

A GPCR ubiquitin-independent MVB/lysosomal sorting pathway is regulated by the adaptor protein complex-3 (AP-3) and ALIX, a noncanonical ESCRT component. AP-3 binds to a PAR1 C-tail–localized, tyrosine-based motif and mediates PAR1 lysosomal degradation. AP-3 also facilitates PAR1 interaction with ALIX, suggesting that AP-3 functions before PAR1 engagement of ALIX and MVB/lysosomal sorting.
Abstract
The sorting of signaling receptors within the endocytic system is important for appropriate cellular responses. After activation, receptors are trafficked to early endosomes and either recycled or sorted to lysosomes and degraded. Most receptors trafficked to lysosomes are modified with ubiquitin and recruited into an endosomal subdomain enriched in hepatocyte growth factor–regulated tyrosine kinase substrate (HRS), a ubiquitin-binding component of the endosomal-sorting complex required for transport (ESCRT) machinery, and then sorted into intraluminal vesicles (ILVs) of multivesicular bodies (MVBs)/lysosomes. However, not all receptors use ubiquitin or the canonical ESCRT machinery to sort to MVBs/lysosomes. This is exemplified by protease-activated receptor-1 (PAR1), a G protein–coupled receptor for thrombin, which sorts to lysosomes independent of ubiquitination and HRS. We recently showed that the adaptor protein ALIX binds to PAR1, recruits ESCRT-III, and mediates receptor sorting to ILVs of MVBs. However, the mechanism that initiates PAR1 sorting at the early endosome is not known. We now report that the adaptor protein complex-3 (AP-3) regulates PAR1 ubiquitin-independent sorting to MVBs through an ALIX-dependent pathway. AP-3 binds to a PAR1 cytoplasmic tail–localized tyrosine-based motif and mediates PAR1 lysosomal degradation independent of ubiquitination. Moreover, AP-3 facilitates PAR1 interaction with ALIX, suggesting that AP-3 functions before PAR1 engagement of ALIX and MVB/lysosomal sorting.
INTRODUCTION
The most abundant class of signaling receptors expressed in the mammalian genome is that of G protein–coupled receptors (GPCRs), which mediate cellular responses to numerous physiological stimuli. GPCRs signal predominantly to heterotrimeric G proteins at the plasma membrane and can also signal from subcellular compartments. Once activated, GPCRs are rapidly desensitized, internalized, and then sorted within the endocytic system and recycled or degraded in lysosomes. The trafficking of GPCRs through the endocytic pathway is critical for controlling the temporal and spatial fidelity of signaling responses. GPCR trafficking facilitates receptor resensitization, signaling through scaffolds on endosomes and signal termination via degradation in lysosomes (Marchese et al., 2008). Moreover, dysregulated GPCR trafficking results in aberrant signaling that contributes to various pathological conditions, including cancer progression (Booden et al., 2004; Li et al., 2004). The precise mechanisms that control trafficking of GPCRs within the endocytic system have not been clearly defined.
After internalization, vesicles containing GPCRs fuse with early endosomes, an endocytic compartment that functions mainly to sort cargo. Internalized GPCRs are then either sorted to tubular sorting endosomes and recycled or sequestered in early endosomes and sorted to lysosomes for degradation. The sorting of most receptors from endosomes to lysosomes occurs via interaction with endosomal-sorting complex required for transport (ESCRT) proteins. The ESCRT machinery comprises four distinct ESCRT complexes that function sequentially to coordinate the sorting of ubiquitinated receptors into intraluminal vesicles (ILVs) of multivesicular bodies (MVBs; Hurley and Hanson, 2010). ESCRT-dependent sorting of ubiquitinated receptors is initiated at an early endosomal subdomain enriched in hepatocyte growth factor–regulated tyrosine kinase substrate (HRS) protein, an ESCRT-0 component, and clathrin (Raiborg et al., 2002). HRS interacts directly with ubiquitinated cargo and ESCRT-I, which also binds to ubiquitin and ESCRT-II. The ESCRT-III complex is then recruited, assembles on endosomal membranes, and facilitates ILV scission. Vps4, an AAA-ATPase, disassembles and recycles ESCRT-III at the MVB and is a process essential for ESCRT function.
Many mammalian GPCRs are modified with ubiquitin and sorted to lysosomes for degradation through the ESCRT pathway. This is best characterized for the G protein–coupled chemokine CXCR4 receptor and protease-activated receptor-2 (PAR2; Marchese et al., 2003; Hasdemir et al., 2007; Malerod et al., 2007; Dores et al., 2012). However, not all GPCRs require direct ubiquitination or all ubiquitin-binding components of the ESCRT machinery for lysosomal sorting. A ubiquitination-deficient mutant of the delta-opioid receptor (DOR) and the calcitonin-like (CL) receptor—a GPCR that is not ubiquitinated—traffic to MVBs/lysosomes like wild-type receptor (Hislop et al., 2004; Cottrell et al., 2007; Henry et al., 2011). Despite not being directly ubiquitinated, DOR and CL receptors sort to lysosomes through an HRS-dependent pathway, suggesting that sorting of receptors is initiated at the HRS-enriched subdomain of the early endosome and mediated by the canonical ESCRT machinery. In contrast to DOR and CL receptor, we previously showed that PAR1, a GPCR for thrombin, sorts to ILVs of MVBs independent of ubiquitin and ubiquitin-binding ESCRT components HRS and Tsg101 (Gullapalli et al., 2006; Wolfe et al., 2007; Dores et al., 2012). We also discovered that the adaptor protein ALIX binds directly to PAR1 via an YPX3L motif, recruits ESCRT-III, and mediates receptor sorting to ILVs of MVBs independent of ubiquitination (Dores et al., 2012). Thus PAR1 bypasses the requirement for ubiquitin and ubiquitin-binding ESCRT components for sorting from endosomes to MVBs/lysosomes. However, the mechanism by which sorting of PAR1 is initiated at the early endosome remains unclear.
The adaptor protein complex-3 (AP-3) is enriched at the early sorting endosome and localizes to tubulovesicular budding profiles (Dell'Angelica et al., 1998; Peden et al., 2004; Theos et al., 2005). In contrast, HRS localizes to a distinct bilayered subdomain of early endosomes that is devoid of AP-3 (Sachse et al., 2002). AP-3 is a heterotetrameric complex comprising β3, δ, μ3, and σ3 adaptin subunits and functions in sorting of lysosomal-associated membrane proteins (LAMPs) from endosomes to lysosomes that accumulate at the limiting membrane of MVBs/lysosomes, as well as CD63, a protein that localizes to ILVs of MVBs (Dell'Angelica et al., 1999; Rous et al., 2002). The β3 adaptin subunit of AP-3 contains a consensus clathrin-binding site, directly interacts with clathrin, and colocalizes with clathrin on endosomal membranes (Dell'Angelica et al., 1998; Peden et al., 2004), whereas the μ3 subunit of AP-3 interacts with tyrosine-based YXXØ motifs, where Y is the critical tyrosine, X is any amino acid, and Ø is a bulky hydrophobic residue, present in the cytoplasmic regions of cargo proteins and mediates lysosomal sorting (Dell'Angelica et al., 1999; Rous et al., 2002). Thus AP-3 regulates sorting of proteins to ILVs of MVBs and lysosomes, but whether the AP-3–enriched subdomain of early endosomes is used by signaling receptors to access ILVs of MVBs is not known.
The adaptor protein ALIX binds to PAR1 and ESCRT-III to facilitate receptor sorting to ILVs of MVBs independent of ubiquitination (Dores et al., 2012). However, the mechanism responsible for initiating ubiquitin-independent MVB/lysosomal sorting of PAR1 at the early endosome remains unknown, since HRS is not essential for PAR1 lysosomal sorting (Gullapalli et al., 2006). Given these observations and the presence of tyrosine sorting motifs in the cytoplasmic tail (C-tail) of PAR1, we examined the function of AP-3. In the present study, we demonstrate that AP-3 binds to a PAR1 C-tail–localized, tyrosine-based motif and regulates lysosomal degradation of the receptor independent of ubiquitination. We further show that AP-3 is required for PAR1 association with ALIX and that a mutant PAR1 defective in AP-3 binding fails to sort to intraluminal membranes—compartments protected from proteinase K. Thus AP-3 regulates PAR1 endosomal sorting before interaction with ALIX and ILV sorting at the MVB.
RESULTS
AP-3 association with activated PAR1
To assess the role of AP-3 in intracellular trafficking of PAR1, we first examined whether the receptor colocalized with endogenous AP-3. HeLa cells stably expressing an N-terminal FLAG-tagged PAR1 were incubated with anti-FLAG antibody at 4°C. Under these conditions only cell surface receptors are labeled with antibody. Cells were then left untreated (0 min) or treated with the agonist peptide SFLLRN for 10 min at 37°C, processed, and immunostained using the SA4 anti–δ-adaptin antibody as described (Berger et al., 2007) and imaged by confocal microscopy. In untreated control cells, PAR1 localized mainly to the cell surface (Figure 1A), whereas endogenous AP-3 appeared as punctae distributed throughout the cytoplasm, consistent with previously reported studies (Peden et al., 2004; Berger et al., 2007). After activation, PAR1 internalized to endocytic vesicles that colocalized with endogenous AP-3 (Figure 1A). The colocalization of PAR1 and AP-3 was verified by determining Pearson's r (Figure 1A). To further confirm the presence of PAR1 and AP-3 on endosomes, we examined colocalization in cells expressing a constitutively active Rab5 Q79L mutant, which perturbs proper transport and fusion of endocytic vesicles, resulting in enlarged endosomes (Stenmark et al., 1994). In unstimulated cells, PAR1 was present mainly on the plasma membrane, whereas endogenous AP-3 localized to the limiting membrane of enlarged endosomes induced by coexpression of the Rab5 Q79L mutant (Figure 1B). After agonist stimulation for 10 min, PAR1 redistributed to enlarged endosomes and exhibited considerable colocalization with AP-3 (Figure 1B). These findings suggest that activated PAR1 sorts to an AP-3–positive endosomal compartment.
FIGURE 1:

PAR1 colocalization and coassociation with AP-3. (A) HeLa cells expressing N-terminal FLAG-tagged PAR1 were incubated with anti-FLAG antibody for 1 h at 4°C to label cell surface receptors and then stimulated for 10 min with 100 μM SFLLRN at 37°C. Cells were fixed, permeabilized, and immunostained for PAR1 (red) and endogenous δ-adaptin (green) using mouse SA4 anti–δ-adaptin antibody and imaged by confocal microscopy. Scale bar, 10 μm. Inset, magnification of the boxed area. The colocalization of PAR1 with AP-3 appears as the yellow color in the merged image and was quantitated by determining Pearson's r from six different cells. The difference in colocalization between untreated and agonist-stimulated PAR1 colocalization with AP-3 was significant (***p < 0.001; n = 6) as determined by Student's t test. (B) HeLa cells expressing FLAG-PAR1 were transiently transfected with Rab5 Q79L mutant tagged with GFP. After transfection, cells were treated and processed as described to detect PAR1 and endogenous AP-3 expression. Arrowheads indicate enlarged Rab5 Q79L–positive endosomes. (C) HeLa cells expressing FLAG-tagged PAR1 were left untreated (0 min) or treated with 100 μM SFLLRN for 10, 20, or 40 min at 37°C. Cells were lysed, and equivalent amounts of cell lysates were used for immunoprecipitation with M2 anti-FLAG antibody or IgG. Immunoprecipitates were resolved by SDS–PAGE and immunoblotted with anti–δ-adaptin antibody to detect endogenous AP-3. Membranes were stripped and reprobed with anti-PAR1 antibody. The asterisk indicates a nonspecific band. Endogenous δ-adaptin expression in total cell lysates was examined as a control. The data (mean ± SD) represent the amount of coprecipitated δ-adaptin normalized to the amount of immunoprecipitated PAR1 at various times of agonist stimulation and was significant, as determined by Student's t test (**p < 0.01; n = 3). These data are representative of at least three independent experiments.
We next determined whether activated PAR1 associated with AP-3 in HeLa cells by coimmunoprecipitation. Cells expressing PAR1 were incubated with agonist for various times at 37°C, lysates were immunoprecipitated, and the amount of endogenous AP-3 associated with the receptor was determined by immunoblotting for the δ-adaptin subunit and quantitated. In contrast to untreated control cells, a marked, approximately threefold increase in endogenous AP-3 associated with PAR1 was observed after 10 min of agonist stimulation, and the association appeared to diminish at 20 min (Figure 1C, lanes 3–5). After 40 min of agonist exposure PAR1 coimmunoprecipitation with endogenous AP-3 was considerably reduced, consistent with substantial degradation of activated PAR1 observed at this time (Figure 1C, lane 6). However, neither PAR1 nor AP-3 was detected in immunoprecipitates from immunoglobulin G (IgG) control (Figure 1C, lanes 1 and 2). These results indicate that activated and internalized PAR1 associates with AP-3 and transits through an AP-3–positive endosomal compartment.
PAR1 lysosomal degradation is regulated by AP-3
To assess AP-3 function in PAR1 lysosomal degradation, we used small interfering RNAs (siRNAs) that target the δ-adaptin subunit of AP-3 to deplete HeLa cells of the endogenous AP-3 complex. The loss of δ-adaptin subunit expression results in degradation of other adaptin subunits and disruption of AP-3 function (Peden et al., 2002). The expression of δ-adaptin was virtually abolished in δ-adaptin siRNA–transfected cells compared to nonspecific siRNA–transfected control cells (Figure 2A). In control siRNA–transfected cells, exposure to agonist for 60 min caused an ∼50% decrease in PAR1 protein, and after 90 min an ∼70% loss of receptor protein was detected (Figure 2A, lanes 1–3). These findings are consistent with the extent of agonist-induced PAR1 degradation typically observed in HeLa cells (Dores et al., 2012). In contrast, activated PAR1 degradation was markedly attenuated in cells lacking endogenous AP-3 (Figure 2A, lanes 4–6); only ∼10–30% loss of receptor protein was observed after incubation with agonist for 60 or 90 min, respectively. These data suggest that AP-3 regulates lysosomal degradation of PAR1.
FIGURE 2:
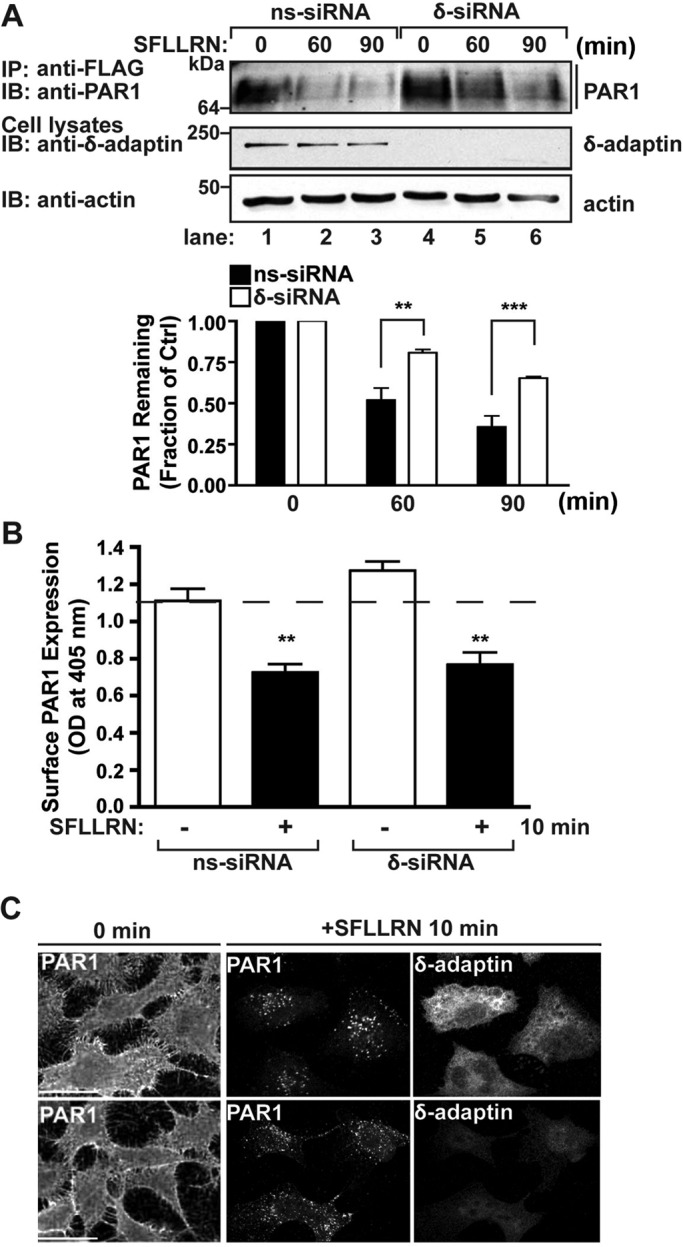
AP-3 regulates agonist-induced PAR1 degradation but not receptor internalization. (A) HeLa cells expressing FLAG-tagged PAR1 were transiently transfected with 50 nM nonspecific (ns) or δ-adaptin–specific siRNAs. Cells were pretreated with 10 μM cycloheximide for 90 min to block de novo protein synthesis, left unstimulated (0 min) or stimulated with 100 μM SFLLRN for 60 or 90 min at 37°C. Equivalent amount of cell lysates were immunoprecipitated with M2 anti-FLAG antibody and examined by immunoblotting with anti-PAR1 antibody. Cell lysates were analyzed for endogenous δ-adaptin and actin as controls. PAR1 degradation was quantitated, and the data (mean ± SD) shown are expressed as the fraction of PAR1 remaining compared with untreated control cells determined at various times from three independent experiments. The differences in PAR1 remaining in control cells vs. δ-adaptin siRNA–treated cells were significant (**p < 0.01, n = 3; and ***p < 0.001, n = 3) as determined by two-way ANOVA. (B) HeLa cells expressing FLAG-tagged PAR1 were transfected with 50 nM ns or δ-adaptin siRNAs. Cells were pretreated with cycloheximide as described, stimulated with 100 μM SFLLRN for 10 min, and fixed, and the amount of PAR1 remaining on the cell surface was quantitated by ELISA. The data shown (mean ± SD) are representative of three separate experiments. In both ns and δ-adaptin siRNA–treated cells, agonist stimulated a significant loss of PAR1 from the cell surface (** p < 0.01; n = 3) as determined by Student's t test. (C) HeLa cells expressing FLAG-tagged PAR1 were transiently transfected with 50 nM ns or δ-adaptin siRNAs. Cell surface PAR1 was prelabeled with anti-PAR1 antibody for 1 h at 4°C, washed, and either left untreated (0 min) or treated with 100 μM SFLLRN for 10 min at 37°C. Cells were fixed, permeabilized, and immunostained for PAR1 or δ-adaptin and imaged by confocal microscopy. Scale bar, 10 μm. The cells shown are representative of many cells examined in multiple independent experiments.
We next examined whether impaired PAR1 degradation observed in AP-3–depleted cells resulted from defects in receptor internalization. PAR1-expressing HeLa cells were incubated in the absence or presence of agonist peptide and fixed, and the amount of receptor remaining on the cell surface was quantitated by enzyme-linked immunosorbent assay (ELISA). In the absence of agonist stimulation, HeLa cells transiently transfected with δ-adaptin–specific siRNAs displayed a modest increase in PAR1 cell surface expression compared with nonspecific siRNA–transfected control cells (Figure 2B). However, agonist induced a substantial and comparable amount of PAR1 internalization in nonspecific and δ-adaptin siRNA–transfected cells (Figure 2B); an ∼40% loss of surface receptor was detected after 10 min of agonist incubation in both conditions. Immunofluorescence microscopy studies also revealed robust internalization of PAR1 after agonist stimulation in control and δ-adaptin–depleted cells (Figure 2C). Taken together, these results indicate that internalization of activated PAR1 is independent of AP-3.
AP-3 regulates GPCR lysosomal degradation independent of ubiquitination
CXCR4 and PAR2 are GPCRs that are directly modified with ubiquitin and use the canonical ESCRT machinery for lysosomal sorting (Marchese et al., 2003; Hasdemir et al., 2007; Malerod et al., 2007; Dores et al., 2012). To examine whether the AP-3–regulated lysosomal degradation pathway of PAR1 is distinct from ubiquitin-dependent GPCR lysosomal sorting, we examined the function of AP-3 in agonist-stimulated degradation of CXCR4 and PAR2. HeLa cells transfected with nonspecific or δ-adaptin–specific siRNAs were incubated with stromal-derived factor-1α (SDF-1α)—the cognate ligand for CXCR4—and the amount of endogenous CXCR4 protein remaining was determined by immunoblotting. Incubation with SDF-1α for 180 min caused a significant ∼50% decrease in CXCR4 protein in control siRNA–transfected cells (Figure 3A). SDF-1α–promoted CXCR4 degradation observed in δ-adaptin–depleted cells was comparable (Figure 3A), indicating that agonist-promoted CXCR4 degradation occurs independent of AP-3. HeLa cells stably expressing FLAG-PAR2 transfected with nonspecific and δ-adaptin–specific siRNAs were stimulated with agonist, and receptor degradation was assessed. Similar to CXCR4, the extent of agonist-promoted PAR2 degradation was comparable in control and δ-adaptin–deficient cells (Figure 3B), suggesting that AP-3 is not required for ubiquitin-dependent GPCR lysosomal degradation.
FIGURE 3:

AP-3 regulates GPCR lysosomal degradation independent of ubiquitination. (A) HeLa cells expressing endogenous CXCR4 were transiently transfected with 50 nM nonspecific (ns) or δ-adaptin siRNAs. Cells were then treated in the absence (–) or presence (+) of 10 nM SDF-1α for 180 min at 37°C. Cell lysates were prepared, and the amount of endogenous CXCR4 remaining was determined by immunoblot and quantitated. The data (mean ± SD) show the extent of CXCR4 degradation and are expressed as the fraction of CXCR4 remaining compared with untreated control cells; they are representative of three independent experiments. (B) HeLa cells expressing FLAG-PAR2 were transfected with 50 nM ns or δ-adaptin siRNAs. Cells were then left unstimulated (–) or stimulated (+) with 100 μM SLIGKV for 120 min, and the amount of PAR2 remaining was determined by immunoblot and quantified. The data (mean ± SD) represent the fraction of PAR2 remaining compared with untreated control cells from three independent experiments. (C) HeLa cells expressing ubiquitination-deficient FLAG-PAR1 0K mutant were transfected with 50 nM ns or δ-adaptin siRNAs. Cells were stimulated with 100 μM SFLLRN for the indicated times, and equivalent amounts of lysates were immunoprecipitated with M2 anti-FLAG antibody. The amount of PAR1 0K remaining was determined by immunoblot and quantified. The data (mean ± SD) represent the fraction of PAR1 0K mutant remaining compared with untreated control cells and are significant (**p < 0.01; n = 3) as determined by two-way ANOVA. Cell lysates were immunoblotted with tubulin, δ-adaptin, or actin antibodies as controls.
To confirm that AP-3 mediates activated PAR1 lysosomal degradation independent of ubiquitination, we examined the capacity of AP-3 to regulate the degradation of a ubiquitination-deficient PAR1 mutant. We previously showed that the PAR1 0K mutant, which contains lysine-to-arginine mutations within the cytoplasmic domains, signals normally but is defective in ubiquitination (Wolfe et al., 2007; Chen et al., 2011). In addition, agonist stimulation of PAR1 0K mutant results in lysosomal sorting and degradation similar to wild-type receptor (Wolfe et al., 2007; Dores et al., 2012). In nonspecific siRNA–transfected control cells, a significant ∼50% decrease in PAR1 0K mutant protein was observed after 60 and 90 min of agonist incubation (Figure 3C, lanes 1–3), consistent with ubiquitin-independent lysosomal sorting of PAR1 (Dores et al., 2012). However, agonist-promoted degradation of PAR1 0K mutant was substantially reduced in cells lacking endogenous AP-3 (Figure 3C, lanes 4–6); only ∼25% loss of receptor protein was observed after prolonged agonist stimulation. These data suggest that AP-3 regulates GPCR lysosomal sorting independent of receptor ubiquitination.
A PAR1 C-tail, tyrosine-based motif mediates AP-3 binding and lysosomal degradation
The μ3 subunit of AP-3 binds directly to tyrosine-based motifs localized within the cytoplasmic domains of transmembrane proteins to facilitate lysosomal sorting (Bonifacino and Traub, 2003). The PAR1 C-tail domain contains two tyrosine-based sorting signals that conform to the YXXØ motif and could potentially mediate AP-3 interaction (Figure 4A). The interaction between μ3-adaptin subunit with tyrosine-based sequences is dependent on the critical tyrosine and a bulky hydrophobic residue at the Y+3 position (Bonifacino and Traub, 2003). Thus we first investigated whether PAR1 tyrosine-based motifs function in receptor lysosomal sorting by examining agonist-stimulated degradation of mutants in which the tyrosine (Y) and hydrophobic leucine (L) residues were mutated to alanines (A; Figure 4A). In HeLa cells stably expressing PAR1 wild type, a 60-min incubation with agonist caused ∼50% loss of receptor protein (Figure 4B, lanes 1 and 2). Agonist-stimulated degradation of the PAR1 AKKAA mutant, in which the critical residues of the tyrosine-based motif Y420KKL423L424 were mutated to A420KKA423A424, was comparable to that for wild-type receptor (Figure 4B, lanes 5 and 6). In contrast, agonist-promoted degradation of the PAR1 ASIA mutant, containing alanine substitutions at the critical tyrosine and leucine residues, was significantly impaired and displayed only a modest ∼23% decrease in receptor protein compared with wild-type receptor after 60 min of agonist stimulation (Figure 4B, lanes 3 and 4). These results indicate that the PAR1 C-tail–proximal tyrosine sorting signal, but not the distal tyrosine-based motif, is important for agonist-stimulated receptor lysosomal degradation.
FIGURE 4:

A PAR1 proximal tyrosine-based motif mediates AP-3 interaction. (A) A schematic of the PAR1 C-tail domain illustrating the proximal YSIL and distal YKKLL tyrosine sorting motifs shaded in gray. The critical residues are underlined. PAR1 tyrosine motif mutants ASIA and AKKAA are shown above the native sequence. (B) HeLa cells expressing comparable amounts of FLAG-tagged PAR1 wild type (WT) or ASIA or AKKAA mutants pretreated with 10 μM cycloheximide were either left unstimulated (–) or stimulated (+) with 100 μM SFLLRN for 60 min at 37°C. Cells were lysed, and PAR1 was immunoprecipitated from equivalent amounts of cell lysates using an M2 anti-FLAG antibody. The amount of PAR1 remaining was examined by immunoblot using an anti-PAR1 antibody and quantitated. The data (mean ± SD) shown are expressed as the fraction of PAR1 remaining compared with untreated control cells representative of three independent experiments. The difference in the amount of PAR1 WT vs. ASIA mutant detected after agonist stimulation was significantly different (***p < 0.001; n = 3) as determined by two-way ANOVA. (C) HeLa cells expressing FLAG-tagged PAR1 WT or ASIA mutant were left unstimulated (0 min) or stimulated with 100 μM SFLLRN for 10 or 20 min at 37°C. Equivalent amounts of cell lysates were immunoprecipitated with M2 anti-FLAG antibody or IgG. Immunoprecipitates were then analyzed for the presence of δ-adaptin subunit. Membranes were stripped and reprobed with antibody to detect PAR1. The amount of endogenous δ-adaptin in total cell lysates was detected by immunoblotting as a control.
We next determined whether PAR1 interacts with AP-3 via its proximal tyrosine-based motif by examining coassociation after agonist stimulation. HeLa cells expressing similar amounts of PAR1 wild type or ASIA mutant were incubated with agonist for 10 or 20 min, receptors were coimmunoprecipitated, and the association of endogenous AP-3 was assessed by immunoblotting. Activation of PAR1 wild type resulted in an increase in coimmunoprecipitated AP-3 that peaked at 10 min and appeared to diminish after 20 min (Figure 4C, lanes 1–3) compared with untreated control cells. In contrast to wild-type PAR1, AP-3 interaction with the PAR1 ASIA mutant was substantially reduced in both control and agonist-stimulated conditions (Figure 4C, lanes 4–6). In addition, neither PAR1 nor δ-adaptin was present in control IgG immunoprecipitates (Figure 4C, lanes 7 and 8). Thus the PAR1 C-tail–proximal, tyrosine-based motif serves as an AP-3–binding site and mediates receptor lysosomal degradation.
PAR1 ASIA mutant defective in AP-3 binding fails to sort to MVBs/lysosomes
To further delineate the mechanism by which disruption of AP-3 binding to the PAR1 proximal tyrosine motif affects intracellular trafficking, we examined PAR1 ASIA mutant sorting to early endosomes and lysosomes after agonist stimulation. Both PAR1 wild type and ASIA mutant localized to the cell surface in the absence of agonist (Figure 5, A and B). After 10 min of agonist stimulation, PAR1 wild type and ASIA mutant redistributed to endocytic vesicles and displayed marked colocalization with the early endosome antigen-1 (EEA1), a marker of early endosomes (Figure 5A). PAR1 colocalization with EEA1 was further evaluated by determining Pearson's r for PAR1 wild type (r = 0.4 ± 0.1, n = 6) and ASIA mutant (r = 0.43 ± 0.05, n = 6) after 10 min of agonist incubation. However, after 20 or 30 min of agonist stimulation, neither PAR1 wild type nor ASIA mutant remained in an EEA1-positive compartment (Figure 5A), indicating that the receptors traffic from the early endosomal compartment. Thus a PAR1 mutant with impaired AP-3 binding displays normal trafficking to early endosomes.
FIGURE 5:

A PAR1 mutant defective in AP-3 binding fails to sort to MVBs/lysosomes. (A) HeLa cells expressing PAR1 wild type (WT) or ASIA mutant were preincubated with anti-FLAG antibody at 4°C to ensure that only surface receptors were labeled. Cells were pretreated with 0.2 mM leupeptin and then either left unstimulated (0 min) or stimulated with 100 μM SFLLRN for the indicated times, fixed, permeabilized, and immunostained with anti-EEA1 antibody and processed for confocal microscopy. Insets, magnifications of boxed areas. Colocalization of PAR1 (red) and EEA1 (green) is indicated by the yellow color in the merged images and was quantitated by determining Pearson's r from six different cells from multiple independent experiments. The difference in PAR1 and EEA1 colocalization detected at 10 min was significant compared with 0 min control cells (***p < 0.001; n = 6) as determined by Student's t test. Scale bar, 10 μm. (B) PAR1 WT or ASIA mutant expressing HeLa cells pretreated with 0.2 mM leupeptin were labeled with anti-FLAG antibody, stimulated with 100 μM SFLLRN for 30 min, immunostained with anti–LAMP-1 and processed for confocal microscopy as described. Insets, magnifications of boxed areas. Colocalization of PAR1 (red) and LAMP-1 (green) is indicated by the yellow color in the merged images and quantitated by determining Pearson's r from six different cells taken from three separate experiments. The difference between PAR1 WT and ASIA with LAMP-1 at 30 min was significant (**p < 0.01; n = 6) as determined by Student's t test. Scale bar, 10 μm.
Next we examined PAR1 ASIA mutant trafficking to lysosomes by examining its colocalization with the lysosomal associated membrane protein-1 (LAMP-1), a protein that resides in late endosomes/lysosomes. As expected, 30 min of incubation with agonist resulted in substantial PAR1 wild-type colocalization with LAMP-1 (Figure 5B). This is consistent with rapid PAR1 transport to lysosomes and degradation (Dores et al., 2012). In contrast, however, activation of the PAR1 ASIA mutant with agonist for 30 min failed to promote substantial colocalization with LAMP-1 (Figure 5B), suggesting that PAR1 ASIA mutant is defective in lysosomal sorting. The difference in receptor colocalization with LAMP-1 was verified by determining Pearson's r for PAR1 wild type (r = 0.23 ± 0.02, n = 6) and ASIA mutant (r = 0.13 ± 0.02, n = 6). At face value, these data suggest that disruption of the PAR1 proximal tyrosine-based motif, an important AP-3–binding site, results in impaired sorting to lysosomes.
To further assess PAR1 ASIA mutant MVB/lysosomal sorting, we examined the capacity of the mutant receptor to remain at the limiting membrane of endosomes or to sort to an internal compartment by evaluating its sensitivity to proteinase K digestion after activation and internalization. Proteinase K protection assays have been used to examine sorting of the epidermal growth factor receptor and PAR1 to ILVs of MVBs (Sabatini and Blobel, 1970; Malerod et al., 2007; Dores et al., 2012). HeLa cells expressing PAR1 wild type or ASIA mutant were incubated in the absence or presence of agonist for 15 min, gently washed, and permeabilized with digitonin. The membrane fractions were isolated, divided into aliquots, and either left untreated or treated with proteinase K or proteinase K supplemented with Triton X-100 detergent. Activation of PAR1 wild type resulted in a significant accumulation of receptor in proteinase K–protected samples compared with cells not treated with agonist (Figure 6A, lanes 2 and 5). PAR1 accumulation was diminished in cells treated with Triton X-100, which allows proteinase K access to internal compartments (Figure 6A, lanes 5 and 6). These findings suggest that activated PAR1 wild type traffics to a protected endosomal compartment and is consistent with its sorting to ILVs of MVBs (Dores et al., 2012). In contrast to wild-type receptor, activated PAR1 ASIA mutant failed to accumulate in compartments resistant to proteinase K digestion (Figure 6A, lanes 8 and 11), suggesting that mutant receptor fails to traffic to protected endosomal compartments. Degradation of the peripheral endosomal–associated protein EEA1 was examined in parallel and was detected only in samples not incubated with proteinase K (Figure 6A), indicating that plasma membrane permeabilization and proteinase K activity remained intact under these conditions.
FIGURE 6:

PAR1 mutant defective in AP-3 binding fails to sort to intralumenal membranes. (A) HeLa cells expressing FLAG-PAR1 wild type (WT) or ASIA mutant were either left untreated (Ctrl) or treated with 100 μM SFLLRN (+SF) for 15 min at 37°C. Membranes were isolated, divided into three fractions, and were left untreated or treated with proteinase K or proteinase K supplemented with 0.1% Triton X-100 detergent. The samples were then immunoblotted for the presence of PAR1 or the peripheral endosomal protein EEA1, a control for plasma membrane permeabilization and proteinase K activity. The data (mean ± SD) are expressed as the fraction of PAR1 remaining after proteinase K treatment as compared with the untreated control (–) from either the Ctrl or +SF-treated samples from three independent experiments and were analyzed by Student's t test (**p < 0.01; n = 3). (B) HeLa cells expressing PAR1 WT or ASIA mutant transiently transfected with Rab5 Q79L mutant were pretreated with 0.2 mM leupeptin, stimulated with agonist, and processed as described. Insets, magnifications of boxed areas. Arrowheads indicate PAR1 WT within enlarged endosomes, and triangles indicate PAR1 ASIA at the limiting membrane of enlarged endosomes. The line scan analysis demonstrates fluorescence intensity within the lumen of the representative endosomes. Normalized fluorescence intensity represents the pixel intensity measured across the dashed gray line, where maximum intensity is set to 100. Normalized distance represents the diameter measured across the dashed gray line, where maxima representing the limiting membrane are labeled as 0 and 100 (white dashed lines).
We next examined PAR1 wild-type and ASIA mutant accumulation in intralumenal membranes of enlarged endosomes induced by Rab5 Q79L mutant coexpression to assess MVB sorting. In untreated control cells, PAR1 wild type and ASIA mutant localized to the cell surface, whereas Rab5 Q79L mutant fused to green fluorescent protein (GFP) was present predominantly at the limiting membrane of enlarged endosomes (Figure 6B). After 20 min of agonist stimulation, PAR1 wild type showed substantial accumulation within the lumen of Rab5 Q79L–positive endosomes (Figure 6B). In contrast, PAR1 ASIA mutant remained at the limiting membrane of enlarged endosomes induced by Rab5 Q79L mutant expression (Figure 6B). A line scan analysis through the center of representative endosomes also indicates that PAR1 wild type localizes to the lumen of Rab5 Q79L–positive endosomes, whereas PAR1 ASIA is restricted to the limiting membrane (Figure 6B). Taken together, these findings suggest that both the PAR1 C-tail–proximal, tyrosine-based motif and AP-3 are important for receptor sorting to MVBs/lysosomes.
AP-3 facilitates PAR1 interaction with ALIX
In recent work, we showed that PAR1 sorts to ILVs of MVBs via direct interaction with ALIX, an adaptor protein that links PAR1 to the ESCRT-III machinery and ILV/MVB sorting, independent of ubiquitination (Dores et al., 2012). To determine the mechanism by which AP-3 regulates PAR1 sorting to lysosomes, we first examined whether AP-3 was necessary for agonist-promoted PAR1 association with ALIX. HeLa cells stably expressing PAR1 were transfected with nonspecific or δ-adaptin–specific siRNAs and then incubated with agonist for various times. PAR1 was immunoprecipitated, and the association of endogenous ALIX was assessed. In control siRNA–transfected cells, agonist-induced a marked increase in PAR1 recruitment of endogenous ALIX at 10 min (Figure 7A, lanes 1 and 2). These findings are consistent with the kinetics of ALIX recruitment to activated PAR1 in HeLa cells as previously reported (Dores et al., 2012). Remarkably, however, in cells deficient in AP-3 expression, agonist activation of PAR1 failed to promote recruitment of endogenous ALIX (Figure 7A, lanes 3 and 4). These results suggest that AP-3 is important for mediating the interaction between PAR1 and ALIX.
FIGURE 7:
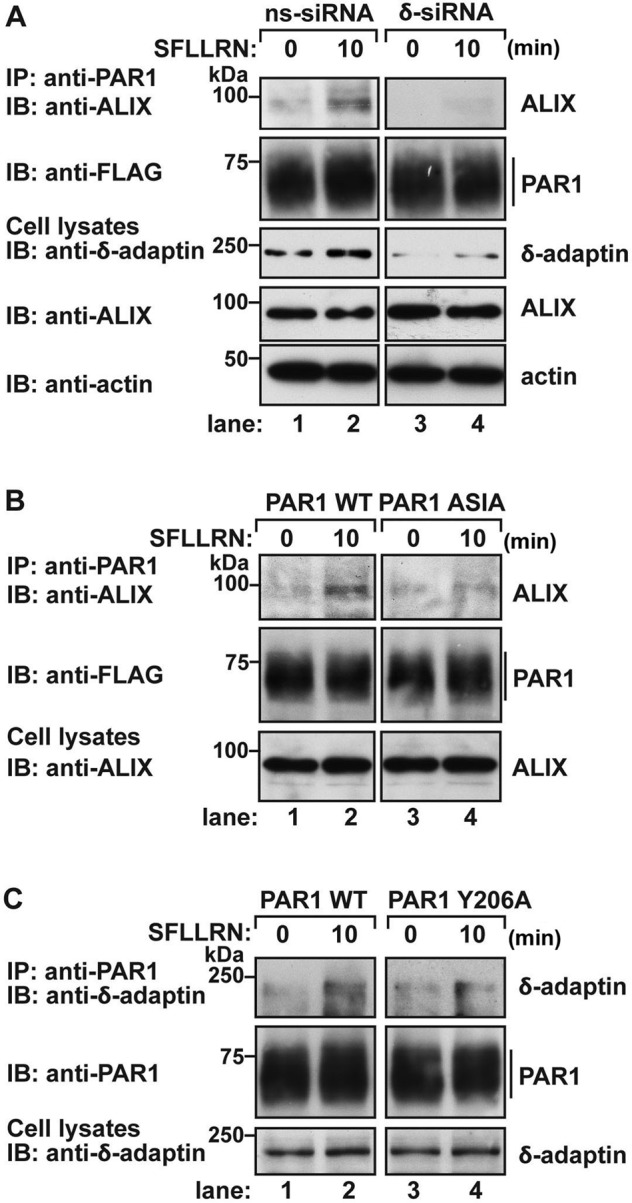
AP-3 mediates PAR1 interaction with ALIX. (A) HeLa cells expressing FLAG-PAR1 wild type (WT) were transfected with 50 nM nonspecific (ns) or δ-adaptin siRNAs. Cells were left unstimulated (0 min) or stimulated with 100 μM SFLLRN for 10 min, and equivalent amounts of cell lysates were immunoprecipitated with anti-PAR1 antibody. Immunoprecipitates were analyzed for the presence of endogenous ALIX and PAR1. The expression of δ-adaptin, ALIX, and actin in total cell lysates was examined by immunoblotting as controls. (B) HeLa cells expressing FLAG-PAR1 WT or ASIA mutant were left untreated (0 min) or treated with 100 μM SFLLRN for 10 min. Equivalent amounts of cell lysates were immunoprecipitated with anti-PAR1 antibody, and the coassociation of endogenous ALIX was assessed by immunoblot. Cell lysates were immunoblotted for ALIX expression as a control. (C) HeLa cells expressing FLAG-PAR1 WT or Y206A mutant were either unstimulated (0 min) or stimulated with 100 μM SFLLRN for 10 min. PAR1 was immunoprecipitated, and the coassociation of endogenous AP-3 was determined by immunoblotting using δ-adaptin antibody. The expression of δ-adaptin in total cell lysates was examined in parallel as a control.
To confirm these findings, we examined whether a PAR1 mutant defective in AP-3 binding retained the capacity to bind to ALIX or whether a mutant PAR1 with impaired ALIX binding interacted with AP-3. HeLa cells stably expressing PAR1 wild type or ASIA mutant were stimulated with agonist for 10 min; PAR1 was immunoprecipitated, and association with ALIX was assessed. Compared to wild-type PAR1, the PAR1 ASIA mutant with impaired AP-3 binding showed minimal coassociation with endogenous ALIX after agonist stimulation (Figure 7B, lanes 2 and 4). These results indicate that disruption of PAR1 interaction with AP-3 prevents binding to ALIX, an adaptor protein important for sorting PAR1 to ILVs of MVBs. To determine whether PAR1 binding to AP-3 is dependent on receptor interaction with ALIX, we examined whether AP-3 associated with the PAR1 Y206A mutant, a receptor variant that cannot bind ALIX (Dores et al., 2012). HeLa cells expressing PAR1 wild type and Y206A mutant were stimulated with agonist and immunoprecipitated with anti-PAR1 antibody, and AP-3 association was examined. Similar to wild-type PAR1, agonist induced a marked increase in AP-3 interaction with PAR1 Y206A mutant after 10 min of agonist stimulation (Figure 7C, lanes 2 and 4). These data indicate that activated PAR1 associates with AP-3 before interaction with ALIX and sorting to ILVs of MVBs.
PAR1 and AP-3 localization is not disrupted by Vps4 E228Q mutant
To determine whether AP-3 localization is dependent on ESCRT function, we examined PAR1 and AP-3 subcellular distribution in cells expressing the Vps4 E228Q mutant. Vps4 is an AAA-ATPase that catalyzes ESCRT-III disassembly and recycling and is essential for ESCRT function (Hurley and Hanson, 2010). HeLa cells expressing PAR1 or PAR2 and either Vps4 wild type or E228Q mutant fused to GFP were incubated with anti-FLAG antibody at 4°C to label the cell surface cohort, stimulated with agonist, and immunostained with SA4 anti–δ-adaptin antibodies. Expression of Vps4 wild type resulted in diffuse distribution throughout the cytoplasm, whereas Vps4 E228Q mutant accumulated in vacuolar-like structures (Figure 8). In the absence of agonist, PAR1 localized to the plasma membrane in Vps4 wild type– and E228Q mutant–expressing cells (Figure 8A). After activation, PAR1 relocalized to endocytic vesicles of normal morphology in Vps4 wild type–expressing cells and showed minimal accumulation in Vps4 E228Q mutant–positive vacuoles (Figure 8A), consistent with previously reported results (Dores et al., 2012). In contrast, activated PAR2 internalized and accumulated predominantly in Vps4 E228Q–positive vacuoles (Figure 8B), consistent with disruption of ubiquitin-dependent MVB sorting of PAR2 (Dores et al., 2012). Of interest, in both Vps4 wild type– and E228Q mutant–expressing cells, AP-3–positive puncta maintained a similar morphology and distribution (Figure 8). Thus the canonical ESCRT machinery does not appear to critically regulate PAR1 or AP-3 subcellular localization as compared with PAR2, a cargo that requires ubiquitin for lysosomal degradation. Taken together, these findings suggest a function for AP-3 in regulation of PAR1 lysosomal sorting that appears distinct from the canonical ubiquitin- and ESCRT-dependent sorting pathway (Figure 9).
FIGURE 8:

PAR1 and AP-3 subcellular localization in Vps4-expressing cells. HeLa cells stably expressing FLAG-PAR1 (A) or FLAG-PAR2 (B) were transiently transfected with Vps4-GFP or Vps4 E228Q-GFP. Cells were incubated with 0.2 mM leupeptin and then incubated with anti-FLAG antibody at 4ºC to label the surface cohort, washed, and either left untreated (0 min) or treated with PAR-specific agonist peptides for the indicated times. Cells were fixed, immunostained for PAR1 and endogenous AP-3, and visualized by confocal microscopy. Cells expressing Vps4 are indicated by arrowheads. The colocalization of PAR (red) with Vps4 (green) is indicated by the yellow color shown in the merged image. Endogenous AP-3 was detected in the same cells and is shown in the adjacent panels. Insets, magnifications of boxed areas. Scale bar, 10 μm.
FIGURE 9:

Model of AP-3 regulation of PAR1 MVB/lysosomal sorting. After internalization, PAR1 traffics to an endosomal sorting compartment and associates with AP-3, an adaptor protein that is known to colocalize with clathrin on endosomes, but whether clathrin is recruited to PAR1 is not known. AP-3 binds to PAR1 via a proximal C-tail, tyrosine-based sorting motif independent of receptor ubiquitination. The interaction between PAR1 and AP-3 retains it at the sorting endosome and facilitates its interaction with ALIX. Thus AP-3 binds to PAR1 before engagement of ALIX. PAR1 interaction with ALIX occurs at the late endosome/MVB. ALIX also recruits ESCRT-III components—charged multivesicular body protein 4 (CHMP4) and CHMP2—to facilitate PAR1 sorting to ILVs of MVBs. The AAA-ATPase Vps4 mediates ESCRT-III disassembly and recycling and is critical for ESCRT function.
DISCUSSION
In the present study, we describe a new function for AP-3 in the sorting of a signaling receptor from early endosomes to MVBs/lysosomes that occurs independent of ubiquitin and the canonical ESCRT machinery. We found that PAR1 and AP-3 coassociate on endocytic vesicles after agonist stimulation. Our studies also show that AP-3 regulates PAR1 lysosomal degradation independent of ubiquitination. In addition, we identified a PAR1 C-tail–localized, tyrosine-based motif that mediates AP-3 interaction and receptor sorting to intralumenal membranes of the sorting endosome—a compartment protected from proteinase K digestion. Remarkably, AP-3 is also required for PAR1 interaction with ALIX, which recruits ESCRT-III to facilitate receptor sorting to ILVs of MVBs (Dores et al., 2012), suggesting that AP-3 functions before PAR1 engagement of ALIX. Moreover, disruption of the canonical ubiquitin- and ESCRT-dependent sorting pathway by coexpression of the Vps4 E228Q mutant failed to perturb AP-3– or PAR1–containing sorting endosomes. Thus AP-3 mediates endocytic sorting of PAR1 through an ALIX-dependent and ubiquitin-independent pathway, indicating that AP-3 regulates a distinct GPCR MVB/lysosomal sorting pathway in mammalian cells (Figure 9).
A critical step in MVB/lysosome sorting is the retention of cargo in early endosomes and subsequent incorporation into ILVs. We previously showed that activated PAR1 sorts to ILVs of MVBs independent of ubiquitination and ubiquitin-binding ESCRT components (Gullapalli et al., 2006; Wolfe et al., 2007; Dores et al., 2012). In recent work, we demonstrated that ALIX binds directly to PAR1 and recruits ESCRT-III to mediate receptor sorting to ILVs of MVBs (Dores et al., 2012). However, the mechanisms responsible for initiating PAR1 sorting at the early sorting endosome remained unclear. In these studies, we show that PAR1 sorting to MVBs/lysosomes and interaction with ALIX is impaired in cells depleted of endogenous AP-3. Similar results were observed with a PAR1 tyrosine motif mutant defective in AP-3 binding. Thus we propose that AP-3 regulates PAR1 sorting at the early endosome before ALIX engagement and ILV sorting at the MVB. This pathway is distinct from the canonical ESCRT-dependent ILV/MVB sorting pathway of ubiquitinated receptors. In the canonical pathway, ubiquitinated receptors are sequestered at the early endosome in an HRS-enriched subdomain coated with clathrin and then sorted into ILVs of MVBs (Raiborg et al., 2002). A similar HRS-dependent mechanism has been implicated in lysosomal sorting of both ubiquitinated and nonubiquitinated GPCRs (Marchese et al., 2003; Hislop et al., 2004; Hasdemir et al., 2007; Henry et al., 2011). However, HRS is not required for PAR1 lysosomal degradation (Gullapalli et al., 2006). Rather than HRS, our findings suggest that AP-3 interacts with PAR1 on endosomes and links the receptor to ALIX to facilitate ILV sorting to MVBs independent of ubiquitination.
AP-3 regulates lysosomal sorting of LAMPs and CD63 by binding directly to C-tail–localized tyrosine motifs (Bonifacino and Traub, 2003). Previous studies reported that fibroblasts derived from mice or Hermansky–Pudlak syndrome patients, which harbor mutations in the β3- or δ-adaptin subunits of AP-3, exhibited reduced AP-3 expression and missorting of LAMP-1, LAMP-2, and CD63 to the cell surface (Dell'Angelica et al., 1999; Peden et al., 2004). In addition, LAMPs and CD63 contain canonical tyrosine-based motifs within their C-tails that mediate interaction with the μ3-adaptin subunit of AP-3 and lysosomal sorting (Williams and Fukuda, 1990; Gough and Fambrough, 1997; Rous et al., 2002). PAR1 contains two C-tail tyrosine-sorting motifs: a proximal motif located 11 residues from the transmembrane domain and a distal motif present at the extreme C-terminus (Figure 4A). In many cases, lysosomal-targeting tyrosine sorting signals are located within 6–13 residues from the transmembrane domain (Bonifacino and Traub, 2003). Consistent with this paradigm, our studies indicate that the PAR1 proximal C-tail tyrosine motif is critical for AP-3–mediated lysosomal sorting, since mutations in the proximal but not the distal motif resulted in impaired lysosomal degradation and AP-3 binding. Thus the PAR1 proximal tyrosine-based motif functions as a dominant signal for AP-3 association and lysosomal sorting.
Previous studies indicated that AP-3 localizes at budding sites of tubule-sorting endosomes containing LAMP-1 and LAMP-2 and defined a new pathway for lysosomal transport of proteins (Peden et al., 2004). In addition to LAMP-1 and LAMP-2, which reside at the limiting membrane of MVBs/lysosomes, AP-3 function is also essential for sorting of CD63, a protein that is enriched in ILVs of MVBs (Dell'Angelica et al., 1999; Peden et al., 2004; Pols and Klumperman, 2009). Of interest, the incorporation of CD63 into ILVs of MVBs is mediated by the sphingolipid ceramide and occurs independent of the ESCRT machinery. CD63 is segregated into endosomal membrane subdomains containing ceramide, which appears to aggregate resulting in inward budding of ILVs (Trajkovic et al., 2008). These findings suggest that distinct trafficking events regulated by AP-3 and subsequent sequestration into a lipid microdomain mediate CD63 sorting into ILVs of MVBs. Different trafficking processes also regulate the trafficking of PAR1 into ILVs of MVBs. Similar to CD63, AP-3 mediates the targeting of PAR1 to MVBs/lysosomes. However, in contrast to CD63, AP-3 facilitates PAR1 interaction with ALIX, which recruits ESCRT-III to mediate receptor sorting to ILVs of MVBs (Dores et al., 2012). Thus AP-3 functions to sort cargo proteins independent of the canonical ESCRT machinery via two different ILV sorting pathways mediated by the sphingolipid ceramide and ALIX/ESCRT-III. Of interest, ALIX also binds to lysobisphosphatidic acid (LBPA), a phospholipid enriched in late endosomes (Matsuo et al., 2004). However, the function of LBPA in PAR1 sorting to ILVs of MVBs remains to be determined.
In addition to PAR1, AP-3 has been shown to regulate trafficking of other mammalian GPCRs. In mouse neuroblastoma cells, the cannabinoid receptor-1 (CB1R) localizes predominantly to intracellular compartments and exhibits partial colocalization with AP-3 (Rozenfeld and Devi, 2008). Moreover, depletion of AP-3 resulted in increased expression of CB1R at the cell surface, suggesting that AP-3 regulates either CB1R endosome to lysosome sorting or retention in endosomes. The mechanism by which AP-3 facilitates trafficking of CB1R is not known, since neither tyrosine nor dileucine sorting motifs are present in the receptor cytoplasmic domains. In addition to CB1R, a function for the neuronal specific AP-3 isoform in recycling of the muscarinic acetylcholine M5 receptor in neurons has been demonstrated (Bendor et al., 2010). However, AP-3 was shown to associate with M5 muscarinic receptor indirectly via AGAP1, an ArfGAP protein that binds directly to the M5 receptor and AP-3. These findings suggest that AP-3 exhibits multiple functions in the regulation of GPCR trafficking in distinct cell types, including endosome-to-lysosome sorting, as well as in receptor recycling.
In summary, our work provides insight into a new mechanism by which MVB sorting of a signaling receptor is initiated at endosomes independent of ubiquitin and HRS, a ubiquitin-binding ESCRT component. We show that the classic adaptor protein AP-3 regulates PAR1 sorting to ILVs of MVBs by facilitating interaction with ALIX, an ESCRT-III–interacting protein. These studies define a new pathway that can be used by signaling receptors to access ILVs of MVBs that is independent of ubiquitination and ubiquitin-binding components of the ESCRT machinery. In addition, we previously showed that activated PAR1 sorts to lysosomes through a pathway that is regulated by sorting nexin-1 (Gullapalli et al., 2006); however, the connection between sorting nexins and AP-3 function in PAR1 trafficking is not known and will be important to determine.
MATERIALS AND METHODS
Reagents and antibodies
The peptide agonists SFLLRN (PAR1 specific) and SLIGKV (PAR2 specific) were synthesized as the carboxyl amide and purified by high-pressure liquid chromatography at the Tufts University Core Facility (Boston, MA). SDF-1α was purchased from Sigma-Aldrich (St. Louis, MO). Rabbit anti-PAR1 antibody was generated against the N-terminal domain of PAR1 encompassing the hirudin-like region as described (Paing et al., 2006). M1 and M2 anti-FLAG and anti–β-actin antibodies were from Sigma-Aldrich. Monoclonal anti-EEA1 and anti–δ-adaptin antibodies were obtained from BD Biosciences (San Jose, CA). Rat monoclonal anti-CXCR4 (2B11) was previously described (Marchese and Benovic, 2001). The monoclonal anti–δ-adaptin (SA4) and LAMP-1 antibodies were obtained from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). Anti–β-tubulin was purchased from Accurate Chemical (Westbury, NY). Anti-ALIX monoclonal antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Horseradish peroxidase–conjugated goat anti-mouse and anti-rabbit were from Bio-Rad (Hercules, CA). Alexa Fluor 488–, 594–, and 647–conjugated goat anti-mouse and anti-rabbit antibodies were obtained from Invitrogen (Carlsbad, CA).
cDNAs and cell lines
A cDNA plasmid encoding PAR1 wild type containing an N-terminal FLAG epitope and various mutants was previously described (Paing et al., 2006; Dores et al., 2012). The N-terminal FLAG-tagged PAR2 cDNA plasmid was previously described (Stalheim et al., 2005). HeLa cells stably expressing FLAG-tagged PAR1 wild type and mutants, as well as FLAG-tagged PAR2 wild type, were generated and maintained as previously described (Paing et al., 2006; Ricks and Trejo, 2009). GFP-tagged Vps4 and Rab5 were previously described (Marchese et al., 2003; Gullapalli et al., 2004).
Cell transfections
Transient transfections with cDNA plasmids cells were performed using Lipofectamine reagent (Invitrogen) according to manufacturer's directions. HeLa cells were transiently transfected with 50 nM nonspecific (ns) siRNA (5′-CTACGTCCAGGAGCGCACC-3′) or δ-subunit SMARTpool siRNAs purchased from Dharmacon (Lafayette, CO) using Lipofectamine 2000 (Invitrogen) for 60 h at 37°C as previously described (Paing et al., 2006; Dores et al., 2012).
PAR1 internalization assays
PAR1 internalization was examined by cell surface ELISA as previously described (Paing et al., 2004).
GPCR degradation assays
CXCR4 degradation was assessed as described previously (Marchese and Benovic, 2001). Briefly, HeLa cells were washed with cold phosphate-buffered saline (PBS), solubilized in 300 μl of 2× sample buffer containing 0.0375 M Tris-HCl, pH 6.5, 8% SDS, 10% glycerol, 5% β-mercaptoethanol, and 0.003% bromophenol blue, and sonicated, and equal amounts of lysates were resolved by SDS–PAGE, transferred, and immunoblotted to detect endogenous CXCR4. Immunoblots were stripped and reprobed with anti-tubulin. PAR1 and PAR2 degradation was assessed from an equivalent amount of cell lysates that were directly analyzed by SDS–PAGE, followed by immunoblotting or by immunoprecipitation, SDS–PAGE, and immunoblotting as previously described (Dores et al., 2012).
Immunofluorescence confocal microscopy
HeLa cells stably expressing PAR1 or PAR2 were processed, fixed, permeabilized, immunostained with species-specific secondary antibodies conjugated to Alexa Fluor 488, 594, or 647, and imaged by confocal microscopy as previously described (Paing et al., 2006; Dores et al., 2012). AP-3 immunostaining was performed essentially as described (Berger et al., 2007). Images shown in Figures 1, 2, and 8 were collected using a FluoView 300 laser scanning confocal imaging system (Olympus, Tokyo, Japan) configured with an IX70 microscope fitted with a PlanApo 60× oil objective. Confocal images (XY-sections at 0.28 μm) were collected sequentially at 800 × 600 resolution with 2× optical zoom using FluoView software. All other images were collected using an Olympus IX81 DSU spinning disk confocal microscope configured with a PlanApo 60× oil objective and Hamamatsu ORCA-ER digital camera (Hamamatsu, Hamamatsu, Japan). Fluorescent images of XY-sections at 0.28 μm were collected sequentially using SlideBook 4.2 software (Intelligent Imaging Innovations, Denver, CO). Pearson's r for quantifying PAR1 colocalization with EEA1, LAMP-1, and AP-3 was calculated for six cells from multiple independent experiments using either SlideBook 4.2 software or ImageJ 1.45 (National Institutes of Health, Bethesda, MD). Line scan analysis was performed using SlideBook 4.2 software, and fluorescence intensity and distance were normalized as previously reported (Henry et al., 2011). The final composite images were created using Photoshop CS (Adobe, San Jose, CA).
Proteinase K protection assays
The proteinase K protection assay was performed as described with minor modifications (Malerod et al., 2007). HeLa cells were plated in six-well culture dishes (4.0 × 105 cells/well), grown overnight at 37°C, and treated with or without agonist. Cells were placed on ice and incubated for 5 min with PBS, harvested, and gently permeabilized using 6.5 μg/ml digitonin. Membranes were collected by centrifugation and resuspended in buffer containing 100 mM K2HPO4/KH2PO4, 5 mM MgCl2, and 250 mM sucrose. Membranes were then divided into three aliquots left untreated, treated with 2.5 ng/ml proteinase K, or treated with proteinase K supplemented with 0.1% Triton-X 100 for 10 min at room temperature. After treatments, samples were diluted with 100 μl of 2× SDS sample buffer containing 20 mM phenylmethylsulfonyl fluoride and analyzed by immunoblot.
Data analysis
Data were analyzed using Prism 4.0 software (GraphPad Software, La Jolla, CA), and statistical significance was determined using Student's t test and two-way analysis of variance (ANOVA) with InStat 3.0 (GraphPad)
Acknowledgments
We thank members of the Trejo laboratory for reagents and advice. This work was supported by National Institutes of Health Grant GM090690 to J.T. M.R.D. is supported by a University of California Tobacco-Related Disease Research Program Postdoctoral Fellowship (UC TRDRP 19FT-0157).
Abbreviations used:
- AP-3
adaptor protein complex-3
- CB1R
cannabinoid receptor-1
- CL
calcitonin-like
- C-tail
cytoplasmic tail
- DOR
delta-opioid receptor
- EEA1
early endosome antigen-1
- ELISA
enzyme-linked immunosorbent assay
- ESCRT
endosomal-sorting complex required for transport
- GFP
green fluorescent protein
- GPCR
G protein–coupled receptor
- HRS
hepatocyte growth factor–regulated tyrosine kinase substrate
- ILV
intraluminal vesicle
- LAMP
lysosomal-associated membrane protein
- MVB
multivesicular body
- PAR
protease-activated receptor
- SDF-1α
stromal-derived factor-1α
- siRNA
small interfering RNA
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-03-0251) on July 25, 2012.
REFERENCES
- Bendor J, Lizardi-Ortiz JE, Westphalen RI, Brandstetter M, Hemmings HC, Jr, Sulzer D, Flajolet M, Greengard P. AGAP1/AP-3-dependent endocytic recycling of M5 muscarinic receptors promotes dopamine release. EMBO J. 2010;29:2813–2826. doi: 10.1038/emboj.2010.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AC, Salazar G, Styers ML, Newell-Litwa KA, Werner E, Maue RA, Corbett AH, Faundez V. The subcellular localization of the Niemann-Pick type C proteins depends on the adaptor complex AP-3. J Cell Sci. 2007;120:3640–3652. doi: 10.1242/jcs.03487. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Booden MA, Ekert L, Der CJ, Trejo J. Persistent signaling by dysregulated thrombin receptor trafficking promotes breast carcinoma cell invasion. Mol Cell Biol. 2004;24:1990–1999. doi: 10.1128/MCB.24.5.1990-1999.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Dores MR, Grimsey N, Canto I, Barker BL, Trejo J. Adaptor protein-2 (AP-2) and epsin-1 mediate protease-activated receptor-1 internalization via phosphorylation- and ubiquitination-dependent sorting signals. J Biol Chem. 2011;286:40760–40770. doi: 10.1074/jbc.M111.299776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell GS, Padilla B, Pikios S, Roosterman D, Steinhoff M, Grady EF, Bunnett NW. Post-endocytic sorting of calcitonin receptor-like receptor and receptor activity-modifying protein 1. J Biol Chem. 2007;282:12260–12271. doi: 10.1074/jbc.M606338200. [DOI] [PubMed] [Google Scholar]
- Dell'Angelica EC, Klumperman J, Stoorvogel W, Bonifacino JS. Association of the AP-3 adaptor complex with clathrin. Science. 1998;280:431–434. doi: 10.1126/science.280.5362.431. [DOI] [PubMed] [Google Scholar]
- Dell'Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell. 1999;3:11–21. doi: 10.1016/s1097-2765(00)80170-7. [DOI] [PubMed] [Google Scholar]
- Dores MR, Chen B, Lin H, Soh UJK, Paing MM, Montagne WA, Meerloo T, Trejo J. ALIX binds a YPX3L motif of the GPCR PAR1 and mediates ubiquitin-independent ESCRT-III/MVB sorting. J Cell Biol. 2012;197:407–419. doi: 10.1083/jcb.201110031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough NR, Fambrough DM. Different steady state subcellular distributions of the three splice variants of lysosome-associated membrane protein LAMP-2 are determined largely by the COOH-terminal amino acid residue. J Cell Biol. 1997;137:1161–1169. doi: 10.1083/jcb.137.5.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullapalli A, Garrett TA, Paing MM, Griffin CT, Yang Y, Trejo J. A role for sorting nexin 2 in epidermal growth factor receptor down-regulation: evidence for distinct functions of sorting nexin 1 and 2 in protein trafficking. Mol Biol Cell. 2004;15:2143–2155. doi: 10.1091/mbc.E03-09-0711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullapalli A, Wolfe BL, Griffin CT, Magnuson T, Trejo J. An essential role for SNX1 in lysosomal sorting of protease-activated receptor-1: evidence for retromer, Hrs and Tsg101 independent functions of sorting nexins. Mol Biol Cell. 2006;17:1228–1238. doi: 10.1091/mbc.E05-09-0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasdemir B, Bunnett NW, Cottrell GS. Hepatocyte growth factor-regulated tyrosine kinase substrate (HRS) mediates post-endocytic trafficking of protease-activated receptor 2 and calcitonin receptor-like receptor. J Biol Chem. 2007;282:29646–29657. doi: 10.1074/jbc.M702974200. [DOI] [PubMed] [Google Scholar]
- Henry AG, White IJ, Marsh M, von Zastrow M, Hislop JN. The role of ubiquitination in lysosomal trafficking of delta-opioid receptors. Traffic. 2011;12:170–184. doi: 10.1111/j.1600-0854.2010.01145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hislop JN, Marley A, Von Zastrow M. Role of mammalian vacuolar protein-sorting proteins in endocytic trafficking of a non-ubiquitinated G protein-coupled receptor to lysosomes. J Biol Chem. 2004;279:22522–22531. doi: 10.1074/jbc.M311062200. [DOI] [PubMed] [Google Scholar]
- Hurley JH, Hanson PI. Membrane budding and scission by the ESCRT machinery: it's all in the neck. Nat Rev Mol Cell Biol. 2010;11:556–566. doi: 10.1038/nrm2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YM, et al. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell. 2004;6:459–469. doi: 10.1016/j.ccr.2004.09.027. [DOI] [PubMed] [Google Scholar]
- Malerod L, Stuffers S, Brech A, Stenmark H. Vps22/EAP30 in ESCRT-II mediates endosomal sorting of growth factor and chemokine receptors destined for lysosomal degradation. Traffic. 2007;8:1617–1629. doi: 10.1111/j.1600-0854.2007.00630.x. [DOI] [PubMed] [Google Scholar]
- Marchese A, Benovic JL. Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J Biol Chem. 2001;276:45509–45512. doi: 10.1074/jbc.C100527200. [DOI] [PubMed] [Google Scholar]
- Marchese A, Paing MM, Temple BRS, Trejo J. G protein-coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmacol Toxicol. 2008;48:601–629. doi: 10.1146/annurev.pharmtox.48.113006.094646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese A, Raiborg C, Santini F, Keen JT, Stenmark H, Benovic JL. The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev Cell. 2003;5:709–722. doi: 10.1016/s1534-5807(03)00321-6. [DOI] [PubMed] [Google Scholar]
- Matsuo H, et al. Role of LBPA and ALIX in multivesicular liposome formation and endosome organization. Science. 2004;303:531–534. doi: 10.1126/science.1092425. [DOI] [PubMed] [Google Scholar]
- Paing MM, Johnston CA, Siderovski DP, Trejo J. Clathrin adaptor AP2 regulates thrombin receptor constitutive internalization and endothelial cell resensitization. Mol Cell Biol. 2006;28:3231–3242. doi: 10.1128/MCB.26.8.3231-3242.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paing MM, Temple BRS, Trejo J. A tyrosine-based sorting signal regulates intracellular trafficking of protease-activated receptor-1: multiple regulatory mechanisms for agonist-induced G protein-coupled receptor internalization. J Biol Chem. 2004;279:21938–21947. doi: 10.1074/jbc.M401672200. [DOI] [PubMed] [Google Scholar]
- Peden AA, Oorschot V, Hesser BA, Austin CD, Scheller RH, Klumperman J. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J Cell Biol. 2004;164:1065–1076. doi: 10.1083/jcb.200311064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peden AA, Rudge RE, Lui WW, Robinson MS. Assembly and function of AP-3 complexes in cells expressing mutant subunits. J Cell Biol. 2002;156:327–336. doi: 10.1083/jcb.200107140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pols MS, Klumperman J. Trafficking and function of the tetraspanin CD63. Exp Cell Res. 2009;315:1584–1592. doi: 10.1016/j.yexcr.2008.09.020. [DOI] [PubMed] [Google Scholar]
- Raiborg C, Bache KG, Gilooly DJ, Madshus IH, Stang E, Stenmark H. Hrs sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nat Cell Biol. 2002;4:394–398. doi: 10.1038/ncb791. [DOI] [PubMed] [Google Scholar]
- Ricks T, Trejo J. Phosphorylation of protease-activated receptor-2 differentially regulates desensitization and internalization. J Biol Chem. 2009;284:34444–34457. doi: 10.1074/jbc.M109.048942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rous BA, Reaves BJ, Ihrke G, Briggs JA, Gray SR, Stephens DJ, Banting G, Luzio JP. Role of adaptor complex AP-3 in targeting wild-type and mutated CD63 to lysosomes. Mol Biol Cell. 2002;13:1071–1082. doi: 10.1091/mbc.01-08-0409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenfeld R, Devi LA. Regulation of CB1 cannabinoid receptor trafficking by the adaptor protein AP-3. FASEB J. 2008;22:2311–2322. doi: 10.1096/fj.07-102731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini DD, Blobel G. Controlled proteolysis of nascent polypeptides in rat liver cell fractions. II. Location of the polypeptides in rough microsomes. J Cell Biol. 1970;45:146–157. doi: 10.1083/jcb.45.1.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachse M, Urbe S, Oorschot V, Strous GJ, Klumperman J. Bilayered clathrin coats on endosomal vacuoles are involved in protein sorting toward lysosomes. Mol Biol Cell. 2002;13:1313–1328. doi: 10.1091/mbc.01-10-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalheim L, Ding Y, Gullapalli A, Paing MM, Wolfe BL, Morris DR, Trejo J. Multiple independent functions of arrestins in regulation of protease-activated receptor-2 signaling and trafficking. Mol Pharm. 2005;67:1–10. doi: 10.1124/mol.104.006072. [DOI] [PubMed] [Google Scholar]
- Stenmark H, Parton RG, Steele-Mortimer O, Lutcke A, Gruenberg J, Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994;13:1287–1296. doi: 10.1002/j.1460-2075.1994.tb06381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theos AC, et al. Functions of adaptor protein (AP)-3 and AP-1 in tyrosinase sorting from endosomes to melanosomes. Mol Biol Cell. 2005;16:5356–5372. doi: 10.1091/mbc.E05-07-0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brugger B, Simons M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319:1244–1247. doi: 10.1126/science.1153124. [DOI] [PubMed] [Google Scholar]
- Williams MA, Fukuda M. Accumulation of membrane glycoproteins in lysosomes requires a tyrosine residue at a particular position in the cytoplasmic tail. J Cell Biol. 1990;111:955–966. doi: 10.1083/jcb.111.3.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe BL, Marchese A, Trejo J. Ubiquitination differentially regulates clathrin-dependent internalization of protease-activated receptor-1. J Cell Biol. 2007;177:905–916. doi: 10.1083/jcb.200610154. [DOI] [PMC free article] [PubMed] [Google Scholar]