

Misfolding mutations in rod opsin cause the blinding disease retinitis pigmentosa. The ER chaperone BiP is required to prevent rod opsin aggregation, and overexpression of BiP can improve mutant rod opsin mobility. This could be important for therapies based on manipulating rod opsin folding and aggregation.
Abstract
Mutations in rod opsin—the light-sensitive protein of rod cells—cause retinitis pigmentosa. Many rod opsin mutations lead to protein misfolding, and therefore it is important to understand the role of molecular chaperones in rod opsin biogenesis. We show that BiP (HSPA5) prevents the aggregation of rod opsin. Cleavage of BiP with the subtilase cytotoxin SubAB results in endoplasmic reticulum (ER) retention and ubiquitylation of wild-type (WT) rod opsin (WT–green fluorescent protein [GFP]) at the ER. Fluorescence recovery after photobleaching reveals that WT-GFP is usually mobile in the ER. By contrast, depletion of BiP activity by treatment with SubAB or coexpression of a BiP ATPase mutant, BiP(T37G), decreases WT-GFP mobility to below that of the misfolding P23H mutant of rod opsin (P23H-GFP), which is retained in the ER and can form cytoplasmic ubiquitylated inclusions. SubAB treatment of P23H-GFP–expressing cells decreases the mobility of the mutant protein further and leads to ubiquitylation throughout the ER. Of interest, BiP overexpression increases the mobility of P23H-GFP, suggesting that it can reduce mutant rod opsin aggregation. Therefore inhibition of BiP function results in aggregation of rod opsin in the ER, which suggests that BiP is important for maintaining the solubility of rod opsin in the ER.
INTRODUCTION
The seven-transmembrane G protein–coupled receptor (GPCR) rhodopsin is responsible for scotopic vision under dim light conditions. The rod opsin apoprotein is synthesized in the inner segment of rod photoreceptor cells before transport to the rod outer segment photosensory cilia. Rhodopsin is formed from rod opsin and the 11-cis-retinal chromophore. Mutations in rod opsin cause the neurodegenerative blindness retinitis pigmentosa (RP; Dryja et al., 1990; Online Mendelian Inheritance in Man MIM: 180380). Dominant mutations in rod opsin account for ∼25% of all autosomal dominant retinitis pigmentosa (ADRP) cases, and >200 point mutations have been identified (www.sph.uth.tmc.edu/Retnet).
Amino acid substitutions in different domains of rod opsin lead to distinct cellular and biochemical defects, which can be classified into several groups (Sung et al., 1993; Mendes et al., 2005). The majority of rod opsin mutations are designated class II because they result in the misfolding of the protein. Heterologous expression of class II rod opsin mutants, such as the common mutation that changes proline 23 to histidine (P23H), revealed that these were retained in the endoplasmic reticulum (ER), suggesting that folding-defective polypeptides were not allowed to traffic through the secretory system (Sung et al., 1991; Kaushal and Khorana, 1994). ER-retained mutant rod opsin is subject to ER-associated degradation (ERAD); if ERAD fails, then mutant rod opsin can spontaneously aggregate and form microscopically visible inclusion bodies (Illing et al., 2002; Saliba et al., 2002).
The cellular factors that facilitate the folding, quality control, and ERAD of rod opsin are beginning to be elucidated. For example, ER degradation–enhancing α-mannosidase-like 1 (EDEM1) and valosin-containing protein (VCP) facilitate the degradation of P23H rod opsin (Kosmaoglou et al., 2009; Griciuc et al., 2010). Whereas calnexin (Cnx) overexpression can stimulate a small improvement in the folding of P23H rod opsin in the presence of 11-cis-retinal (Noorwez et al., 2009), experiments on loss of Cnx activity revealed that it is not required for the P23H rod opsin ERAD or the biogenesis of wild-type rod opsin (Kosmaoglou and Cheetham, 2008; Kraus et al., 2010). This is different from Drosophila rhodopsin, Rh1, which requires a form of Cnx for correct folding (Rosenbaum et al., 2006). Indeed, although Drosophila genetics identified several Rh1 rhodopsin–specific chaperones, such as NinaA (Baker et al., 1994), the chaperones that facilitate normal mammalian rod opsin biogenesis and mediate the folding of wild-type rod opsin are less well understood.
Recently Gorbatyuk et al. (2010) demonstrated that overexpression of BiP (HSPA5), the form of Hsp70 within the lumen of the ER, could suppress retinal degeneration in the P23H rat model of ADRP. However, this was the result of alleviating ER stress and suppressing apoptosis rather than promoting of P23H rod opsin folding.
BiP participates in numerous processes, such as protein folding and oligomerization (Haas and Wabl, 1983), prevention of nonnative polypeptide aggregation (Puig and Gilbert, 1994), and preparation of terminally misfolded polypeptides for retrotranslocation and degradation in the cytosol (Molinari et al., 2002; Kabani et al., 2003; Sorgjerd et al., 2006). The cellular levels of BiP are tightly controlled and have been shown to increase during differentiation due to a rise in the ER cargo load or following induction of the unfolded protein response (UPR). BiP also plays an important role in fine tuning the UPR (Pincus et al., 2010). Of interest, P23H rod opsin expression activates the UPR and alters the expression of BiP (Lin et al., 2007).
The depletion of BiP is lethal for early embryonic cells; however, BiP+/− mice with 50% of the BiP WT level were viable but had increased expression of other ER chaperones (Luo et al., 2006). Knock-in mice that express BiP with a deleted ER retention signal (KDEL) were not embryonic lethal but died several hours after birth. These mutant BiP mice showed disordered layer formation in the cerebral cortex and cerebellum and died of respiratory failure. Secretory cells, such as epithelial alveolar or Cajal–Retzius cells, were most affected by deletion of the KDEL sequence, suggesting that BiP function is critical in secretory cells (Mimura et al., 2007). These mice died before their photoreceptors developed, so it was not possible to examine the effect of BiP depletion on these sensory neurons that synthesize ∼10 million rod opsin molecules per day in their ER (Palczewski, 2012).
Subtilase cytotoxin (SubAB) is a recently characterized bacterial AB5 toxin, which was discovered in a highly virulent Shiga-toxigenic Escherichia coli strain responsible for an outbreak of hemolytic uremic syndrome in Australia (Paton et al., 2004). The catalytic subunit of the toxin is a subtilase family serine protease with unique and exquisite specificity for BiP, cleaving the chaperone at a dileucine motif in the loop that connects the protein-binding and ATPase domains (Paton et al., 2006). Here we exploit SubAB-mediated BiP cleavage to investigate the effect of BiP depletion on rod opsin biogenesis and show that BiP is required to prevent both wild-type and mutant rod opsin from aggregating. Furthermore, BiP overexpression improves the mobility of mutant rod opsin, suggesting a potential mechanism for the observed enhanced photoreceptor survival.
RESULTS
SubAB causes cleavage of BiP and activation of the UPR
To confirm that SubAB cleaved BiP in an SK-N-SH cell culture model of rod opsin processing (Mendes and Cheetham, 2008; Kosmaoglou et al., 2009), we applied SubAB to the cells at a concentration of 1.5 μg/ml, which was previously reported as optimal for efficient BiP cleavage (Paton et al., 2006), over a range of times and analyzed BiP cleavage by Western blotting (Supplemental Figure S1A). BiP cleavage was evident after 1 h of treatment and persisted over 18 h after addition of SubAB, with the same efficiency. The cleavage of BiP gave rise to a C-terminal antibody-reactive species (Paton et al., 2006), which was detected at ∼28 kDa, albeit at very low levels (Supplemental Figure S1A, asterisk). This band did not increase in intensity with longer incubation periods, and its level never reached the level of uncleaved BiP (the ∼75-kDa band) in untreated cells. Semiquantitative Western blotting revealed that SubAB treatment resulted in cleavage of >90% of the BiP (Supplemental Figure S1B).
SubAB affects rod opsin traffic
To test the hypothesis that BiP is important for rod opsin folding, we investigated the effect of SubAB-mediated cleavage of BiP on wild-type (WT) rod opsin traffic by immunofluorescence (Figure 1). WT rod opsin tagged at its C-terminus with green fluorescent protein (WT-GFP) was transfected in SK-N-SH cells, and after transfection the cells were allowed to recover for 2 h before treatment with SubAB for 18 h (Figure 1A). WT-GFP trafficked efficiently to the plasma membrane of SK-N-SH cells. By contrast, treatment with SubAB resulted in the ER retention of WT rod opsin protein. Some of the WT-GFP was still able to traffic to the plasma membrane of transfected cells, but the majority was observed in intracellular compartments and colocalized with Cnx (Figure 1A).
FIGURE 1:
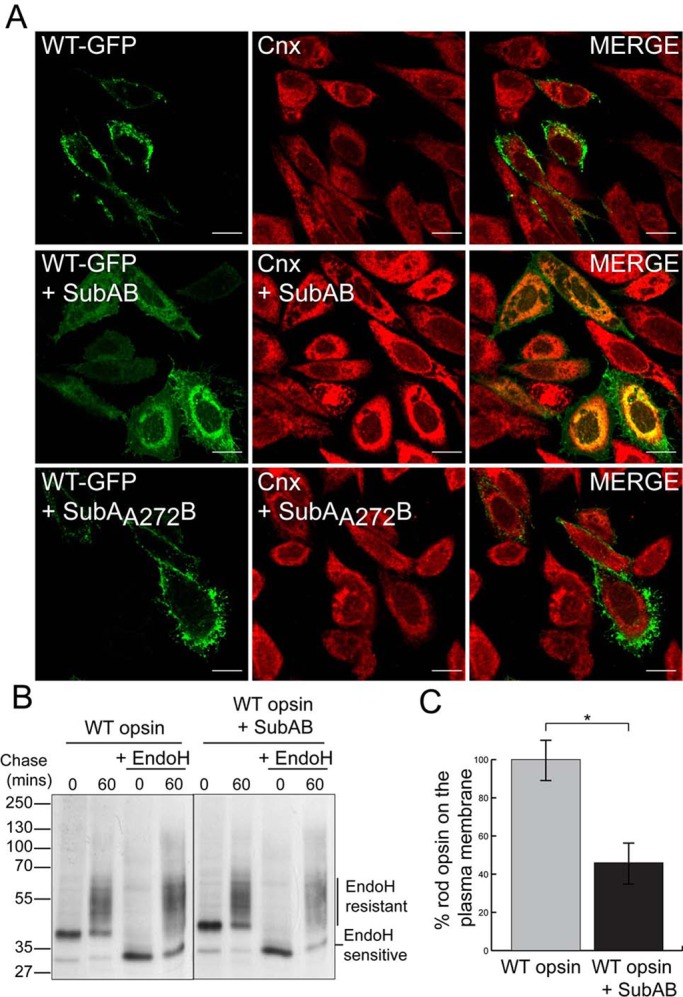
SubAB-mediated cleavage of BiP impairs WT rod opsin traffic. (A) SK-N-SH cells were transfected with WT rod opsin-GFP (WT-GFP) and were left untreated or treated with 1.5 μg/ml SubAB or SubAA272B for 18 h before fixation, as indicated. Cnx antibody (Sigma-Aldrich) was used at 1:600. The cells were fixed with 4% PFA and permeabilized and were analyzed by confocal microscopy using the intrinsic GFP fluorescence and fluorescence from anti-rabbit Alexa 594 secondary antibodies. The scanning intensity levels between control and SubAB treated cells were kept the same. Scale bar, 10 μm. (B) SubAB reduces the amount of EndoH-resistant WT rod opsin. Immunoprecipitated (IP) material from SK-N-SH cells was transfected with WT rod opsin, untreated or treated with SubAB during depletion, labeling, and chase-time intervals, as indicated. Cells were pulsed for 20 min with [35S]methionine/cysteine and were chased in unlabeled methionine media for up to 60 min. The 1D4 IPs were digested with 1500 U of EndoH overnight, as indicated. Digests were analyzed by SDS–PAGE and were visualized by autoradiography. Exposures have been adjusted to show similar levels at time 0. (C) SubAB results in a decrease in cell surface expression of WT rod opsin. “In-cell Western” analysis was performed on WT rod opsin–transfected SK-N-SH cells untreated or treated with SubAB for 16 h. Cells were fixed with 3% PFA and immunostained with 4D2 mAb to the extracellular N-terminus of rod opsin. The fluorescence on the plasma membrane (nonpermeabilized) was determined as a percentage of total fluorescence (permeabilized with 0.025% Triton); n = 3. The data were normalized to untreated WT rod opsin traffic to compensate for difference in expression level; *p < 0.05; p = 0.017.
We observed that SubAB treatment led to an increase in Cnx staining, independent of rod opsin transfection (Supplemental Figure S1C). SubAB treatment induces the UPR (Luo et al., 2006; Wolfson et al., 2008; Pincus et al., 2010), but Western blotting revealed that there was no increase in Cnx levels (Supplemental Figure S1D), suggesting that the increase in Cnx staining may be due to epitope availability or other reactivity in this antibody. BiP is important for many cellular processes, and therefore we wanted to test that cell death resulting from BiP depletion did not confound our investigation. Lactate dehydrogenase (LDH) assays confirmed that SubAB only caused significant cell death after 44 h, and therefore we restricted our assays to 2 or 18 h of treatment where no significant cell death was observed (Supplemental Figure S2A). The expression of rod opsin did not make the cells more susceptible to SubAB at these time points (Supplemental Figure S2B).
The SubAB mutant SubAA272B, which lacks the protease activity of the wild-type toxin (Paton et al., 2004, 2006), had no effect on WT rod opsin traffic or Cnx staining (Figure 1A). These data suggest that reduction of BiP by SubAB-mediated cleavage is responsible for WT rod opsin ER retention.
SubAB-mediated cleavage of BiP interferes with the maturation of WT rod opsin
To confirm that BiP reduction was leading to WT rod opsin ER retention, we monitored the development of endoglycosidase H (EndoH) glycosidase resistance, and hence traffic beyond the ER, using a pulse-chase assay (Figure 1B). SubAB treatment led to a reduction in the translation of WT rod opsin, presumably as a result of protein kinase–like ER kinase (PERK) activation and eukaryotic translation initiation factor 2 subunit α (eIF2α) phosphorylation as part of the UPR. When exposures were matched to normalize the level of WT rod opsin, however, the amount of EndoH-resistant glycoforms was reduced in the presence of SubAB after a 60-min chase (Figure 1B).
To confirm reduced traffic of WT rod opsin in the presence of SubAB, we examined permeabilized and nonpermeabilized cells stained with an antibody to the extracellular domain of rod opsin (4D2). Imaging under the same conditions showed reduced rod opsin on the plasma membrane and increased ER retention after SubAB treatment (Supplemental Figure S3). A quantitative “in-cell Western” assay using 4D2 on nonpermeabilized and permeablilized cells and a LI-COR Odyssey imager (Kosmaoglou et al., 2009; Meimaridou et al., 2011) confirmed that SubAB treatment reduced the percentage of WT rod opsin that trafficked to the plasma membrane by almost 60% (Figure 1C). Therefore SubAB treatment leads to the ER retention of a significant proportion of WT rod opsin and prevents traffic to the Golgi or plasma membrane.
BiP and rod opsin coimmunoprecipitate from retina
The requirement for BiP in WT rod opsin traffic beyond the ER suggested that BiP could be an important chaperone for rod opsin biogenesis. We used immunoprecipitation of WT rod opsin to confirm that BiP can form a complex with WT rod opsin (Figure 2). SK-N-SH cells transfected with untagged WT rod opsin were immunoprecipitated with rod opsin antibody 1D4 in the presence or absence of SubAB. A small amount of endogenous BiP could be recovered with rod opsin, whereas SubAB treatment led to BiP cleavage and abolished any detectable coimmunoprecipitation (Figure 2A). Furthermore, overexpression of BiP with WT rod opsin led to an increase in the coimmunoprecipitation of BiP with WT rod opsin (Figure 2B).
FIGURE 2:
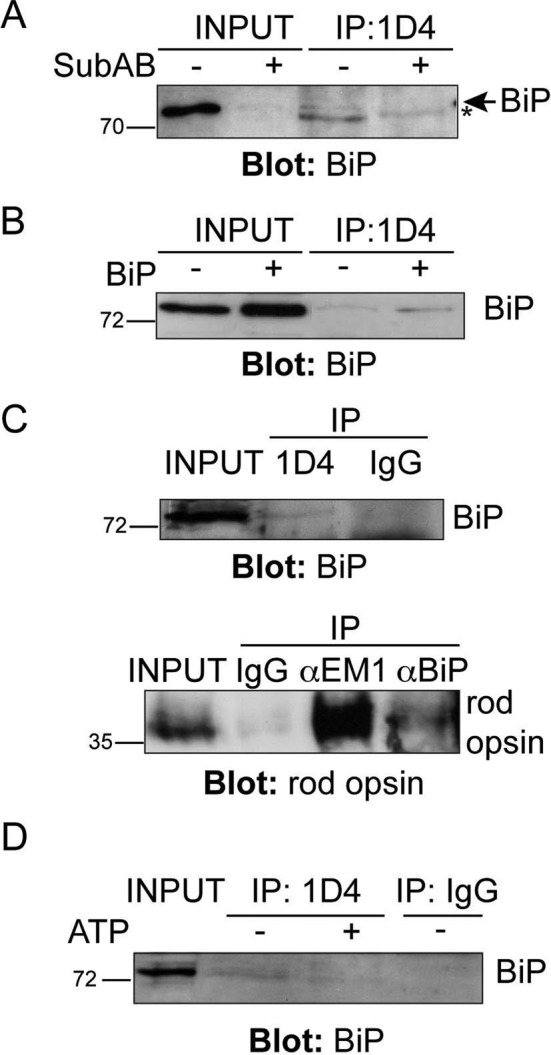
BiP and WT rod opsin are present in a complex. (A, B) Coimmunoprecipitation (CoIP) of BiP with rod opsin antibody 1D4. Western blot showing BiP immunoreactivity of 1D4 IPs; input loading was 12.5% of IP loading. SK-N-SH cells were transfected with WT rod opsin and treated with SubAB, as indicated (A). The asterisk highlights a nonspecific reactivity migrating a little faster than BiP. (B) Transfection of WT rod opsin with BiP (+BiP) led to enhanced BiP pull down. (C) CoIP of BiP and rod opsin from porcine retina. Reciprocal IP of BiP with 1D4 anti–rod opsin antibody (top, input loading was 0.4% of IP loading) and 1D4 immunoreactive rod opsin with BiP (αBiP) or EDEM1 (αEM1) antibody (bottom, input loading was 0.14% of IP loading) but not with control nonspecific antibody (IgG). (D) ATP reduces BiP recovery with 1D4 from porcine retina lysate. A 5 mM ATP wash (+ATP), as indicated, in the IP reduced the recovery of BiP with rod opsin antibody 1D4; input loading was 0.4% of IP loading. The position of molecular weight markers is highlighted on the left in kilodaltons.
To test whether BiP and rod opsin form a physiological complex in porcine retina, we performed coimmunoprecipitation with antibodies against rhodopsin and BiP. Immunoprecipitation with 1D4 reproducibly recovered a BiP immunoreactive band at ∼75 kDa, which was not detected with control antibody precipitation (Figure 2C). Similarly, immunoprecipitation with antibodies against BiP revealed a rhodopsin-immunoreactive species at ∼35 kDa (Figure 2C), although the recovery was less than with an antibody to another rhodopsin chaperone, EDEM1 (Kosmaoglou et al., 2009). When an ATP wash was included in the 1D4 immunoprecipitation there was a reduction in the amount of BiP that was recovered with 1D4 (Figure 2D), suggesting that BiP is binding rhodopsin as a client that is released on chaperone nucleotide exchange. These data confirm that rhodopsin and BiP are present in a complex in retina, presumably a transient complex that forms during rod opsin biogenesis.
SubAB treatment stimulates ubiquitin decoration of WT and P23H rod opsin at the ER
Cytoplasmic inclusions of misfolded, aggregated rod opsin recruit ubiquitin, and rod opsin is ubiquitylated (Illing et al., 2002; Saliba et al., 2002). Therefore we tested whether loss of BiP resulted in enhanced misfolding and ubiquitylation of rod opsin. SK-N-SH cells were transfected with myc-ubiquitin and treated with SubAB. In the absence of SubAB, ubiquitin was diffusely distributed throughout the cytoplasm and nucleus (Supplemental Figure S4). SubAB treatment resulted in a slight shift in ubiquitin staining to the nuclear envelope and ER, as previously reported (Lass et al., 2008; Supplemental Figure S4). In the presence of WT rod opsin, however, there was a strong, SubAB-dependent relocalization of the ubiquitin to decorate the ER (Figure 3A).
FIGURE 3:
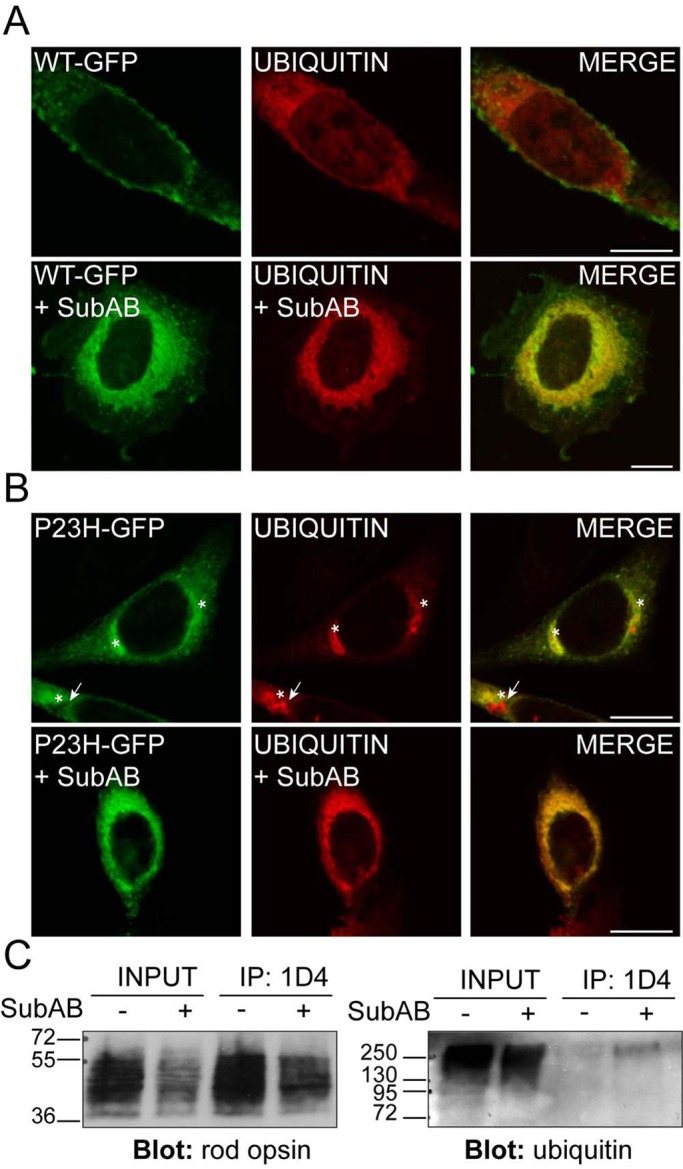
SubAB-mediated cleavage of BiP enhances rod opsin ubiquitylation. SK-N-SH cells were transfected with WT rod opsin-GFP (WT-GFP, green; A) or P23H rod opsin-GFP (P23H-GFP, green; B) with ubiquitin (UBIQUITIN, red). Two hours after transfection, cells either had a change of media or were treated with SubAB, as indicated, for 18 h before fixation. Anti-myc antibody (9E10) was used to detect ubiquitin localization. Asterisk highlights rod opsin inclusions; arrow highlights ubiquitin-positive, rod opsin–negative inclusion. Scale bar, 10 μm. (C) SubAB enhances WT rod opsin ubiquitylation. The 1D4 IP of WT rod opsin–transfected SK-N-SH cell lysates in the presence or absence of SubAB, as indicated, was Western blotted for rod opsin (left) or ubiquitin (right). Input loading was 12% of the IP loading. The position of molecular weight markers is highlighted on the left in kilodaltons.
P23H rod opsin is a mutant misfolding variant that is retained in the ER and forms cytoplasmic, ubiquitylated inclusions (Illing et al., 2002; Saliba et al., 2002). In untreated cells, ubiquitin was exclusively recruited to P23H-GFP inclusions and not the P23H rod opsin that was retained with the ER (Figure 3B). Conversely, SubAB treatment resulted in the recruitment of ubiquitin to the ER, as shown by a more widespread perinuclear localization and reticular pattern (Figure 3B). In addition, SubAB treatment resulted in the formation of occasional rod opsin–negative inclusions that were identified by ubiquitin staining (Figure 3B). This is similar to the ubiquitylation of endogenous proteins observed in cells expressing a truncated version of Cnx, short Cnx (Kosmaoglou and Cheetham, 2008). Therefore the depletion of BiP potentially stimulates the ubquitylation of both WT and mutant rod opsin at the ER (Figure 3, A and B).
To confirm that WT rod opsin was ubiquitylated in response to SubAB treatment, we immunoprecipitated WT rod opsin and Western blotted with antibodies to ubiquitin (Figure 3C). Despite lower levels of rod opsin after SubAB treatment, there was a clear enrichment of a high–molecular weight smear of ubiquitin immunoreactivity immunoprecipitated with 1D4 from the SubAB-treated cells, confirming that ubiquitin was associated with rod opsin, probably as a result of rod opsin ubiquitylation.
Reduced mobility of WT-GFP in the ER after SubAB treatment
The enhanced ER retention and ubiquitylation of rod opsin suggested that there might be enhanced aggregation in the absence of BiP. To test this hypothesis, we used fluorescence recovery after photobleaching (FRAP) to compare the diffusional mobility of WT and P23H rod opsin in ER membranes. FRAP has been used extensively to study protein mobility, and indirectly protein aggregation, within the ER—for example, with a temperature-sensitive mutant of vesicular stomatitis virus G (VSVG) (Nehls et al., 2000) and, more recently, P23H rod opsin in Xenopus rod outer segments (Haeri and Knox, 2012). The presence of WT-GFP on the plasma membrane meant that the bleaching had to be confined to the ER, and therefore a 2-μm square of the ER was selected and photobleached and recovery monitored (Supplemental Figure S5). Fluorescence recovered to between 70 and 80% of prebleach levels within 1 min of photobleaching of WT-GFP in control untreated cells. The recovery for WT-GFP was rapid and did not appear to involve gross changes in the fine architecture of the ER. By contrast, P23H-GFP only recovered 50–55% fluorescence in 1 min, confirming that the mutant rod opsin was less mobile within the ER, corresponding to its well-documented misfolding. When WT-GFP was treated with SubAB for either 2 or 18 h there was a drastic reduction of fluorescence recovery; only 50% of the fluorescence was recovered within the same time frame, similar to the untreated P23H rod opsin and much lower than untreated WT-GFP (Figure 4).
FIGURE 4:
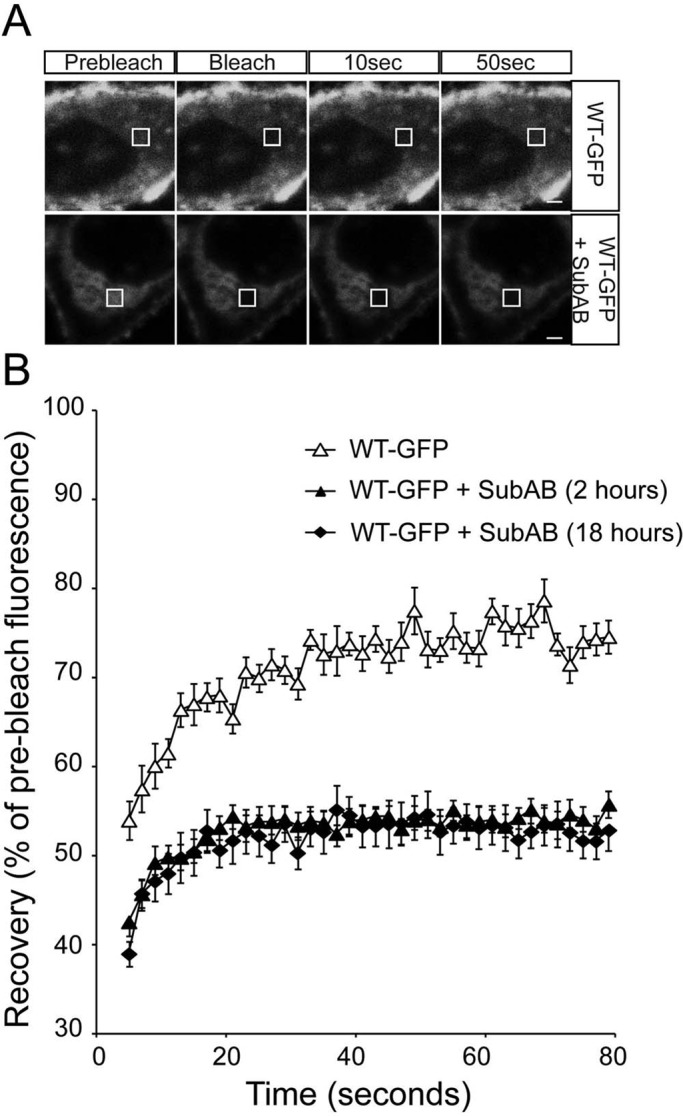
ER-localized WT rod opsin mobility is reduced by SubAB treatment. (A) Representative images of live-cell FRAP analysis of control WT-GFP and in the presence of 1.5 μg/ml SubAB for 18 h, as indicated. A 2 × 2 μm area of the ER corresponding to the boxed area was photobleached, and the cells were imaged every 2 s for up to 80 s; images are shown prebleach, immediately postbleach, and at 10- and 50-s recovery. Scale bars, 2 μm. (B) Graphical representation of recovery after photobleach for WT-GFP (open triangles) and WT-GFP in the presence of SubAB for 2 h (closed triangles) or 18 h (closed diamonds), as indicated. Fluorescence intensities for the 2 × 2 μm area were normalized to prebleach levels at 100%; error bars, SE; n ≥ 12.
It was possible that these changes could be mediated by gross changes in ER morphology or the aggregation of other ER proteins. Therefore we used several fluorescently tagged control proteins to monitor the general properties of the ER: yellow fluorescent protein (YFP)–HSJ1b(274–324), GFP-Sec61β, and signal sequence-GFP-KDEL. HSJ1b is targeted to the cytoplasmic face of the ER by geranyl-geranylation (Chapple and Cheetham, 2003). We used the C-terminal 50 amino acids of HSJ1b fused to YFP (YFP-HSJ1b(274–324)) to monitor the effects of SubAB on ER membranes independent of any direct association with BiP. This fusion protein contains only the isoprenylation motif and ER targeting sequences of HSJ1b and not the cochaperone or client binding domains of the chaperone (J. P. C., S. S. N., and M. E. C., unpublished data). YFP-HSJ1b(274–324) was targeted to the ER of cells; high levels of expression led to a strong effect on cell morphology resembling organized smooth ER–like stacked ER cisternae (OSER) (Snapp et al., 2003), and therefore only low-expressing cells were imaged (Supplemental Figure S6). After photobleaching, YFP-HSJ1b(274–324) fluorescence recovered rapidly to >90% of the prebleach level and higher than the recovery of WT-GFP (Supplemental Figure S5). Of importance, unlike WT-GFP, SubAB treatment had no effect on YFP-HSJ1b(274–324) recovery (Supplemental Figure S6). Sec61β is a small ER transmembrane protein that is part of the ER translocon, and GFP-Sec61β is targeted to the ER membrane (Voeltz et al., 2006). GFP-Sec61β recovered to ∼85% of prebleach level and was not affected by SubAB treatment (Supplemental Figure S7). To control for changes in the ER lumen, we used GFP-KDEL, a fusion of the ER targeting sequence of mouse calreticulin (17 amino acids) with EGFP and an ER retention signal peptide, KDEL, at its C terminus (Bannai et al., 2004). This soluble lumenal form of GFP was highly mobile and could diffuse rapidly in the ER lumen (Dayel et al., 1999); it did not bleach effectively under the conditions used for rod opsin. Therefore for this fusion protein we measured the number of full-intensity bleach iterations required to bleach 40% of the prebleach fluorescence as an estimate of mobility. This revealed that SubAB did not significantly slow the mobility of GFP-KDEL (Supplemental Figure S8). Collectively, these control data show that SubAB does not alter the mobility of components of the three ER compartments in which rod opsin is present (cytoplasmic face, transmembrane, and lumen), which suggests the effects on rod opsin are specific. Therefore BiP may have a role in maintaining the solubility of WT rod opsin in ER membranes.
An ATPase mutant of BiP reduces WT rod opsin mobility in the ER
To confirm that the changes in rod opsin biogenesis after SubAB treatment correlated specifically with a reduction of BiP function and not another unknown effect of SubAB activity, we investigated the effect of an ATPase-deficient form of BiP, BiP(T37G). Like all Hsp70 proteins, BiP is an ATPase that undergoes cycles of client protein binding and release dependent on nucleotide hydrolysis and exchange. The T37G substitution in BiP severely inhibits its ATPase activity. This mutant BiP can still bind immunoglobulin heavy chains, but it cannot release the heavy chains on the addition of ATP (Gaut and Hendershot, 1993). WT-GFP with overexpressed wild-type BiP trafficked to the plasma membrane and recovered fluorescence as before and the same as without BiP overexpression (Figure 5). In the presence of overexpressed BiP(T37G), however, WT-GFP was retained within the ER similar to SubAB treatment (Figure 5A). Furthermore, recovery of fluorescence after photobleaching was dramatically reduced in the presence of BiP(T37G) (Figure 5B). Of importance, BiP(T37G) had no effect on the recovery of YFP-HSJ1b(274–324) (Supplemental Figure S9), GFP-Sec61β (Supplemental Figure S10), or GFP-KDEL (Supplemental Figure S8), supporting the hypothesis that these effects are specific to BiP client proteins.
FIGURE 5:
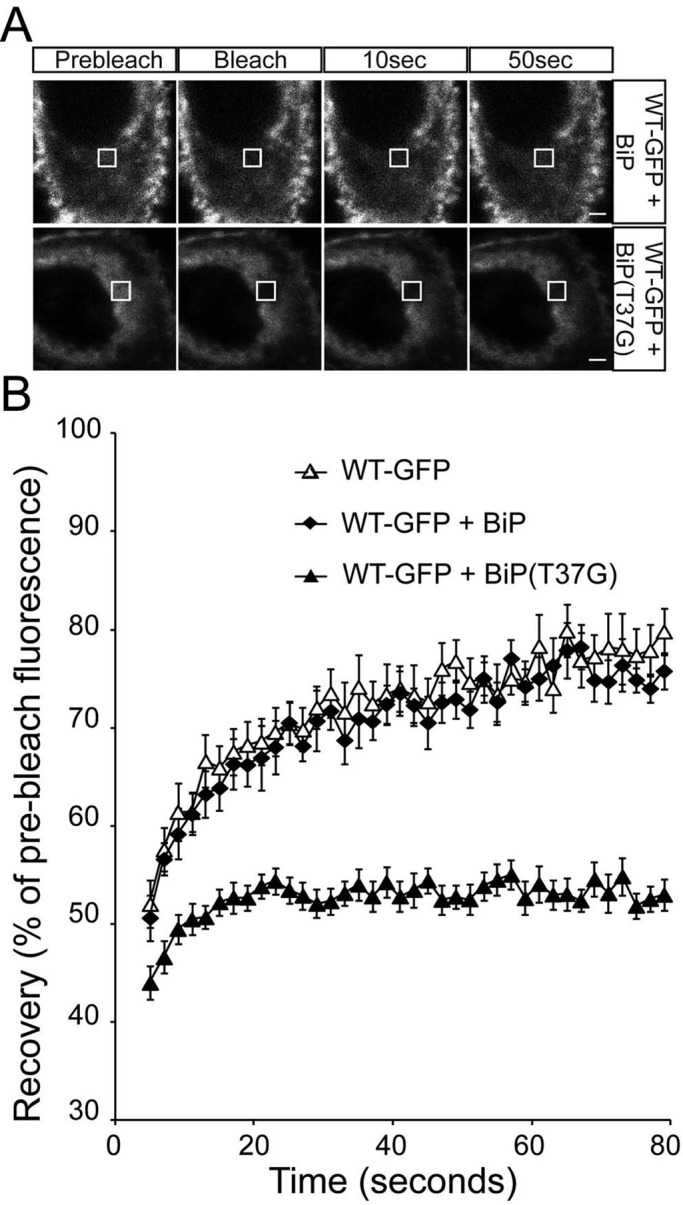
ER-localized WT rod opsin has reduced mobility in the presence of BiP(T37G). (A) Representative images of live-cell FRAP analysis of WT-GFP cotransfected with BiP or BiP(T37G), as indicated. A 2 × 2 μm area of the ER corresponding to the boxed area was photobleached, and the cells were imaged every 2 s for up to 80 s; images are shown prebleach, immediately postbleach, and at 10- and 50-s recovery. Scale bars,2 μm. (B) Graphical representation of recovery after photobleach for WT-GFP alone (open triangles) and transfected with WT-BiP (closed diamonds) and BiP(T37G) (closed triangles). Fluorescence intensities for the 2 × 2 μm area were normalized to prebleach levels at 100%. Error bars, SE; n ≥ 12.
The effect of BiP depletion and overexpression on P23H rod opsin
BiP depletion with SubAB did not affect P23H rod opsin localization in the ER but did appear to alter ubiquitylation status (Figure 3B), suggesting potential changes in P23H within the ER. Therefore we examined P23H-GFP mobility using FRAP. SubAB treatment reduced the already low level of P23H-GFP recovery further, from 55 to 45% (Figure 6), suggesting further aggregation of P23H-GFP within the ER upon BiP depletion. Cotransfection with the BiP “trap” mutant BiP(T37G) also led to a reduction in the P23H-GFP mobile fraction (Figure 7). Of importance, overexpression of control BiP improved the mobility of P23H-GFP, and recovery was restored close to the level of WT-GFP (Figure 7). There was no change in P23H localization with BiP overexpression. Therefore, although there was an improvement of P23H rod opsin mobility within the ER, the protein was still not capable of folding sufficiently close to the native state to exit the ER and traffic to the plasma membrane.
FIGURE 6:
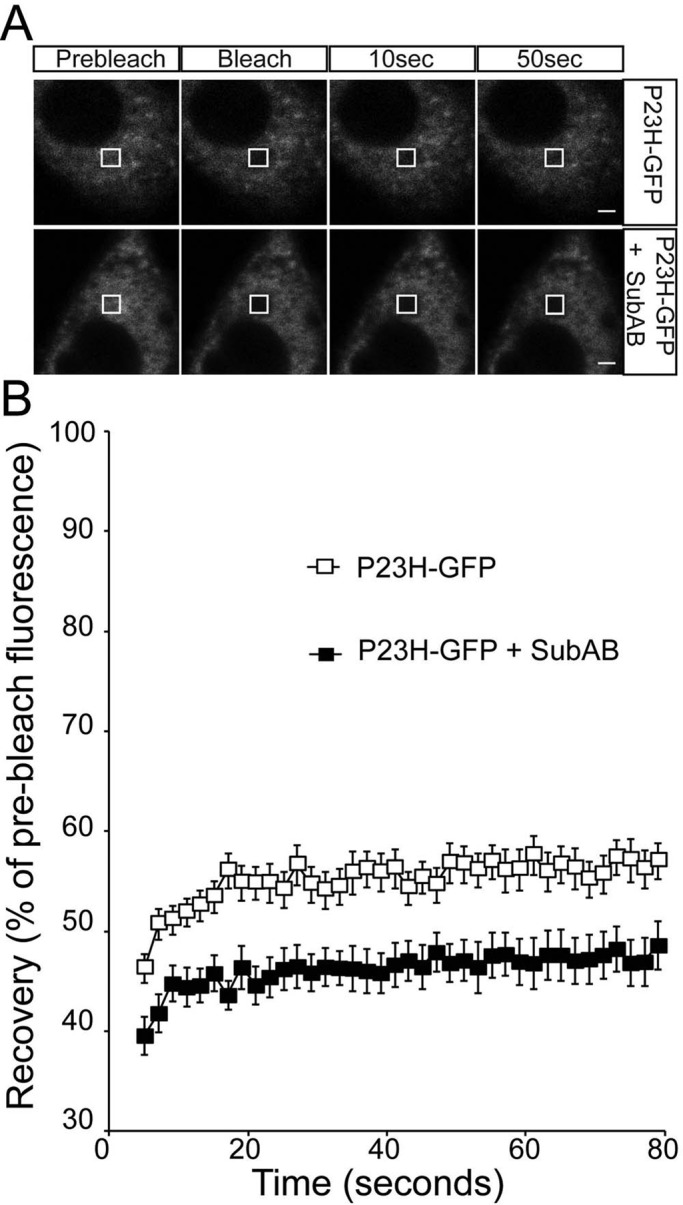
ER-localized P23H rod opsin mobility is reduced by SubAB treatment. (A) Representative images of live-cell FRAP analysis of control P23H-GFP and in the presence of 1.5 μg/ml SubAB for 18 h, as indicated. A 2 × 2 μm area of the ER corresponding to the boxed area was photobleached, and the cells were imaged every 2 s for up to 80 s; images are shown prebleach, immediately postbleach, and at 10- and 50-s recovery. Scale bars, 2 μm. (B) Graphical representation of recovery after photobleach for P23H-GFP (open squares) and P23H-GFP in the presence of SubAB for 18 h (closed squares), as indicated. Fluorescence intensities for the 2 × 2 μm area were normalized to prebleach levels at 100%. Error bars, SE; n ≥ 12.
FIGURE 7:
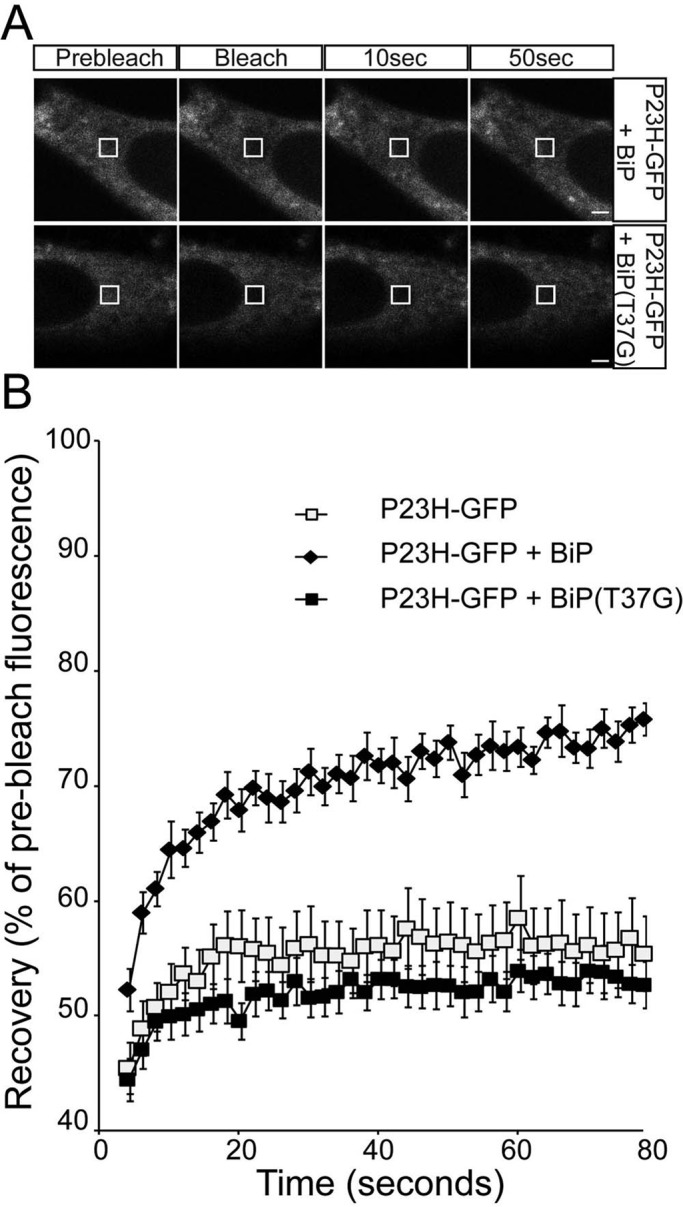
ER-localized P23H rod opsin has increased mobility with BiP overexpression. (A) Representative images of live-cell FRAP analysis of P23H-GFP cotransfected with BiP or BiP(T37G), as indicated. A 2 × 2 μm area of the ER corresponding to the boxed area was photobleached, and the cells were imaged every 2 s for up to 80 s; images are shown prebleach, immediately postbleach, and at 10- and 50-s recovery. Scale bars, 2 μm. (B) Graphical representation of recovery after photobleach for P23H-GFP alone (open squares) and transfected with WT-BiP (closed diamonds) and BiP(T37G) (closed squares). Fluorescence intensities for the 2 × 2 μm area were normalized to prebleach levels at 100%. Error bars, SE; n ≥ 12.
DISCUSSION
The data presented here suggest that BiP is involved in the normal processing of rod opsin and can affect mutant rod opsin mobility. Rod opsin is the archetypal seven-transmembrane G protein–coupled receptor, and this might suggest a role for BiP in the biogenesis of other GPCRs. Here we used the subtilase toxin SubAB as a tool to reduce BiP activity. The cleavage of BiP to <10% of its original level in cells treated with SubAB is a powerful way to preferentially and rapidly reduce expression of this highly abundant ER chaperone. BiP is a component of the cellular translocation machinery. In assisting nascent polypeptides during translocation, 10% functional BiP appears to be sufficient in maintaining efficiency and sealing of the translocon pore, as extensive cell death was not observed over 18–24 h of SubAB treatment. The translocation of rod opsin did not appear to be affected by BiP depletion, as some WT protein trafficked to the plasma membrane, indicative of passage through the secretory pathway. In addition, the colocalization of both WT and P23H rod opsin with the ER marker Cnx indicated functional ER import. Although absence of BiP in yeast (Kar2p) results in the inactivation of the translocation machinery (Vogel et al., 1990), this does not appear to be the case in mammalian cells, in which lumenal components were not required for protein translocation (Bulleid and Freedman, 1988). In addition, an in vitro system for monitoring the targeting and translocation of rod opsin revealed that ATP was dispensable for targeting and translocation, whereas GTP was essential (Kanner et al., 2003).
Because there did not appear to be a defect in rod opsin translocation after BiP depletion, its role as a chaperone assisting in the proper folding and maturation of rod opsin was investigated. SubAB-mediated depletion caused a stalling in the biogenesis of rod opsin in the ER. In the absence of functional BiP, the WT rod opsin protein might attempt to fold unaided, resulting in its aggregation via exposed hydrophobic sequences in the lumenal or transmembrane domains. It is not clear which domains in rod opsin BiP binds, although we believe that BiP is most likely acting via the rod opsin intradiscal loops (which correspond to the extracellular or ER lumenal domains). This interaction is probably transient, as the binding efficiency is low—for example, much lower than that of another interactor, EDEM1 (Kosmaoglou et al., 2009)—but serves to prevent these domains from aggregating before forming a near-native structure of the intradiscal “plug” that brings the transmembrane helices together.
In addition, BiP could maintain WT rod opsin in an intermediate conformation until further chaperones and folding factors are recruited to the complex to complete folding. Of interest, upon BiP depletion the WT rod opsin appears to misfold to such a degree that ubiquitin is recruited to the ER with high efficiency. Previous data showed that P23H rod opsin is ubiquitylated in inclusions (Saliba et al., 2002). However, BiP depletion also resulted in enhanced ubiquitylation at the ER of P23H rod opsin–expressing cells. ER-retained P23H rod opsin is not usually observed to recruit ubiquitin to the ER, suggesting that any ubiquitylated protein is removed from the ER and degraded as part of ERAD. This suggests that without functional BiP, rod opsin cannot fold correctly and also cannot be efficiently dislocated into the cytosol for proteasomal degradation. Ubiquitin ligases found on the cytoplasmic face of the ER are likely to tag the misfolded cytoplasmic domains of rod opsin for degradation, and this may become more pronounced if the dislocation machinery is compromised by the loss of BiP. The mobility of P23H rod opsin, which is already misfolded, ER retained, and coimmunoprecipitates with BiP (unpublished data), was reduced further on SubAB treatment, and this might reflect a requirement for BiP to maintain misfolded rod opsin in a dislocation-competent state. The failure to dislocate could be a consequence of rod opsin aggregation in the ER without BiP activity, as an aggregated transmembrane protein might not be able to pass back across the ER membrane.
Other chaperones are likely to be involved in WT rod opsin biogenesis in the ER and mutant rod opsin quality control—for example, EDEM1 and VCP (Kosmaoglou et al., 2009; Griciuc et al., 2010). The depletion of BiP and subsequent activation of the UPR will up-regulate these factors, and yet their up-regulation could not rescue the enhanced misfolding and aggregation caused by loss of BiP activity. It is unlikely that the increased expression of any of these factors led to the retention of WT rod opsin in the ER, as treatment with tunicamycin, which is a potent inducer of the UPR, does not affect the biogenesis and traffic of WT rod opsin (Kaushal et al., 1994; Saliba et al., 2002). Furthermore, the effects on WT rod opsin mobility were observed within 2 h of SubAB treatment. The overexpression of an ATPase mutant of BiP also resulted in a loss of rod opsin mobility and traffic from the ER, supporting a direct role for BiP in rod opsin biogenesis.
A recent report showed that BiP overexpression can reduce photoreceptor cell death in the P23H rod opsin rat model of ADRP. This neuroprotection was suggested to be mediated by a prosurvival mechanism related to reduced ER stress and not a profolding effect on the mutant protein (Gorbatyuk et al., 2010). Similarly, we observed that BiP overexpression did not enhance P23H rod opsin traffic in SK-N-SH cells, but it did improve the mobility of P23H rod opsin. Therefore, whereas BiP is required to maintain rod opsin in a folding- and/or dislocation-competent state, it cannot promote correct folding of rod opsin. BiP overexpression may enable an enhanced or sustained ability to mediate mutant rod opsin ERAD that could reduce ER stress and enhance photoreceptor survival (Gorbatyuk et al., 2010). Determining the other factors that can promote rod opsin folding and/or degradation will be important for targeted therapies for rhodopsin ADRP.
MATERIALS AND METHODS
Materials
The SubAB toxin and SubAA272B were purified from recombinant E. coli as hexahistidine-tagged fusion proteins by nickel-nitriloacetic acid chromatography, as described previously (Paton et al., 2004; Talbot et al., 2005). Primary antibodies 1D4 and 4D2 against rod opsin were a gift from Robert Molday (Department of Biochemistry and Molecular Biology, University of British Columbia, Vancouver, Canada). Antibodies to Cnx (C4731) and BiP (G8918 and SAB4501452) were from Sigma-Aldrich (Poole, United Kingdom). Lipofectamine and Plus reagent were purchased from Invitrogen (Paisley, United Kingdom). Protease Inhibitor Cocktail in dimethyl sulfoxide for mammalian cell extracts was purchased from Sigma-Aldrich. The bicinchoninic acid protein assay kit was from Pierce (Thermo Scientific, Cramlington, United Kingdom), and the LDH assay kit was purchased from Roche (Basel, Switzerland). Rod opsin constructs, untagged rod opsin in pMT3 and rod opsin-GFP, were as described previously (Saliba et al., 2002). WT-BiP and BiP(T37G) expression plasmids were a gift from Linda Hendershot (St Jude's Children's Research Hospital, Memphis, TN). YFP-HSJ1b(274–324) was created by introducing a BamHI site, in-frame with YFP, 150 base pairs from the end of the HSJ1b open reading frame (Chapple and Cheetham, 2003) before cloning into pEYFP-C3 (Clontech, Mountain View, CA). The GFP-KDEL plasmid was a gift from Hiroko Bannai (BSI, Riken, Japan), and the pAc-GFPC1-Sec61β plasmid was purchased from Addgene (Cambridge, MA).
Cell seeding, transfection, and immunocytochemistry
SK-N-SH cells were maintained, transfected, and imaged essentially as described previously (Kosmaoglou et al., 2009). Briefly, 24 h after transfection, cells were fixed in 4% paraformaldehyde (PFA) and were either not permeabilized or permeabilized with Triton X-100 (0.5%) in phosphate-buffered saline (PBS; 137 mM sodium chloride, 2.7 mM potassium chloride, 8.1 mM di-sodium hydrogen phosphate, 1.5 mM potassium dihydrogen phosphate; OXOID, Basingstoke, Hampshire, United Kingdom). The slides were washed twice with PBS and blocked for 1 h with PBS containing 3% (wt/vol) bovine serum albumin and 10% (vol/vol) normal donkey serum. Anti-myc (Sigma-Aldrich) antibody (1:1000), anti-Cnx primary antibody (1:600; Sigma-Aldrich), or 4D2 (1:100) primary antibody in blocking buffer was added for 1 h. The slides were then washed twice in PBS, and donkey anti-mouse Cy3 (Jackson ImmunoResearch Europe, Newmarket, United Kingdom) was used at 1:100 in blocking buffer for 1 h. Cells were washed twice with PBS and once with 4′,6-diamidino-2-phenylindole (Sigma-Aldrich) at a concentration of 2 μg/ml in PBS before mounting in fluorescent mounting medium (Dako, Glostrup, Denmark). Fluorescence was observed on a Zeiss LSM 510 or LSM 700 confocal laser-scanning microscope (Carl Zeiss, Jena, Germany) for image acquisition. Images were exported from the LSM browser to Adobe Photoshop (Adobe, San Jose, CA) for figure preparation and annotation in Adobe Illustrator.
Western blotting, immunoprecipitation, and pulse-chase analysis
For Western blotting of BiP, SK-N-SH cell monolayers were washed twice in Hank's balanced salt solution (HBSS) on ice and were lysed for at least 15 min at 4°C in 1% (wt/vol) Triton X-100 in PBS. Lysis buffer was supplemented with 2% (vol/vol) mammalian protease inhibitor cocktail (Sigma-Aldrich). Cell lysates were centrifuged at 14,000 rpm in a microcentrifuge for 15 min at 4ºC. A volume of 5× modified Laemmli sample buffer was added to the soluble fraction to a final concentration of 1× (Kosmaoglou et al., 2009).
Immunoprecipitation of rod opsin or BiP from SK-N-SH cells or porcine retinas was essentially as described (Kosmaoglou et al., 2009). Cells or retinas were lysed in 1% (wt/vol) DM lysis buffer (1% [wt/vol] n-dodecyl-β-d-maltoside [DM; Sigma-Aldrich] in PBS) with 2% (vol/vol) mammalian protease inhibitor cocktail and 2% (vol/vol) phosphatase inhibitors (Sigma-Aldrich). The lysate was precleared and incubated with mouse 1D4 antibody to the C-terminus of rod opsin or with BiP antibody overnight at 4ºC using an end-over-end rotor. The next morning, protein G–Sepharose beads (GE Healthcare, Buckinghamshire, United Kingdom) or Dynabeads (Invitrogen) were washed 3× with 1 ml of DM lysis buffer, followed by centrifugation at 2000 rpm for 2 min in a microfuge, and after each wash the supernatant was removed and discarded. After the last wash the beads were resuspended to make a 50% slurry. A 50-μl amount of protein G–Sepharose or 40 μl of protein G Dynabeads was added to each sample containing lysate and antibody for a period up to 3 h at 4 ºC using an end-over-end rotor. At the end of the incubation the bead-antibody-protein complexes were washed 5× in 1 ml of DM lysis buffer, and the supernatant was discarded after each wash. For the “ATP wash,” 1% DM in PBS with 5 mM ATP and 2 mM MgCl2 was used. The complexes were eluted from the beads with 60 μl of 5× modified Laemmli sample buffer. Samples for rod opsin immunoblotting were not heated. Samples for BiP immunoblotting were heated at 95ºC for 5 min.
Pulse-chase analysis was performed as described previously (Kosmaoglou et al., 2009). The lysates were then precleared for 2 h with prewashed protein G–Sepharose beads. At the end of the preclearing, lysates were centrifuged at 14,000 rpm in a microcentrifuge for 2 min, and the supernatants were removed and placed in clean tubes for incubation with 1D4 mAb to rod opsin for overnight incubation at 4ºC without rotation. For the deglycosylation reactions 15 μg of total protein in DM soluble cell lysate and 5× Laemmli sample treatment buffer was digested with EndoH (New England BioLabs, Hitchin, United Kingdom). Digestions were carried out overnight at 37ºC before resolving by SDS–PAGE.
In-cell Western analysis
In-cell Western analysis was essentially as described previously (Kosmaoglou et al., 2009; Meimaridou et al., 2011). Briefly, SK-N-SH cells were transfected with WT rod opsin and fixed in 3% PFA prepared in HBSS for 15 min. Nonspecific binding was blocked using Odyssey Blocking Buffer (LI-COR, Cambridge, United Kingdom). For total rod opsin levels, cells were permeabilized with 0.025% Triton X-100. Rod opsin immunoreactivity was detected with 4D2 (1:100) and secondary antibody (1:10,000; LI-COR). The cells were scanned using an Odyssey Imager (LI-COR). The percentage of total rod opsin reaching the plasma membrane was determined by dividing the nonpermeabilized signal by the total, and the values were normalized to untreated levels to compensate for differences in level of expression. Data were processed in Excel (Microsoft, Redmond, WA), and statistical analysis was performed using a two-tailed paired Student's t test.
Lactate dehydrogenase cell death assay
LDH assays were performed essentially as described (Mendes and Cheetham, 2008). Briefly, SK-N-SH cells were seeded on a 96-well dish at a density of 5 × 103 cells/well. Twenty-four hours later cells were transfected with GFP-tagged rod opsin or GFP or left untransfected. After 2-h recovery the cells were treated with 1.5 μg/ml SubAB for 2, 18, and 44 h. The LDH assay was performed following the manufacturer's instructions (Roche). The 96-well plate was centrifuged at 250 × g for 10 min, and 100 μl/well of supernatant was removed and transferred to a new 96-well plate. The reaction mixture, composed of 250 μl of diaphorase/NAD+ and 11.25 ml of dye solution containing iodotetrazolium chloride and sodium lactate, was freshly prepared, and 100 μl/well was added to the supernatant. After 30 min of light-protected incubation at 22°C, absorbance was measured at 492 nm using a Safire microplate reader (Tecan, Männedorf, Switzerland). Data were processed in Excel, and statistical analysis was performed using a two-tailed paired Student's t test.
FRAP analysis
FRAP was performed essentially as described (Meimaridou et al., 2011). SK-N-SH cells were seeded on MatTek slide dishes (MatTek, Ashland, MA) at a density of 600,000 cells/dish and after 24 h were transfected with 500 ng of either WT or P23H rod opsin-GFP and 500 ng of empty vector, using 8 μl of Plus and 4 μl of Lipofectamine (Invitrogen), according to the manufacturer's instructions. DMEM-F12 containing 20% fetal bovine serum was added at the end of the 3-h period. After a recovery time of 2 h the cells were treated with 1.5 μg/ml SubAB for 2 or 18 h as indicated. SubAB was readministered after the 18-h period to maintain the cleavage of BiP during the FRAP image acquisition. FRAP analyses were performed at 37ºC on the temperature-controlled stage of a Zeiss LSM510 confocal microscope. Low-expressing, large-inclusion-free cells were selected and quickly Z-scanned to identify the middle of each cell. Bleaching of a defined region of interest (ROI) within the ER of an individual cell (indicated by boxes in Figures 4–7) was performed using a 488-nm laser line at 100% power and transmission over five iterations. Image acquisition was performed by scanning the entire field at <4% laser power, and gain and offset were used to maximize the dynamic range within the ROI without saturation. Recovery was monitored over 40 cycles of imaging with a 2-s interval between each acquisition. Fluorescence intensity in regions of interest was quantified using Zeiss Zen software. The intensity values were normalized to the average of the two prebleach values obtained. The normalized values were used to construct the graphs in Figures 4–7 and Supplemental Figures S5–S7, S9, and S10 using Excel. For the KDEL-GFP experiment, repeated bleaching of the defined ROI within the ER of an individual cell was performed using a 488-nm laser line at 100% power and transmission in groups of 10 iterations until ROI fluorescence dropped to 40%. The average number of iterations needed to bleach to 40% was used to construct the graph in Supplemental Figure S8. Images were exported from LSM browser to Adobe Photoshop for figure preparation and annotation in Adobe Illustrator.
Supplementary Material
Acknowledgments
We thank Linda Hendershot for the gift of WT-BiP and BiP(T37G) expression plasmids, Hiroko Bannai for the GFP-KDEL plasmid, and Robert Molday for the 1D4 and 4D2 rhodopsin antibodies. This work was supported by Fight for Sight and the Wellcome Trust. D.A. and M.K. are Fight for Sight Prize students.
Abbreviations used:
- ADRP
autosomal dominant retinitis pigmentosa
- Cnx
calnexin
- DM
n-dodecyl-β-d-maltoside
- ER
endoplasmic reticulum
- ERAD
endoplasmic reticulum–associated degradation
- GFP
green fluorescent protein
- GPCR
G protein–coupled receptor
- UPR
unfolded protein response
- YFP
yellow fluorescent protein
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-02-0168) on August 1, 2012.
*These authors contributed equally.
REFERENCES
- Baker EK, Colley NJ, Zuker CS. The cyclophilin homolog NinaA functions as a chaperone, forming a stable complex in vivo with its protein target rhodopsin. EMBO J. 1994;13:4886–4895. doi: 10.1002/j.1460-2075.1994.tb06816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannai H, Inoue T, Nakayama T, Hattori M, Mikoshiba K. Kinesin dependent, rapid, bi-directional transport of ER sub-compartment in dendrites of hippocampal neurons. J Cell Sci. 2004;117:163–175. doi: 10.1242/jcs.00854. [DOI] [PubMed] [Google Scholar]
- Bulleid NJ, Freedman RB. Defective co-translational formation of disulphide bonds in protein disulphide-isomerase-deficient microsomes. Nature. 1988;335:649–651. doi: 10.1038/335649a0. [DOI] [PubMed] [Google Scholar]
- Chapple JP, Cheetham ME. The chaperone environment at the cytoplasmic face of the endoplasmic reticulum can modulate rhodopsin processing and inclusion formation. J Biol Chem. 2003;278:19087–19094. doi: 10.1074/jbc.M212349200. [DOI] [PubMed] [Google Scholar]
- Dayel MJ, Hom EF, Verkman AS. Diffusion of green fluorescent protein in the aqueous-phase lumen of endoplasmic reticulum. Biophys J. 1999;76:2843–2851. doi: 10.1016/S0006-3495(99)77438-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryja TP, McGee TL, Hahn LB, Cowley GS, Olsson JE, Reichel E, Sandberg MA, Berson EL. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N Engl J Med. 1990;323:1302–1307. doi: 10.1056/NEJM199011083231903. [DOI] [PubMed] [Google Scholar]
- Gaut JR, Hendershot LM. Mutations within the nucleotide binding site of immunoglobulin-binding protein inhibit ATPase activity and interfere with release of immunoglobulin heavy chain. J Biol Chem. 1993;268:7248–7255. [PubMed] [Google Scholar]
- Gorbatyuk MS, Knox T, LaVail MM, Gorbatyuk OS, Noorwez SM, Hauswirth WW, Lin JH, Muzyczka N, Lewin AS. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci USA. 2010;107:5961–5966. doi: 10.1073/pnas.0911991107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griciuc A, Aron L, Piccoli G, Ueffing M. Clearance of rhodopsin(P23H) aggregates requires the ERAD effector VCP. Biochim Biophys Acta. 2010;1803:424–434. doi: 10.1016/j.bbamcr.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Haas IG, Wabl M. Immunoglobulin heavy chain binding protein. Nature. 1983;306:387–389. doi: 10.1038/306387a0. [DOI] [PubMed] [Google Scholar]
- Haeri M, Knox BE. Rhodopsin mutant P23H destabilizes rod photoreceptor disk membranes. PLoS One. 2012;7:e30101. doi: 10.1371/journal.pone.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illing ME, Rajan RS, Bence NF, Kopito RR. A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J Biol Chem. 2002;277:34150–34160. doi: 10.1074/jbc.M204955200. [DOI] [PubMed] [Google Scholar]
- Kabani M, Kelley SS, Morrow MW, Montgomery DL, Sivendran R, Rose MD, Gierasch LM, Brodsky JL. Dependence of endoplasmic reticulum-associated degradation on the peptide binding domain and concentration of BiP. Mol Biol Cell. 2003;14:3437–3448. doi: 10.1091/mbc.E02-12-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanner EM, Friedlander M, Simon SM. Co-translational targeting and translocation of the amino terminus of opsin across the endoplasmic membrane requires GTP but not ATP. J Biol Chem. 2003;278:7920–7926. doi: 10.1074/jbc.M207462200. [DOI] [PubMed] [Google Scholar]
- Kaushal S, Khorana HG. Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry. 1994;33:6121–6128. doi: 10.1021/bi00186a011. [DOI] [PubMed] [Google Scholar]
- Kaushal S, Ridge KD, Khorana HG. Structure and function in rhodopsin: the role of asparagine-linked glycosylation. Proc Natl Acad Sci USA. 1994;91:4024–4028. doi: 10.1073/pnas.91.9.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmaoglou M, Cheetham ME. Calnexin is not essential for mammalian rod opsin biogenesis. Mol Vis. 2008;14:2466–2474. [PMC free article] [PubMed] [Google Scholar]
- Kosmaoglou M, Kanuga N, Aguila M, Garriga P, Cheetham ME. A dual role for EDEM1 in the processing of rod opsin. J Cell Sci. 2009;122:4465–4472. doi: 10.1242/jcs.055228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus A, et al. Calnexin deficiency leads to dysmyelination. J Biol Chem. 2010;285:18928–18938. doi: 10.1074/jbc.M110.107201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lass A, Kujawa M, McConnell E, Paton AW, Paton JC, Wojcik C. Decreased ER-associated degradation of alpha-TCR induced by Grp78 depletion with the SubAB cytotoxin. Int J Biochem Cell Biol. 2008;40:2865–2879. doi: 10.1016/j.biocel.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Mao C, Lee B, Lee AS. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol. 2006;26:5688–5697. doi: 10.1128/MCB.00779-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meimaridou E, Gooljar SB, Ramnarace N, Anthonypillai L, Clark AJ, Chapple JP. The cytosolic chaperone Hsc70 promotes traffic to the cell surface of intracellular retained melanocortin-4 receptor mutants. Mol Endocrinol. 2011;25:1650–1660. doi: 10.1210/me.2011-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes HF, Cheetham ME. Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum Mol Genet. 2008;17:3043–3054. doi: 10.1093/hmg/ddn202. [DOI] [PubMed] [Google Scholar]
- Mendes HF, van der Spuy J, Chapple JP, Cheetham ME. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med. 2005;11:177–185. doi: 10.1016/j.molmed.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Mimura N, et al. Aberrant quality control in the endoplasmic reticulum impairs the biosynthesis of pulmonary surfactant in mice expressing mutant BiP. Cell Death Differ. 2007;14:1475–1485. doi: 10.1038/sj.cdd.4402151. [DOI] [PubMed] [Google Scholar]
- Molinari M, Galli C, Piccaluga V, Pieren M, Paganetti P. Sequential assistance of molecular chaperones and transient formation of covalent complexes during protein degradation from the ER. J Cell Biol. 2002;158:247–257. doi: 10.1083/jcb.200204122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehls S, et al. Dynamics and retention of misfolded proteins in native ER membranes. Nat Cell Biol. 2000;2:288–295. doi: 10.1038/35010558. [DOI] [PubMed] [Google Scholar]
- Noorwez SM, Sama RR, Kaushal S. Calnexin improves the folding efficiency of mutant rhodopsin in the presence of pharmacological chaperone 11-cis-retinal. J Biol Chem. 2009;284:33333–33342. doi: 10.1074/jbc.M109.043364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K. Chemistry and biology of vision. J Biol Chem. 2012;287:1612–1619. doi: 10.1074/jbc.R111.301150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton AW, Beddoe T, Thorpe CM, Whisstock JC, Wilce MC, Rossjohn J, Talbot UM, Paton JC. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature. 2006;443:548–552. doi: 10.1038/nature05124. [DOI] [PubMed] [Google Scholar]
- Paton AW, Srimanote P, Talbot UM, Wang H, Paton JC. A new family of potent AB(5) cytotoxins produced by Shiga toxigenic Escherichia coli. J Exp Med. 2004;200:35–46. doi: 10.1084/jem.20040392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincus D, Chevalier MW, Aragon T, van Anken E, Vidal SE, El-Samad H, Walter P. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol. 2010;8:e1000415. doi: 10.1371/journal.pbio.1000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig A, Gilbert HF. Anti-chaperone behavior of BiP during the protein disulfide isomerase-catalyzed refolding of reduced denatured lysozyme. J Biol Chem. 1994;269:25889–25896. [PubMed] [Google Scholar]
- Rosenbaum EE, Hardie RC, Colley NJ. Calnexin is essential for rhodopsin maturation, Ca2+ regulation, photoreceptor cell survival. Neuron. 2006;49:229–241. doi: 10.1016/j.neuron.2005.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba RS, Munro PM, Luthert PJ, Cheetham ME. The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J Cell Sci. 2002;115:2907–2918. doi: 10.1242/jcs.115.14.2907. [DOI] [PubMed] [Google Scholar]
- Snapp EL, Hegde RS, Francolini M, Lombardo F, Colombo S, Pedrazzini E, Borgese N, Lippincott-Schwartz J. Formation of stacked ER cisternae by low affinity protein interactions. J Cell Biol. 2003;163:257–269. doi: 10.1083/jcb.200306020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorgjerd K, Ghafouri B, Jonsson BH, Kelly JW, Blond SY, Hammarstrom P. Retention of misfolded mutant transthyretin by the chaperone BiP/GRP78 mitigates amyloidogenesis. J Mol Biol. 2006;356:469–482. doi: 10.1016/j.jmb.2005.11.051. [DOI] [PubMed] [Google Scholar]
- Sung CH, Davenport CM, Nathans J. Rhodopsin mutations responsible for autosomal dominant retinitis pigmentosa. Clustering of functional classes along the polypeptide chain. J Biol Chem. 1993;268:26645–26649. [PubMed] [Google Scholar]
- Sung CH, Schneider BG, Agarwal N, Papermaster DS, Nathans J. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 1991;88:8840–8844. doi: 10.1073/pnas.88.19.8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot UM, Paton JC, Paton AW. Protective immunization of mice with an active-site mutant of subtilase cytotoxin of Shiga toxin-producing Escherichia coli. Infect Immun. 2005;73:4432–4436. doi: 10.1128/IAI.73.7.4432-4436.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124:573–586. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- Vogel JP, Misra LM, Rose MD. Loss of BiP/GRP78 function blocks translocation of secretory proteins in yeast. J Cell Biol. 1990;110:1885–1895. doi: 10.1083/jcb.110.6.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson JJ, May KL, Thorpe CM, Jandhyala DM, Paton JC, Paton AW. Subtilase cytotoxin activates PERK, IRE1 and ATF6 endoplasmic reticulum stress-signalling pathways. Cell Microbiol. 2008;10:1775–1786. doi: 10.1111/j.1462-5822.2008.01164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.