dendritic length as compared with unexposed controls (p<0.02) (Figures 6b and c). Moreover, SD after PSS exposure significantly increased the total dendritic length as compared with exposed-untreated animals (p<0.0065).
Figure 6.

Effect of post-exposure SD on dendritic morphology in the dentate gyrus granule cells: top line: the behavioral procedure used for the unexposed and PSS-exposed rats. Vertical arrow represents total SD procedure. (a) Sholl-analysis for intersections per 25-μm radial unit distance from unexposed-untreated controls (N=7), unexposed rats treated with SD (N=8), exposed-untreated rats (N=8) or exposed rats treated with SD (N=8). Inset—the AUC, representing distance from soma. (b) Quantitative analysis of total dendritic length (μm) of dentate gyrus granule cells from the suprapyramidal blade. (c) Computer-generated plots of reconstructions and photomicrographs of the dendritic tree from granule cells. (d) Quantification of overall spine density per 10 μm of dendritic granule cells. (e) Photomicrographs showing representative Golgi–Cox-impregnated dendritic spines. Neurons from exposed animals treated with SD had significantly more dendritic intersections within each sphere at Sholl radii 10–85 μm than did neurons from the exposed-untreated group. Moreover, exposed animals treated with SD exhibited significantly greater total dendritic length as compared with exposed-untreated animals. The spine density along the dentate granule cells was significantly increased in exposed animals treated with SD as compared with exposed-untreated animals. Results displayed as mean±SEM. CON, unexposed control; PSS, predator scent stress; SD, sleep deprivation.
Sholl-analysis
Sholl-analysis for intersections per 25-μm radial unit distance showed that neurons from exposed animals treated with SD had significantly more dendritic intersections within each sphere at Sholl radii of 10–85 μm than did neurons from untreated animals exposed to PSS (Bonferroni post-hoc p<0.05) (Figure 6a). Moreover, neurons from exposed animals treated with SD had significantly more dendritic intersections within each sphere at Sholl radii of 10–35 μm than did neurons from unexposed-untreated animals (Bonferroni post-hoc p<0.05). Three-way ANOVA showed a significant radius of shell effect (F(10, 308)=86.3, p<0.0001), an exposure–treatment interaction effect (F(1, 308)=11.7, p<0.001), and a treatment-radius of shell interaction effect (F(10, 308)=2.5, p<0.0075). Figure 6a inset depicts the AUCs.
Spine density
Quantitative analysis of spine density per 10-μm of dendrite revealed that there were significantly fewer spines along the DG cells in the exposed-untreated animals as compared with unexposed-untreated controls (p<0.0003) (Figures 6d and e). The spine density along the dentate granule cells was significantly increased in animals treated with SD post-exposure as compared with exposed-untreated animals (Bonferroni post-hoc: p<0.0001).
MIFE and EPI Prevented Post-Exposure SD-Attenuated Behavioral Stress Responses
As shown in Table 2, one-way ANOVA revealed significant effects in terms of time spent in open arms (F(3, 29)=5.2, p<0.006), open arm entries (F(3, 29)=7.2, p<0.001), total arms activity (F(3, 29)=8.6, p<0.0005), and anxiety index (F(3, 29)=5.8, p<0.0085). Bonferroni test confirmed that the exposed-SD group (PSS-SD-Vehicle) exhibited a significant increase in overall time spent in the open arms and in open arm entries and a significantly decreased anxiety index compared with the MIFE-exposed-SD (p<0.02, p<0.008, and p<0.015, respectively), EPI-exposed-SD (p<0.025, p<0.007, and p<0.008, respectively), and exposed-vehicle groups (p<0.01, p<0.007, and p<000035, respectively). Moreover, exposed-SD and EPI-exposed-SD groups exhibited a significant increase in total activity as compared with the MIFE-exposed-SD (p<0.0001 and p<0.015) or exposed-vehicle (p<0.0025 and p<0.001, respectively).
Table 2. Effects of Mifepristone and Epinephrine Administration Post-Exposure and Pre-SD on Behavioral Stress Responses.

Startle response
ANOVA revealed significant effects between groups (F(3, 29)=5.4, p<0.006). Bonferroni test confirmed that the exposed-SD group showed a significantly decreased mean startle amplitude compared with the MIFE-exposed-SD (p<0.02), EPI-exposed-SD (p<0.02), and exposed-vehicle group (p<0.0025).
Startle habituation
One-way ANOVA revealed significant effects between groups (F(3, 29)=5.5, p<0.005). Bonferroni test confirmed that the exposed-SD group exhibited significantly increased startle habituation compared with the MIFE-exposed-SD (p<0.03), EPI-exposed-SD (p<0.015), and exposed vehicle groups (p<0.01).
Freezing response to trauma-cue exposure
The trauma-cue elicited significant exposure–treatment interaction (F(1, 29)=19.8 p<0.0001). Bonferroni test confirmed that the exposed-SD group showed a significantly decreased freezing response to trauma-cue compared with the MIFE-exposed-SD (p<0.001), EPI-exposed-SD (p<0.002), and exposed-vehicle groups (p<0.002) (Table 1).
Relative prevalence rates according to CBC
There were significant differences in the prevalence rates of individuals displaying MBR among groups (Pearson χ2=16.5, df=3, p<0.0009). The prevalence of MBR individuals among PSS-SD-vehicle animals was 62.5% of the total population (N=5/8), whereas no MBR individuals were identified in the exposed-SD+MIFE or EPI and the exposed group treated with vehicle alone (Figure 7b). There were no significant differences in the prevalence of either PBR (Figure 7c) or EBR among groups (Figure 7a). The prevalence of EBR animals among PSS-SD animals treated with MIFE or EPI was 50 and 66.6% of the total population (N=4/8 and N=4/6, respectively) and differed significantly from the PSS-SD groups (χ2=7.5, p<0.0065; χ2=5.3, p<0.0025, respectively), in which there were no EBR individuals.
Figure 7.

Effects of MIFE and EPI administration post-exposure and pre-SD on relative prevalence rates according to CBC classification: top line: the behavioral procedure used for the unexposed and PSS-exposed rats. Vertical arrow represents total SD procedure. Administration of MIFE or EPI post-exposure and pre-SD increased the prevalence of PTSD-like behavioral responses (EBR) (a) relative to the vehicle treatment group and concomitantly decreased the prevalence of minimal behavioral responders (b). No differences were observed in the prevalence of PBRs (c). CON, unexposed control; EBR, extreme behavioral response; EPI, epinephrine; MBR, minimal behavioral response; MIFE, mifepristone; PBR, partial behavioral response; PSS, predator scent stress; SD, sleep deprivation.
MIFE and EPI Injections Post-Exposure SD Changed Expression of Hippocampal GR-Immunoreactive Cells
In the hippocampal subregions CA1 (Figure 8a) and DG (Figure 8b), there were significant differences between groups (F(3, 12)=8.0, p<0.005 and F(3, 12)=9.4, p<0.0025, respectively). Bonferroni test confirmed that post-exposure SD significantly decreased expression of GR-IR cells in the CA1 and DG hippocampus subregions 7 days later, compared with exposed-SD groups treated with MIFE or EPI, or the exposed group treated with vehicle alone (CA1: p<0.01, p<0.008, and p<0.007; DG: p<0.035, p<0.009 and p<0.015, respectively). No differences were observed in the CA3 area.
Figure 8.

Effects of MIFE and EPI administration post-exposure and pre-SD on GR immunoreactivity in the hippocampus: top line: the behavioral procedure used for the unexposed and PSS-exposed rats. Vertical arrow represents total SD procedure. The quantitative analysis of GR immunostaining in the hippocampus subregions CA1 (a), and DG (b) of exposed rats treated with vehicle (N=4), exposed-vehicle rats treated with SD (N=4), exposed–SD rats treated with MIFE (N=4) or exposed-SD rats treated with EPI (N=3). On the right are representative photographs of GR immunoreactivity for each area. Photographs were acquired at × 20 (Scale bar, 100 μm) and × 40 magnification (Scale bar, 50 μm). The cells in red were GR positive. All data represent group mean±SEM. CA1, cornu ammonis 1; CON, unexposed control; DG, dentate gyrus; EPI, epinephrine; MIFE, mifepristone; PSS, predator scent stress; SD, sleep deprivation. The color reproduction of this figure is available on the Neuropyschopharmacology journal online.
MIFE and EPI Injections Post-Exposure SD Decreased Cytostructure of DG Granule Cells
Total dendritic length
One-way ANOVA revealed significant differences between groups (F(3, 12)=6.4, p<0.0002). Bonferroni test confirmed that MIFE and EPI post-PSS-SD significantly decreased the total dendritic length as compared with the PSS-SD group (p<0.015 and p<0.0035, respectively) (Figures 9b and c). Moreover, SD after PSS exposure significantly increased the total dendritic length as compared with exposed-vehicle animals (p<0.002).
Figure 9.

Effects of MIFE and EPI administration post-exposure and pre-SD on dendritic morphology in the DG granule cells: top line: the behavioral procedure used for the unexposed and PSS-exposed rats. Vertical arrow represents total SD procedure. (a) Sholl-analysis for intersections per 25-μm radial unit distance from exposed rats treated with vehicle (N=4), exposed-vehicle rats treated with SD (N=4), exposed-SD rats treated with MIFE (N=4) or exposed-SD rats treated with EPI (N=3). Inset—the AUC, representing distance from soma. (b) Quantitative analysis of total dendritic length (μm) of DG granule cells from the suprapyramidal blade. (c) Computer-generated plots of reconstructions and photomicrographs of the dendritic tree from granule cells. (d) Quantification of overall spine density per 10 μm of dendritic granule cells. (e) Photomicrographs showing representative Golgi–Cox-impregnated dendritic spines. Neurons from exposed animals treated with SD had significantly more dendritic intersections within each sphere at Sholl radii 35–60 μm than did neurons from the exposed-SD group treated with MIFE or EPI. Moreover, exposed animals treated with SD exhibited significantly greater total dendritic length as compared with exposed-SD animals treated with MIFE or EPI. The spine density along the dentate granule cells was significantly increased in exposed animals treated with SD as compared with exposed-SD animals treated with MIFE or EPI. Results displayed as mean±SEM. CON, unexposed control; EPI, epinephrine; MIFE, mifepristone; PSS, predator scent stress; SD, sleep deprivation. *p<0.05 vs PSS-Vehicle, PSS-SD-Mif, PSS-SD-Epi; #p<0.05 vs PSS-SD-Mif, PSS-SD-Epi.
Sholl-analysis
Sholl-analysis for intersections per 25-μm radial unit distance showed that neurons from exposed animals treated with SD had significantly more dendritic intersections within each sphere at Sholl radii of 10–85 μm than did neurons from all other groups (Bonferroni post-hoc p<0.05) (Figure 9a). Two-way ANOVA showed a significant group effect (F(3, 121)=9.06, p<0.0001) and radius of shell effect (F(10, 121)=77.5, p<0.0001). Figure 9a inset depicts the AUCs.
Spine density
Quantitative analysis of spine density per 10-μm of dendrite revealed that there were significantly fewer spines along the DG cells in the exposed-SD animals treated with MIFE and EPI as compared with exposed-SD controls (p<0.0003) (Figures 9d and e). The spine density along the dentate granule cells was significantly increased in animals treated with SD post-exposure as compared with exposed-vehicle animals (Bonferroni post-hoc: p<0.0001).
DISCUSSION
Convergent evidence has accumulated that sleep serves as an off-line period in which newly encoded hippocampus-dependent memories are gradually adapted to pre-existing knowledge networks (Born and Wilhelm, 2011). As memories are integral to PTSD-related symptoms, we evaluated the effects of post-exposure SD on behavioral responses to stress in a controlled, prospective animal model. As the results show, SD proved to be a highly effective intervention for the attenuation of stress-induced behavioral effects, when initiated in the aftermath of stress exposure. Compared with exposed controls, treated animals displayed significantly reduced behavioral disruption and significantly attenuated physiological, molecular, and morphological responses to the stressor. The abolition of this ameliorative effect by MIFE and EPI indicates that the beneficent anxiolytic effects are mediated (to a significant degree, at least) by the HPA-axis and adrenergic activity. Furthermore, the equally resounding effect on freezing response to the neutral reminder of the stressor indicates that memory-related processes were affected by SD. This is perhaps not surprising, because sleep has been shown to improve learning and memory processes (Diekelmann and Born, 2010; Gais and Born, 2004; Gais et al, 2000; Stickgold, 2005; Walker and Stickgold, 2006) on the one hand, and on the other hand, memory-related factors are intimately involved in post-traumatic sequelae.
Six hours of SD after PSS exposure resulted in a statistically significant moderation of behavior patterns representing stress-induced anxiety, avoidance, and hyperarousal responses on the EPM and ASR tests. A resounding overall shift in the prevalence rates of animals fulfilling criteria for EBR, which were effectively reduced to nil, was mirrored by a concomitant increase in minimal behavioral responders. Freezing responses to the late (day 8) neutral trauma-cue were markedly attenuated (16.4% of time freezing in the treatment group as compared with 75.8% for untreated controls). As memory is required to bridge the time interval between stress exposure and trauma-cue, and because the SD procedure intentionally spanned the time-frame within which memory consolidation processes take place at the cellular level (McGaugh, 2000), the reduction in freezing responses suggests that memory-related processes were affected. In other words, post-exposure SD may affect traumatic memory consolidation and thereby effectively ameliorate long-term, stress-induced, PTSD-like behavioral disruptions. These results are consistent with previous studies that assessed the effects of SD on contextual fear conditioning and memory consolidation (Gais et al, 2000; Graves et al, 2003; Hagewoud et al, 2011; Wagner et al, 2001, 2006).
Memory consolidation requires gene transcription and de novo protein synthesis (Abel and Lattal, 2001; Abel et al, 1998; Davis and Squire, 1984; Dudai, 1996; Flexner et al, 1965; McGaugh, 2000) in the hippocampus and associated structures, and is modulated by the complex interplay between the neuro-hormones and neurotransmitters released by two interacting effector systems—the HPA-axis and sympathetic-adrenergic systems (Roozendaal et al, 2006a, 2006b, 2009). Radio-telemetrically collected cardiovascular data showed that the sympathetic outflows to the HR were strongly activated during the PSS response in controls, whilst post-exposure SD abolished the PSS-induced tachycardia and increased parasympathetic activity. In other words, in response to post-exposure SD, the autonomic balance of cardiovascular regulation shifted to a more robust parasympathetic dominance. These results conform to findings in clinical studies of one night of SD, which demonstrated slightly increased HR variability and baroreceptor reflex sensitivity, suggesting an increase in cardiac vagal regulation (Pagani et al, 2009) and reduced muscle sympathetic efferent nerve activity (Kato et al, 2000).
The response of the HPA-axis in post-exposure sleep-deprived animals displayed a distinctly different pattern of response from untreated controls, as reflected by urinary corticosterone levels. The exposed-untreated group was characterized by raised levels maintained at a plateau throughout the 6 days of follow-up. In contrast, SD 1 h after real or sham stress exposure caused corticosterone levels to increase in a rapid spike to values higher than the plateau (above), followed by a rapid decline within the next 12 h, in both groups, presumably resulting from transient enhancement of negative feedback in the wake of the initial spike. The biphasic corticosterone response induced by SD conforms to the adaptive pattern of the neuroendocrine stress response. These results are in line with several studies reporting an elevation of cortisol levels during one night of total SD (Balbo et al, 2010; Baumgartner et al, 1990; Bouhuys et al, 1990; Sgoifo et al, 2006; Voderholzer et al, 2004; von Treuer et al, 1996; Weibel et al, 1995; Weitzman et al, 1983; Yamaguchi et al, 1978). Moreover, these data also support our previous findings suggesting that a fault in the initial adaptive endogenous response of the HPA-axis unfavorably alters the trajectory of trauma exposure (Cohen et al, 2008, 2011b; Kozlovsky et al, 2009b; Zohar et al, 2011).
However, if the elevated corticosterone levels 1 h after the SD procedure are indeed ‘stress-induced', it is surprising that the mean HR was not elevated. It is important to remember that, whereas HR was assessed directly and in real-time, corticosterone was assessed in the urine and thus indirectly reflects systemic corticosterone levels collected in the bladder over several hours, that is, with a significant delay. Furthermore, HR findings indicating decreased sympathetic outflow after SD could be related to fatigue and sleepiness. Taken together, the integration effects between the sympathetic-adrenergic system and corticosteroids on memory consolidation seem to be an important element in behavioral adaptation following stress exposure.
The pattern of gene expression for GR in the hippocampus paralleled the gross neuroendocrine response pattern and was characterized by a consistent upregulation of GR expression in exposed-untreated rats. In the CA1 area, predominant nuclear localization of GR was still observed 8 days after the stress exposure in exposed-untreated animals as compared with the exposed-SD group. SD corrected this, eliciting a relative downregulation of GR in CA1 and DG areas. This effect could prevent the severe adverse effects of prolonged, excessive activation of GR (Kozlovsky et al, 2009a) on hippocampal morphology (Sapolsky, 1992). These findings suggest that post-exposure SD may reduce the peripheral and central long-term hyperactivity within the HPA-axis, which is associated with adaptive stress responses.
On the morphological level, SD was associated with an increase in total dendritic length and spine density in granule cells in the DG, alongside increased DG dendritic arborization and complexity of the dendritic tree, with significantly more dendritic intersections within each sphere at Sholl radii 10–85 μm as compared with the exposed-untreated group. Overall, post-exposure SD was associated with obtunded GR levels in the hippocampus and with enhanced dendritic growth and increased spine density, which together provide the infrastructural basis required for the observed attenuation of the physiological and behavioral stress responses.
We hypothesize that post-exposure SD disrupts sleep-dependent processes of neural reactivation assumed to be necessary for synaptic and network changes underlying the consolidation of new memory traces via a biphasic, hyper-activation of the HPA-axis and attenuation of sympathetic-adrenergic tone, and in this manner interfered with traumatic memory consolidation processes.
To assess this hypothesis, the effects of pharmacological manipulation using MIFE and EPI were examined. A single bolus of systemic treatment with MIFE or EPI was sufficient to prevent the influence of SD on memory consolidation. Single treatments with MIFE, GR antagonist, or EPI, injected immediately after stress exposure were associated with significantly poorer long-term outcome than for exposed-SD vehicle controls. Both MIFE and EPI treatments were associated with a far greater degree of behavioral disruption in the EPM and ASR tests, reflected by a pronounced increase in prevalence rates of EBR and in trauma-cue freezing responses, relative to the exposed-SD vehicle group. These anxiogenic-like effects were accompanied by a significant upregulation of GR-IR cells. Moreover, microscopy imaging of GR-IR after PSS exposure in the hippocampus area demonstrated subregion-specific differences in GR translocation patterns. In exposed-SD animals, the majority of the GR-immunoreactions were found in the cytoplasm of CA1 pyramidal cells, whereas some were dispersely present in the nucleus. Administration of MIFE or EPI immediately after PSS exposure increased the nuclear GR-IR, whereas decreasing the dispersed cytoplasmic compartment GR-IR. In the DG area, a considerable amount of GR-IR was present in the nucleus in all groups, as previously described (Sarabdjitsingh et al, 2009). These results suggest that acute activation of GR (HPA-axis) during stress exposure is necessary for stress adaptation.
Glucocorticoid activation may trigger gene transcription and protein synthesis through interactions with specific DNA sequences known as glucocorticoid-responsive elements in their promoter regions, or with other transcription factors (such as nuclear factor-kB and CREB and phosphorylation of ERK1, ERK2 and CREB), which are essential for memory processes as well as for neuronal plasticity and connectivity (Jin et al, 2007; Kida et al, 2002; McGaugh et al, 2002).
These findings are supported by previous studies examining the role of GCs in susceptibility to ‘PTSD-like behaviors' (Cohen et al, 2006b, 2008; Zohar et al, 2011). These studies demonstrated a greater susceptibility to experimentally induced PTSD-like behavioral changes in rats with a hypoactive and hypo-reactive HPA axis, that is, Lewis strain, compared with a rat strain with a hyper-responsive HPA-axis, that is, Fischer rats. Exogenous administration of cortisol to Lewis rats before the stressor effectively decreased the prevalence of subsequent extreme behavioral disruption (Cohen et al, 2006b). Further study examined the effect of a single intervention with high-dose corticosterone 1 h post-exposure and showed a significant reduction in the incidence of PTSD-like behaviors and improved resilience to subsequent trauma (Cohen et al, 2008).
On the morphological level, MIFE- and EPI-treated animals clearly demonstrated significantly lower dendritic complexity, lower total dendritic length, fewer branches, and lower spine density along DG dendrites 8 days after exposure, as compared with exposed-SD animals. As the dendritic arbor is responsible for receiving and consolidating neuronal information input (Sorra and Harris, 2000; Vessey and Karra, 2007), the reduced dendritic arbor in the MIFE- and EPI-treated animals can have considerable consequences for the functional properties of cells and neuronal circuitry, including decreased synaptic plasticity and synaptic strength, and impaired stabilization of synaptic connectivity, which may in turn lead to vulnerability to psychopathology.
EPI has also been shown repeatedly to be involved in memory reinforcement of different behavioral tasks (Cahill et al, 1994; McGaugh, 1989). Post-training administration of EPI to humans enhances memory consolidation for emotionally arousing material (Cahill and Alkire, 2003), whereas blockage of (nor)adrenergic function selectively impairs this (Cahill et al, 1994; Hurlemann et al, 2005; van Stegeren et al, 1998). Clinical studies suggest that enhanced noradrenergic activity during trauma may augment the encoding of the memory (O'Donnell et al, 2004). Enhanced noradrenergic activity and elevated levels of norepinephrine in the cerebrospinal fluid correlate with severity of symptoms of PTSD (Debiec et al, 2011; Geracioti et al, 2001; Strawn and Geracioti, 2008). High levels of norepinephrine release during exposure to a traumatic event have been proposed to result in over-consolidation of the traumatic memory, thereby leading to PTSD (Pitman, 1989). Recent studies indicate that the combined action of norepinephrine and glucocorticoid hormones potently affects memory function (McGaugh and Roozendaal, 2002; Roozendaal et al, 2006b). Noradrenergic activation of the basolateral amygdala is required for the adrenal steroids to influence hippocampal memory storage (Quirarte et al, 1997; Roozendaal et al, 1999), and glucocorticoids seem to exert a permissive action on the efficacy of the noradrenergic system (de Kloet et al, 1991; Roozendaal et al, 2006b, 2009).
The results of this study suggest that prevention of sleep in the early aftermath of stress exposure may be beneficial in attenuating traumatic, stress-related sequelae. Post-exposure SD impairs hippocampus-dependent traumatic memory formation and consolidation, a mechanism possibly pertinent to the development of PTSD.
The above findings, although interesting, must be tempered by the limitations of the study: we must take care not to be too literal in interpreting animal models and methods. It would be presumptuous (and spurious) to assume that the ‘criteria' applied in this study, in fact reflect psychophysiological parameters in the life of the rat, commensurate with the criteria for PTSD in humans. Moreover, whether sleep loss per se or the mildly stressful nature of gentle handling caused the increased activity of the neuroendocrine stress system is a question for which there is no simple answer. Future studies are required to examine this.
Conclusions
SD throughout the first hours after stress exposure might represent a simple, yet effective, intervention for the secondary prevention of stress-induced pathologies. Further studies will be required to examine whether SD in the immediate aftermath of traumatic events represents an avenue for secondary prevention of stress-related clinical disorders.
Acknowledgments
We are grateful for funding from the Israel Academy of Science and Humanities grant (416/09) and the Ministry of Health (3-0000-6086) grant to HC.
The authors declare no conflict of interest.
References
- Abel T, Lattal KM. Molecular mechanisms of memory acquisition, consolidation and retrieval. Curr Opin Neurobiol. 2001;11:180–187. doi: 10.1016/s0959-4388(00)00194-x. [DOI] [PubMed] [Google Scholar]
- Abel T, Martin KC, Bartsch D, Kandel ER. Memory suppressor genes: inhibitory constraints on the storage of long-term memory. Science. 1998;279:338–341. doi: 10.1126/science.279.5349.338. [DOI] [PubMed] [Google Scholar]
- Aerni A, Traber R, Hock C, Roozendaal B, Schelling G, Papassotiropoulos A, et al. Low-dose cortisol for symptoms of posttraumatic stress disorder. Am J Psychiatry. 2004;161:1488–1490. doi: 10.1176/appi.ajp.161.8.1488. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders—(DSMIV-TR) Washington, DC; 2004. [Google Scholar]
- Balbo M, Leproult R, Van Cauter E. Impact of sleep and its disturbances on hypothalamo-pituitary-adrenal axis activity. Int J Endocrinol. 2010;2010:759234. doi: 10.1155/2010/759234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner A, Riemann D, Berger M. Neuroendocrinological investigations during sleep deprivation in depression. II. Longitudinal measurement of thyrotropin, TH, cortisol, prolactin, GH, and LH during sleep and sleep deprivation. Biol Psychiatry. 1990;28:569–587. doi: 10.1016/0006-3223(90)90395-i. [DOI] [PubMed] [Google Scholar]
- Born J, Rasch B, Gais S. Sleep to remember. Neuroscientist. 2006;12:410–424. doi: 10.1177/1073858406292647. [DOI] [PubMed] [Google Scholar]
- Born J, Wilhelm I. System consolidation of memory during sleep. Psychol Res. 2011;76:192–203. doi: 10.1007/s00426-011-0335-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhuys AL, Flentge F, Van den Hoofdakker RH. Effects of total sleep deprivation on urinary cortisol, self-rated arousal, and mood in depressed patients. Psychiatry Res. 1990;34:149–162. doi: 10.1016/0165-1781(90)90016-x. [DOI] [PubMed] [Google Scholar]
- Brennan F, Ottenweller J, Seifu Y, Zhu G, Servatius R. Persistent stress-induced elevations of urinary corticosterone in rats. Physiol Behav. 2000;71:441–446. doi: 10.1016/s0031-9384(00)00365-6. [DOI] [PubMed] [Google Scholar]
- Bryant RA, Creamer M, O'Donnell M, Silove D, McFarlane AC. A study of the protective function of acute morphine administration on subsequent posttraumatic stress disorder. Biol Psychiatry. 2009;65:488–440. doi: 10.1016/j.biopsych.2008.10.032. [DOI] [PubMed] [Google Scholar]
- Cahill L, Alkire MT. Epinephrine enhancement of human memory consolidation: interaction with arousal at encoding. Neurobiol Learn Mem. 2003;79:194–198. doi: 10.1016/s1074-7427(02)00036-9. [DOI] [PubMed] [Google Scholar]
- Cahill L, Prins B, Weber M, McGaugh J. Beta-adrenergic activation and memory for emotional events. Nature. 1994;20:702–704. doi: 10.1038/371702a0. [DOI] [PubMed] [Google Scholar]
- Cohen H, Joseph Z, Matar M. The relevance of differential response to trauma in an animal model of post-traumatic stress disorder. Biol Psychiatry. 2003;53:463–473. doi: 10.1016/s0006-3223(02)01909-1. [DOI] [PubMed] [Google Scholar]
- Cohen H, Kaplan Z, Koresh O, Matar MA, Geva AB, Zohar J. Early post-stressor intervention with propranolol is ineffective in preventing posttraumatic stress responses in an animal model for PTSD. Eur Neuropsychopharmacol. 2011a;21:230–240. doi: 10.1016/j.euroneuro.2010.11.011. [DOI] [PubMed] [Google Scholar]
- Cohen H, Kaplan Z, Matar M, Loewenthal U, Kozlovsky N, Zohar J. Anisomycin, a protein synthesis inhibitor, disrupts traumatic memory consolidation and attenuates post traumatic stress response in rats. Biol Psychiatry. 2006a;60:767–776. doi: 10.1016/j.biopsych.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Cohen H, Kozlovsky N, Alona C, Matar MA, Joseph Z. Animal model for PTSD: from clinical concept to translational research. Neuropharmacology. 2012;62:715–724. doi: 10.1016/j.neuropharm.2011.04.023. [DOI] [PubMed] [Google Scholar]
- Cohen H, Kozlovsky N, Matar MA, Zohar J, Kaplan Z. The characteristic long-term upregulation of hippocampal NF-kappaB complex in PTSD-like behavioral stress response is normalized by high-dose corticosterone and pyrrolidine dithiocarbamate administered immediately after exposure. Neuropsychopharmacology. 2011b;36:2286–2302. doi: 10.1038/npp.2011.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen H, Matar MA, Buskila D, Kaplan Z, Zohar J. Early post-stressor intervention with high-dose corticosterone attenuates posttraumatic stress response in an animal model of posttraumatic stress disorder. Biol Psychiatry. 2008;64:708–717. doi: 10.1016/j.biopsych.2008.05.025. [DOI] [PubMed] [Google Scholar]
- Cohen H, Matar MA, Zohar J.2011cThe ‘cut-off behavioral criteria' method—modeling clinical diagnostic criteria in animal studies of PTSDIn: Gouild TD (ed).Mood and Anxiety Related Phenotypes in Mice: Characterization using Behavioral TestsVol II.Humana Press c/o Springer Science, New-York; 185–208. [Google Scholar]
- Cohen H, Zohar J. An animal model of posttraumatic stress disorder: the use of cut-off behavioral criteria. Ann NY Acad Sci. 2004;1032:167–178. doi: 10.1196/annals.1314.014. [DOI] [PubMed] [Google Scholar]
- Cohen H, Zohar J, Gidron Y, Matar AM, Belkind D, Loewenthal U, et al. Blunted HPA axis response to stress influences susceptibility to posttraumatic stress response in rats. Biol Psychiatry. 2006b;59:1208–1218. doi: 10.1016/j.biopsych.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Cohen H, Zohar J, Matar MA, Kaplan Z, Geva AB. Unsupervised fuzzy clustering analysis supports behavioral cutoff criteria in an animal model of posttraumatic stress disorder. Biol Psychiatry. 2005;58:640–650. doi: 10.1016/j.biopsych.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Cohen H, Zohar J, Matar MA, Zeev K, Loewenthal U, Richter-Levin G. Setting apart the affected: the use of behavioral criteria in animal models of post traumatic stress disorder. Neuropsychopharmacology. 2004;29:1962–1970. doi: 10.1038/sj.npp.1300523. [DOI] [PubMed] [Google Scholar]
- Davis HP, Squire LR. Protein synthesis and memory: a review. Psychol Bull. 1984;96:518–559. [PubMed] [Google Scholar]
- de Kloet ER, Joels M, Oitzl M, Sutanto W. Implication of brain corticosteroid receptor diversity for the adaptation syndrome concept. Methods Achiev Exp Pathol. 1991;14:104–132. [PubMed] [Google Scholar]
- de Quervain D, Roozendaal B, McGaugh J. Stress and glucocorticoids impair retrieval of long-term spatial memory. Nature. 1998;394:787–790. doi: 10.1038/29542. [DOI] [PubMed] [Google Scholar]
- de Quervain D, Roozendaal B, Nitsch R, McGaugh J, Hock C. Acute cortisone administration impairs retrieval of long-term declarative memory in humans. Nat Neurosci. 2000;3:313–314. doi: 10.1038/73873. [DOI] [PubMed] [Google Scholar]
- Debiec J, Bush DE, LeDoux JE. Noradrenergic enhancement of reconsolidation in the amygdala impairs extinction of conditioned fear in rats—a possible mechanism for the persistence of traumatic memories in PTSD. Depress Anxiety. 2011;28:186–193. doi: 10.1002/da.20803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekelmann S, Born J. The memory function of sleep. Nat Rev Neurosci. 2010;11:114–126. doi: 10.1038/nrn2762. [DOI] [PubMed] [Google Scholar]
- Dudai Y. Consolidation: fragility on the road to the engram. Neuron. 1996;17:367–370. doi: 10.1016/s0896-6273(00)80168-3. [DOI] [PubMed] [Google Scholar]
- Fahlm O, Sornmo L. Software for QRS detection in ambulatory monitiring—a review. Med Biol Eng Comput. 1984;22:289–297. doi: 10.1007/BF02442095. [DOI] [PubMed] [Google Scholar]
- Flexner LB, Flexner JB, De La Haba G, Roberts RB. Loss of memory as related to inhibition of cerebral protein synthesis. J Neurochem. 1965;12:535–541. doi: 10.1111/j.1471-4159.1965.tb04246.x. [DOI] [PubMed] [Google Scholar]
- Gais S, Born J. Declarative memory consolidation: mechanisms acting during human sleep. Learn Mem. 2004;11:679–685. doi: 10.1101/lm.80504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gais S, Plihal W, Wagner U, Born J. Early sleep triggers memory for early visual discrimination skills. Nat Neurosci. 2000;3:1335–1339. doi: 10.1038/81881. [DOI] [PubMed] [Google Scholar]
- Geracioti TD, Jr, Baker DG, Ekhator NN, West SA, Hill KK, Bruce AB, et al. CSF norepinephrine concentrations in posttraumatic stress disorder. Am J Psychiatry. 2001;158:1227–1230. doi: 10.1176/appi.ajp.158.8.1227. [DOI] [PubMed] [Google Scholar]
- Graves LA, Heller EA, Pack AI, Abel T. Sleep deprivation selectively impairs memory consolidation for contextual fear conditioning. Learn Mem. 2003;10:168–176. doi: 10.1101/lm.48803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green EJ, Juraska JM. The dendritic morphology of hippocampal dentate granule cells varies with their position in the granule cell layer: a quantitative Golgi study. Exp Brain Res. 1985;59:582–586. doi: 10.1007/BF00261350. [DOI] [PubMed] [Google Scholar]
- Grippo AJ, Lamb DG, Carter CS, Porges SW. Cardiac regulation in the socially monogamous prairie vole. Physiol Behav. 2007;90:386–393. doi: 10.1016/j.physbeh.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagewoud R, Bultsma LJ, Barf RP, Koolhaas JM, Meerlo P. Sleep deprivation impairs contextual fear conditioning and attenuates subsequent behavioural, endocrine and neuronal responses. J Sleep Res. 2011;20:259–266. doi: 10.1111/j.1365-2869.2010.00895.x. [DOI] [PubMed] [Google Scholar]
- Hagewoud R, Whitcomb SN, Heeringa AN, Havekes R, Koolhaas JM, Meerlo P. A time for learning and a time for sleep: the effect of sleep deprivation on contextual fear conditioning at different times of the day. Sleep. 2010;33:1315–1322. doi: 10.1093/sleep/33.10.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlemann R, Hawellek B, Matusch A, Kolsch H, Wollersen H, Madea B, et al. Noradrenergic modulation of emotion-induced forgetting and remembering. J Neurosci. 2005;25:6343–6349. doi: 10.1523/JNEUROSCI.0228-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin XC, Lu YF, Yang XF, Ma L, Li BM. Glucocorticoid receptors in the basolateral nucleus of amygdala are required for postreactivation reconsolidation of auditory fear memory. Eur J Neurosci. 2007;25:3702–3712. doi: 10.1111/j.1460-9568.2007.05621.x. [DOI] [PubMed] [Google Scholar]
- Kato M, Phillips BG, Sigurdsson G, Narkiewicz K, Pesek CA, Somers VK. Effects of sleep deprivation on neural circulatory control. Hypertension. 2000;35:1173–1175. doi: 10.1161/01.hyp.35.5.1173. [DOI] [PubMed] [Google Scholar]
- Kavushansky A, Vouimba RM, Cohen H, Richter-Levin G. Activity and plasticity in the CA1, the dentate gyrus, and the amygdala following controllable vs. uncontrollable water stress. Hippocampus. 2006;16:35–42. doi: 10.1002/hipo.20130. [DOI] [PubMed] [Google Scholar]
- Kida S, Josselyn SA, de Ortiz SP, Kogan JH, Chevere I, Masushige S, et al. CREB required for the stability of new and reactivated fear memories. Nat Neurosci. 2002;5:348–355. doi: 10.1038/nn819. [DOI] [PubMed] [Google Scholar]
- Kopp C, Longordo F, Nicholson JR, Luthi A. Insufficient sleep reversibly alters bidirectional synaptic plasticity and NMDA receptor function. J Neurosci. 2006;26:12456–12465. doi: 10.1523/JNEUROSCI.2702-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlovsky N, Matar MA, Kaplan Z, Zohar J, Cohen H. A distinct pattern of intracellular glucocorticoid-related responses is associated with extreme behavioral response to stress in an animal model of post-traumatic stress disorder. Eur Neuropsychopharmacol. 2009a;19:759–771. doi: 10.1016/j.euroneuro.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Kozlovsky N, Matar MA, Kaplan Z, Zohar J, Cohen H. The role of the galaninergic system in modulating stress-related responses in an animal model of posttraumatic stress disorder. Biol Psychiatry. 2009b;65:383–391. doi: 10.1016/j.biopsych.2008.10.034. [DOI] [PubMed] [Google Scholar]
- Longordo F, Kopp C, Luthi A. Consequences of sleep deprivation on neurotransmitter receptor expression and function. Eur J Neurosci. 2009;29:1810–1819. doi: 10.1111/j.1460-9568.2009.06719.x. [DOI] [PubMed] [Google Scholar]
- Ma XM, Huang JP, Kim EJ, Zhu Q, Kuchel GA, Mains RE, et al. Kalirin-7, an important component of excitatory synapses, is regulated by estradiol in hippocampal neurons. Hippocampus. 2010;21:166–677. doi: 10.1002/hipo.20780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquet P. Sleep on it! Nat Neurosci. 2000;3:1235–1236. doi: 10.1038/81750. [DOI] [PubMed] [Google Scholar]
- Maquet P. The role of sleep in learning and memory. Science. 2001;294:1048–1052. doi: 10.1126/science.1062856. [DOI] [PubMed] [Google Scholar]
- McGaugh J. Involvement of hormonal and neuromodulatory systems in the regulation of memory storage. Annu Rev Neurosci. 1989;12:255–287. doi: 10.1146/annurev.ne.12.030189.001351. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. Memory—a century of consolidation. Science. 2000;287:248–251. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- McGaugh JL, McIntyre CK, Power AE. Amygdala modulation of memory consolidation: interaction with other brain systems. Neurobiol Learning Memory. 2002;78:539–552. doi: 10.1006/nlme.2002.4082. [DOI] [PubMed] [Google Scholar]
- McGaugh JL, Roozendaal B. Role of adrenal stress hormones in forming lasting memories in the brain. Curr Opin Neurobiol. 2002;12:205–210. doi: 10.1016/s0959-4388(02)00306-9. [DOI] [PubMed] [Google Scholar]
- Moldovan M, Constantinescu AO, Balseanu A, Oprescu N, Zagrean L, Popa-Wagner A. Sleep deprivation attenuates experimental stroke severity in rats. Exp Neurol. 2010;222:135–143. doi: 10.1016/j.expneurol.2009.12.023. [DOI] [PubMed] [Google Scholar]
- O'Donnell T, Hegadoren K, Coupland N. Noradrenergic mechanisms in the pathophysiology of post-traumatic stress disorder. Neuropsychobiology. 2004;50:273–283. doi: 10.1159/000080952. [DOI] [PubMed] [Google Scholar]
- Pagani M, Pizzinelli P, Traon AP, Ferreri C, Beltrami S, Bareille MP, et al. Hemodynamic, autonomic and baroreflex changes after one night sleep deprivation in healthy volunteers. Auton Neurosci. 2009;145:76–80. doi: 10.1016/j.autneu.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C.2005The Rat Brain in Stereotaxic Coordinates5th ednElsevier Academic Press: Burlington, MA [Google Scholar]
- Peigneux P, Laureys S, Delbeuck X, Maquet P. Sleeping brain, learning brain. The role of sleep for memory systems. Neuroreport. 2001;12:A111–A124. doi: 10.1097/00001756-200112210-00001. [DOI] [PubMed] [Google Scholar]
- Pitman RK. Post-traumatic stress disorder, hormones, and memory. Biol Psychiatry. 1989;26:221–223. doi: 10.1016/0006-3223(89)90033-4. [DOI] [PubMed] [Google Scholar]
- Pitman RK, Milad MR, Igoe SA, Vangel MG, Orr SP, Tsareva A, et al. Systemic mifepristone blocks reconsolidation of cue-conditioned fear; propranolol prevents this effect. Behav Neurosci. 2011;125:632–638. doi: 10.1037/a0024364. [DOI] [PubMed] [Google Scholar]
- Pitman RK, Sanders KM, Zusman RM, Healy AR, Cheema F, Lasko NB, et al. Pilot study of secondary prevention of posttraumatic stress disorder with propranolol. Biol Psychiatry. 2002;51:189–192. doi: 10.1016/s0006-3223(01)01279-3. [DOI] [PubMed] [Google Scholar]
- Quirarte GL, Roozendaal B, McGaugh JL. Glucocorticoid enhancement of memory storage involves noradrenergic activation in the basolateral amygdala. Proc Natl Acad Sci USA. 1997;94:14048–14053. doi: 10.1073/pnas.94.25.14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redila VA, Christie BR. Exercise-induced changes in dendritic structure and complexity in the adult hippocampal dentate gyrus. Neuroscience. 2006;137:1299–1307. doi: 10.1016/j.neuroscience.2005.10.050. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Hui GK, Hui IR, Berlau DJ, McGaugh JL, Weinberger NM. Basolateral amygdala noradrenergic activity mediates corticosterone-induced enhancement of auditory fear conditioning. Neurobiol Learn Mem. 2006a;86:249–255. doi: 10.1016/j.nlm.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, McEwen BS, Chattarji S. Stress, memory and the amygdala. Nat Rev Neurosci. 2009;10:423–433. doi: 10.1038/nrn2651. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Nguyen BT, Power AE, McGaugh JL. Basolateral amygdala noradrenergic influence enables enhancement of memory consolidation induced by hippocampal glucocorticoid receptor activation. Proc Natl Acad Sci USA. 1999;96:11642–11647. doi: 10.1073/pnas.96.20.11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, Okuda S, Van der Zee EA, McGaugh JL. Glucocorticoid enhancement of memory requires arousal-induced noradrenergic activation in the basolateral amygdala. Proc Natl Acad Sci USA. 2006b;103:6741–6746. doi: 10.1073/pnas.0601874103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM. Do glucocorticoid concentrations rise with age in the rat. Neurobiol Aging. 1992;13:171–174. doi: 10.1016/0197-4580(92)90025-s. [DOI] [PubMed] [Google Scholar]
- Sarabdjitsingh RA, Meijer OC, Schaaf MJ, de Kloet ER. Subregion-specific differences in translocation patterns of mineralocorticoid and glucocorticoid receptors in rat hippocampus. Brain Res. 2009;1249:43–53. doi: 10.1016/j.brainres.2008.10.048. [DOI] [PubMed] [Google Scholar]
- Schelling G. Effects of stress hormones on traumatic memory formation and the development of posttraumatic stress disorder in critically ill patients. Neurobiol Learn Mem. 2002;78:596–609. doi: 10.1006/nlme.2002.4083. [DOI] [PubMed] [Google Scholar]
- Schelling G, Briegel J, Roozendaal B, Stoll C, Rothenhausler HB, Kapfhammer HP. The effect of stress doses of hydrocortisone during septic shock on posttraumatic stress disorder in survivors. Biol Psychiatry. 2001;50:978–985. doi: 10.1016/s0006-3223(01)01270-7. [DOI] [PubMed] [Google Scholar]
- Schelling G, Kilger E, Roozendaal B, de Quervain DJ, Briegel J, Dagge A, et al. Stress doses of hydrocortisone, traumatic memories, and symptoms of posttraumatic stress disorder in patients after cardiac surgery: a randomized study. Biol Psychiatry. 2004;55:627–633. doi: 10.1016/j.biopsych.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Schelling G, Richter M, Roozendaal B, Rothenhausler HB, Krauseneck T, Stoll C, et al. Exposure to high stress in the intensive care unit may have negative effects on health-related quality-of-life outcomes after cardiac surgery. Crit Care Med. 2003;31:1971–1980. doi: 10.1097/01.CCM.0000069512.10544.40. [DOI] [PubMed] [Google Scholar]
- Schelling G, Stoll C, Kapfhammer HP, Rothenhausler HB, Krauseneck T, Durst K, et al. The effect of stress doses of hydrocortisone during septic shock on posttraumatic stress disorder and health-related quality of life in survivors. Crit Care Med. 1999;27:2678–2683. doi: 10.1097/00003246-199912000-00012. [DOI] [PubMed] [Google Scholar]
- Sgoifo A, Buwalda B, Roos M, Costoli T, Merati G, Meerlo P. Effects of sleep deprivation on cardiac autonomic and pituitary-adrenocortical stress reactivity in rats. Psychoneuroendocrinology. 2006;31:197–208. doi: 10.1016/j.psyneuen.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Sholl DA. The measurable parameters of the cerebral cortex and their significance in its organization. Prog Neurobiol. 1956;2:324–333. [PubMed] [Google Scholar]
- Sorra KE, Harris KM. Overview on the structure, composition, function, development, and plasticity of hippocampal dendritic spines. Hippocampus. 2000;10:501–511. doi: 10.1002/1098-1063(2000)10:5<501::AID-HIPO1>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Stickgold R. Sleep-dependent memory consolidation. Nature. 2005;437:1272–1278. doi: 10.1038/nature04286. [DOI] [PubMed] [Google Scholar]
- Strawn J, Geracioti JT. Noradrenergic dysfunction and the psychopharmacology of posttraumatic stress disorder. Depress Anxiety. 2008;25:260–271. doi: 10.1002/da.20292. [DOI] [PubMed] [Google Scholar]
- Tobler I, Jaggi K. Sleep and EEG spectra in the Syrian hamster (Mesocricetus auratus) under baseline conditions and following sleep deprivation. J Comp Physiol A. 1987;161:449–459. doi: 10.1007/BF00603970. [DOI] [PubMed] [Google Scholar]
- Vaiva G, Ducrocq F, Jezequel K, Averland B, Lestavel P, Brunet A, et al. Immediate treatment with propranolol decreases posttraumatic stress disorder two months after trauma. Biol Psychiatry. 2003;54:947–949. doi: 10.1016/s0006-3223(03)00412-8. [DOI] [PubMed] [Google Scholar]
- van Stegeren AH, Everaerd W, Cahill L, McGaugh JL. Memory for emotional events: differential effects of centrally versus peripherally acting beta-blocking agents, Psychopharmacology (Berlin) 1998;138:305–310. doi: 10.1007/s002130050675. [DOI] [PubMed] [Google Scholar]
- Vessey JP, Karra D. More than just synaptic building blocks: scaffolding proteins of the post-synaptic density regulate dendritic patterning. J Neurochem. 2007;102:324–332. doi: 10.1111/j.1471-4159.2007.04662.x. [DOI] [PubMed] [Google Scholar]
- Voderholzer U, Hohagen F, Klein T, Jungnickel J, Kirschbaum C, Berger M, et al. Impact of sleep deprivation and subsequent recovery sleep on cortisol in unmedicated depressed patients. Am J Psychiatry. 2004;161:1404–1410. doi: 10.1176/appi.ajp.161.8.1404. [DOI] [PubMed] [Google Scholar]
- von Treuer K, Norman TR, Armstrong SM. Overnight human plasma melatonin, cortisol, prolactin, TSH, under conditions of normal sleep, sleep deprivation, and sleep recovery. J Pineal Res. 1996;20:7–14. doi: 10.1111/j.1600-079x.1996.tb00232.x. [DOI] [PubMed] [Google Scholar]
- Wagner U, Gais S, Born J. Emotional memory formation is enhanced across sleep intervals with high amounts of rapid eye movement sleep. Learn Mem. 2001;8:112–119. doi: 10.1101/lm.36801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner U, Hallschmid M, Rasch B, Born J. Brief sleep after learning keeps emotional memories alive for years. Biol Psychiatry. 2006;60:788–790. doi: 10.1016/j.biopsych.2006.03.061. [DOI] [PubMed] [Google Scholar]
- Walker MP, Stickgold R. Sleep, memory, and plasticity. Annu Rev Psychol. 2006;57:139–166. doi: 10.1146/annurev.psych.56.091103.070307. [DOI] [PubMed] [Google Scholar]
- Weibel L, Follenius M, Spiegel K, Ehrhart J, Brandenberger G. Comparative effect of night and daytime sleep on the 24-hour cortisol secretory profile. Sleep. 1995;18:549–556. [PubMed] [Google Scholar]
- Weinberger N, Gold P, Sternberg D. Epinephrine enables Pavlovian conditioning under anesthesia. Science. 1984;223:605–607. doi: 10.1126/science.6695173. [DOI] [PubMed] [Google Scholar]
- Weitzman ED, Zimmerman JC, Czeisler CA, Ronda J. Cortisol secretion is inhibited during sleep in normal man. J Clin Endocrinol Metab. 1983;56:352–358. doi: 10.1210/jcem-56-2-352. [DOI] [PubMed] [Google Scholar]
- Yamaguchi N, Maeda K, Kuromaru S. The effects of sleep deprivation on the circadian rhythm of plasma cortisol levels in depressive patients. Folia Psychiatr Neurol Jpn. 1978;32:479–487. doi: 10.1111/j.1440-1819.1978.tb00155.x. [DOI] [PubMed] [Google Scholar]
- Zohar J, Yahalom H, Kozlovsky N, Cwikel-Hamzany S, Matar MA, Kaplan Z, et al. High dose hydrocortisone immediately after trauma may alter the trajectory of PTSD: interplay between clinical and animal studies. Eur Neuropsychopharmacol. 2011;21:796–809. doi: 10.1016/j.euroneuro.2011.06.001. [DOI] [PubMed] [Google Scholar]