

A 221-entry NESdb database produces data sets of true- and false-positive nuclear export signals (NES). Analysis of these data sets leads to identification of a set of sequence and structural properties that distinguishes true NESs from peptides without export capability that merely conform to the NES consensus sequences.
Abstract
We compiled >200 nuclear export signal (NES)–containing CRM1 cargoes in a database named NESdb. We analyzed the sequences and three-dimensional structures of natural, experimentally identified NESs and of false-positive NESs that were generated from the database in order to identify properties that might distinguish the two groups of sequences. Analyses of amino acid frequencies, sequence logos, and agreement with existing NES consensus sequences revealed strong preferences for the Φ1-X3-Φ2-X2-Φ3-X-Φ4 pattern and for negatively charged amino acids in the nonhydrophobic positions of experimentally identified NESs but not of false positives. Strong preferences against certain hydrophobic amino acids in the hydrophobic positions were also revealed. These findings led to a new and more precise NES consensus. More important, three-dimensional structures are now available for 68 NESs within 56 different cargo proteins. Analyses of these structures showed that experimentally identified NESs are more likely than the false positives to adopt α-helical conformations that transition to loops at their C-termini and more likely to be surface accessible within their protein domains or be present in disordered or unobserved parts of the structures. Such distinguishing features for real NESs might be useful in future NES prediction efforts. Finally, we also tested CRM1-binding of 40 NESs that were found in the 56 structures. We found that 16 of the NES peptides did not bind CRM1, hence illustrating how NESs are easily misidentified.
INTRODUCTION
Transport of proteins between the nucleus and the cytoplasm is mostly mediated by transport factors in the karyopherin-β family, which are also known as importins and exportins (Görlich and Kutay, 1999; Conti and Izaurralde, 2001; Weis, 2003; Tran et al., 2007). The direction of nuclear–cytoplasmic transport is dictated by nuclear targeting signals within the cargo proteins. Nuclear localization signals (NLSs) direct proteins into the nucleus, whereas nuclear export signals (NESs) direct export of proteins from the nucleus to the cytoplasm (Dingwall et al., 1982, 1988; Kalderon et al., 1984; Lanford and Butel, 1984; Fischer et al., 1995; Wen et al., 1995; Lee et al., 2006). Two classes of NLS—known as the classic NLSs and the PY-NLS—and one class of NES—known as the leucine-rich or classic NES—have been characterized (for reviews see Chook and Blobel, 2001; Chook and Süel, 2011; Lange et al., 2007; Xu et al., 2010; Marfori et al., 2011).
NESs were first identified in the proteins HIV-1 Rev and cyclical AMP-dependent protein kinase inhibitor (PKIα; Fischer et al., 1995; Wen et al., 1995). Because these early export signals (Rev 75LPPLERLTL83; PKIα 38LALKLAGLDL47) are rich in leucine residues, they are often called leucine-rich NESs. Since then, NESs have been identified in >200 proteins, and many contain not specifically leucine but more generally hydrophobic patterns. NESs are peptides that are 8–15 residues long and conform loosely to the widely used traditional consensus of Φ1-X2,3-Φ2-X2,3-Φ3-X-Φ4, where Φn represents Leu, Val, Ile, Phe, or Met and X can be any amino acid (Bogerd et al., 1996; Henderson and Eleftheriou, 2000; Engelsma et al., 2004; la Cour et al., 2004; Kutay and Güttinger, 2005). NESs bind directly to the export karyopherin CRM1 (also known as exportin 1), which escorts cargo proteins through the nuclear pore complex (Fornerod et al., 1997; Fukuda et al., 1997; Neville et al., 1997; Ossareh-Nazari et al., 1997; Richards et al., 1997; Stade et al., 1997). Crystal structures of CRM1 bound to three different NESs showed that the hydrophobic positions or Φn of the NESs dock into five hydrophobic pockets within a narrow groove on the convex surface of CRM1 (Dong et al., 2009a, b; Monecke et al., 2009; Güttler et al., 2010). Bound NESs of the cargoes snurportin 1 (SNUPN) and PKIα adopt combined α-helix-loop structures, whereas the NES of HIV-1 Rev binds CRM1 in a mostly loop-like conformation (Dong et al., 2009a, b; Monecke et al., 2009; Güttler et al., 2010). Although individual NESs may adopt different conformations, the NES-binding grooves in the CRM1 structures seem to remain conformationally invariant (Güttler et al., 2010).
NES-containing proteins have been discovered in a wide range of organisms, and CRM1 cargoes appear to possess diverse cellular functions, which control many normal cellular processes and disease states of cells (Lim and Wang, 2006; Stauber et al., 2007; Zemp and Kutay, 2007; Suhasini and Reddy, 2009; Ding et al., 2010; Emami, 2011; Güttler and Görlich, 2011; Turner et al., 2012). Therefore identification of NESs in the genome is important to decipher sequence features that underlie protein functions. However, successful identification or prediction of NESs has been hindered by the very broad traditional Φ1-X2,3-Φ2-X2,3-Φ3-X-Φ4 consensus sequence that describes the NES (la Cour et al., 2004; Fu et al., 2011). The Φ1–Φ3 portion of this consensus sequence describes the ubiquitous two-turn amphipathic helix. Matching sequences can be found in most helix-containing proteins, and many of these sequences may be part of hydrophobic cores that are not accessible for CRM1 binding. Despite its breadth, the traditional NES consensus lacks sensitivity, as it described only ∼60% of the experimentally identified NESs (la Cour et al., 2004). To improve sensitivity of the traditional NES consensus, Kosugi et al. (2008) expanded the consensus with two additional variations of hydrophobic spacings and with an expanded vocabulary of hydrophobic residues. More recently, Güttler et al. (2010; Güttler and Görlich, 2011) suggested a structure-based NES consensus with five instead of four hydrophobic positions. However, all three sets of available NES consensus sequences describe many protein segments that do not function as NESs. It is clear that the use of consensus sequences alone falls short of the goal of accurate NES prediction. The shortcomings of sequence-based prediction is illustrated by a new NES predictor named NESsential, in which incorporation of metafeatures such as predicted disorder propensity and solvent accessibility helped to distinguish real and false-positive NESs (Fu et al., 2011).
More than 200 NES-containing CRM1 cargoes have been reported in the literature, and we compiled the majority of these experimentally identified, NES-containing proteins into a database named NESdb (Xu et al., 2012). The database provides large data sets of “experimentally identified NESs,” or “experimental NESs”; “negative,” or non-NES sequences; and “false positives,” or non-NES sequences that match the existing NES consensus sequences. These data sets provide the opportunity to conduct sequence and structural analyses to uncover sequence and structural properties that are unique to experimentally identified NESs. Such properties might be useful to distinguish real from false-positive NESs and to improve prediction efforts in the future.
la Cour et al. (2003, 2004) made the first attempt to systematically analyze NES properties of 67 NESs that were collected in a database named NESbase. Their study revealed interesting NES properties, such as overrepresentation of acidic residues in nonhydrophobic positions of NESs, preference for α-helical structure, and tendency of NESs to be located in highly flexible and surface-exposed regions. They also developed the first NES predictor, named NetNES (la Cour et al., 2004), which is a sequence-based program that combines neural network and hidden Markov models. Recently Fu et al. (2011) presented the new and improved NES predictor NESsential, which uses a different machine learning algorithm (Support Vector Machine) and incorporates folding context such as predicted disorder scores and solvent accessibilities into its feature set. Fu et al. (2011) also presented a list of 70 additional CRM1 cargoes, and their analysis of the combined set of NESs focused on preferences for negatively charged residues and disorder tendencies of the sequences.
In light of the significantly larger and more up-to-date NESdb database (Xu et al., 2012), recently available CRM1-NES structures (Dong et al., 2009a, b; Monecke et al., 2009; Güttler et al., 2010), and the fast-growing Protein Data Bank (PDB; Berman et al., 2000), a comprehensive analysis of the sequences and, more important, the three-dimensional (3D) structures of this large collection of natural NESs is needed to corroborate and expand our current understanding of the properties of these signals. Here we performed sequence and structural analyses of 234 NESs from 200 CRM1 cargoes that were collected in NESdb. Like previous studies (la Cour et al., 2004; Fu et al., 2011), we also examined the amino acid contents, predicted secondary structures, and disorder scores of NESs. Unlike these previous studies, we identified 56 NES-containing cargoes in the PDB that provided structural data and allowed analysis of secondary structures, crystallographic B-factors, exposed surface areas, and locations of NESs in these 3D structures in the context of the folded cargoes or cargo domains. Structural analysis revealed conformational, disorder, and surface accessibility features that are different in experimentally identified NESs versus false-positive sequences. In addition, we also tested CRM1 binding of 40 NESs that were found in the 56 structures and showed that 16 of the NES peptides did not bind CRM1, hence illustrating how NESs can be easily misidentified. In summary, we identified a set of sequence and structural properties that distinguish experimentally determined NESs from false-positive NESs. This set of features might be useful as filters to increase the accuracy of NES prediction in the future.
RESULTS
CRM1 cargoes in the NESdb database
Most NES-containing CRM1 cargoes were identified on individual bases by experiments such as cellular mislocalization after leptomycin B (LMB) treatment and nuclear export assays in cells. We compiled a comprehensive NES database named NESdb by manually curating the published literature for such experimentally validated CRM1 cargoes (Xu et al., 2012). Proteins in NESdb must show experimental evidence of nuclear export by CRM1 and must contain at least one experimentally identified NES. Acceptable experimental evidence for CRM1-mediated nuclear export includes LMB sensitivity, CRM1 binding, competition with other known CRM1 cargoes, and loss of CRM1 binding or nuclear export activity upon mutation of key NES residues.
NESdb contains 221 NES-containing proteins that were reported up to December 2011 (Xu et al., 2012). An average of 16 new CRM1 cargoes were reported each year for the last 10 years. Ninety-five percent of the proteins in NESdb were reported to accumulate in the nucleus upon treatment with LMB, which covalently modifies a reactive cysteine in the NES-binding site of CRM1 and prevents NES binding (Kudo et al., 1998, 1999). Sixty-eight percent of the NESdb proteins were reported to contain at least one peptide capable of targeting a reporter protein out of the nucleus, and 24% of NESdb proteins were tested and shown to bind CRM1 in vitro.
CRM1 cargoes in NESdb come from a variety of organisms (Figure 1A). Seventy-seven are from the animal kingdom, of which most are human proteins and others are mouse, rat, frog, chicken, or insect proteins. There are 17 fungi proteins: 12 from Saccharomyces cerevisiae and 5 from Schizosaccharomyces pombe. Surprisingly few S. cerevisiae cargoes have been reported, even though CRM1 is essential in the organism. The slower state of discovery here may be due to LMB insensitivity of the S. cerevisiae CRM1, which contains a threonine in place of the reactive cysteine for LMB attachment. The S. pombe CRM1 contains a reactive cysteine for LMB modification, and Matsuyama et al. (2006) found that 285 S. pombe proteins were mislocalized by LMB. We have not incorporated this list of 285 S. pombe proteins in NESdb, since most of their NESs remain unmapped.
FIGURE 1:
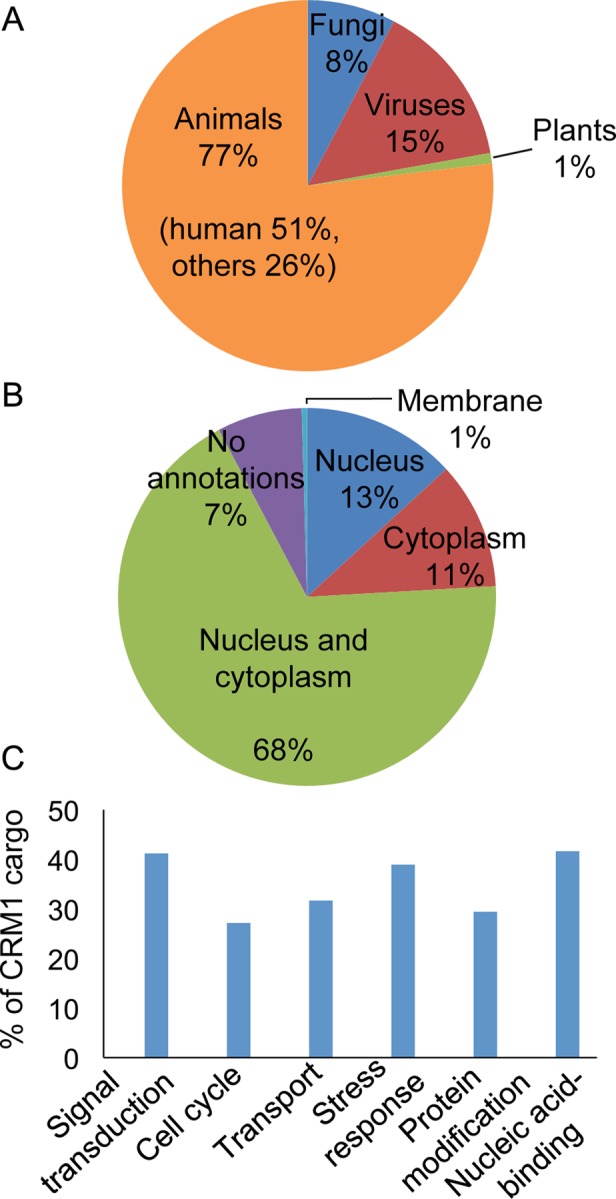
Overview of CRM1 cargoes in NESdb. (A) The organisms from which CRM1 cargoes come. (B) The cellular localizations of CRM1 cargoes as defined by Gene Ontology (GO) annotations. (C) Cellular functions and biological processes in which CRM1 cargoes participate, as listed in their GO annotations.
We used Gene Ontology annotations of the CRM1 cargoes to obtain information about their cellular localizations and functions (Ashburner et al., 2000). Sixty-eight percent of the NES-containing proteins shuttle between the nucleus and the cytoplasm; 13% are found primarily in the nucleus and 11% primarily in the cytoplasm (Figure 1B). Finally, CRM1 cargoes participate in many different cellular processes. Almost half are nucleic acid–binding proteins, and others participate in stress response, signal transduction, cell cycle, transcriptional regulation, and transport (Figure 1C).
NES sequences in NESdb
We annotated a total of 268 NES sequences for the 221 CRM1 cargoes in NESdb, since several cargoes contain multiple NESs. Locations of all 268 NESs within the CRM1 cargoes were assigned based on experimental information described in the original publications. Experiments to define NESs include NES mutations that abolish LMB-sensitive nuclear export, in vitro CRM1 binding, and/or the ability of the NES peptide to target reporter proteins out of the nucleus. CRM1-binding or nuclear export assays were performed for the isolated NES peptide or for the full-length cargo protein. However, because many peptides in globular proteins match the very broad NES consensus, it is difficult to accurately identify NESs. There are several instances where sequences were initially reported as NESs but subsequent experiments challenged their validity. Therefore we grouped the 268 NES sequences in the NESdb into two lists. The first list is named “NESs” and contains 242 experimentally identified NESs with no conflicting experimental evidence. We refer to NESs in this list as “experimentally identified NESs” or “experimental NESs.” The second list is named “NESs in doubt” and contains 24 sequences that were reported as NESs but about which subsequent studies cast doubt on their status as NESs. Some of the sequences in this list failed in subsequent studies to bind CRM1 in the context of a protein domain (e.g., Stat1 and c-Abl; McBride and Reich, 2003; Hantschel et al., 2005), and others did not bind CRM1 even as peptides (examples include Dcps, APRIL, and Gal3; see later discussion). We refer to the previously identified NESs in this list as “misidentified NESs.” Beyond the experimental NES sequences, the remaining sequences in the CRM1 cargoes are assumed to be negative or non-NES sequences.
For this work, we define an NES as a short peptide of 15 amino acids. An NES may be shorter than 15 amino acids if it is located at the N-terminus of the protein. NES residues are numbered 1–15 and usually contain four hydrophobic positions labeled Φ1, Φ2, Φ3, and Φ4. NESs are aligned at their C-terminus with the last hydrophobic or Φ4 residue, defined as position 15. Negative NESs are also defined as 15 residues long and generated using a sliding window protocol along non-NES sequences of the cargoes. For example, if an experimental NES is not located within residues 1–16, then two negative sequences, residues 1–15 and 2–16, can be generated from this segment. Taken together, we generated >100,000 negative sequences from NESdb entries. The following analyses were conducted with 234 experimental NESs from 200 cargoes that share <40% sequence identity to another NESdb protein.
Among the 234 experimental NESs, 153 match the traditional consensus sequence of Φ1-X2,3-Φ2-X2,3-Φ3-X-Φ4 (Φn = L, V, I, F, or M) and 208 match the expanded Kosugi consensus sequences (class 1a, Φ1-X3-Φ2-X2-Φ3-X-Φ4; class 1b, Φ1-X2-Φ2-X2-Φ3-X-Φ4; class 1c, Φ1-X3-Φ2-X3-Φ3-X-Φ4; class 1d, Φ1-X2-Φ2-X3-Φ3-X-Φ4; class 2, Φ1-X-Φ2-X2-Φ3-X-Φ4; class 3, Φ1-X2-Φ2-X3-Φ3-X2-Φ4; where Φn = L, V, I, F, or M, and A, C, T, and W can be one of the Φn). Among the >100,000 negative sequences, 1069 fit the traditional consensus and 5550 fit the Kosugi consensus (Table 1). Negative sequences that match the NES consensus sequences are termed false positives. The Kosugi consensus achieved a higher recall rate (fraction of experimental NESs recovered by consensus) than the traditional consensus (89 vs. 65%), but its precision rate (fraction of consensus fitting sequences that are indeed experimental NESs) is lower (Table 1). Owing to the overwhelmingly large number of false positives, the precision rates of both the traditional and Kosugi consensus are quite low, at 12 and 4%, respectively (Table 1).
TABLE 1:
Performance of different sets of NES consensus sequences.
| Traditional consensus | Kosugi consensus | Structure-based consensus (5-Φs) | Xu consensus (new) | |
|---|---|---|---|---|
| Recall | 65% (153 of 234) | 89% (208 of 234) | 51% (121 of 234) | 84% (197 of 234) |
| Precision | 12% (153 of 1222) | 4% (208 of 5758) | 9% (121 of 1292) | 6% (197 of 3100) |
Sequence logos of the NESs
We aligned the 234 experimentally identified NES sequences in NESdb at their position 15 and generated a sequence logo to display patterns of the sequence alignment (Figure 2A). The height of a stack of letters in a sequence logo indicates functional conservation, which is not preservation but the fit to a particular pattern. Here similar functional constraints on evolutionarily unrelated NES sequences caused them to converge to similar patterns. The sequence logo in Figure 2A showed four highly conserved positions, as indicated by the height of the stack of letters at positions 6, 10, 13, and 15. Leucine is the most frequently found amino acid at these functionally conserved locations. These four highly conserved positions produce the Φ1-X3-Φ2-X2-Φ3-X-Φ4 pattern, which must be the most prevalent hydrophobic pattern that describes experimentally identified NESs. Positions Φ3 and Φ4 of this prevalent pattern are dominated by the five traditional hydrophobic residues Leu, Ile, Val, Met, and Phe. On further inspection, the sequence logo revealed that position 13 is also occupied by nonhydrophobic amino acids, suggesting spacing options other than the Φ3-X-Φ4 spacing that is stipulated by the traditional consensus. Negatively charged residues, especially glutamates, are found at relatively high frequency at intervening positions between the Φs. la Cour et al. (2004) noticed a strong prevalence of Glu, Asp, and Ser at positions that are not occupied by hydrophobic residues. Fu et al. (2011) also reported a preference for acidic residues, albeit to a lesser degree.
FIGURE 2:
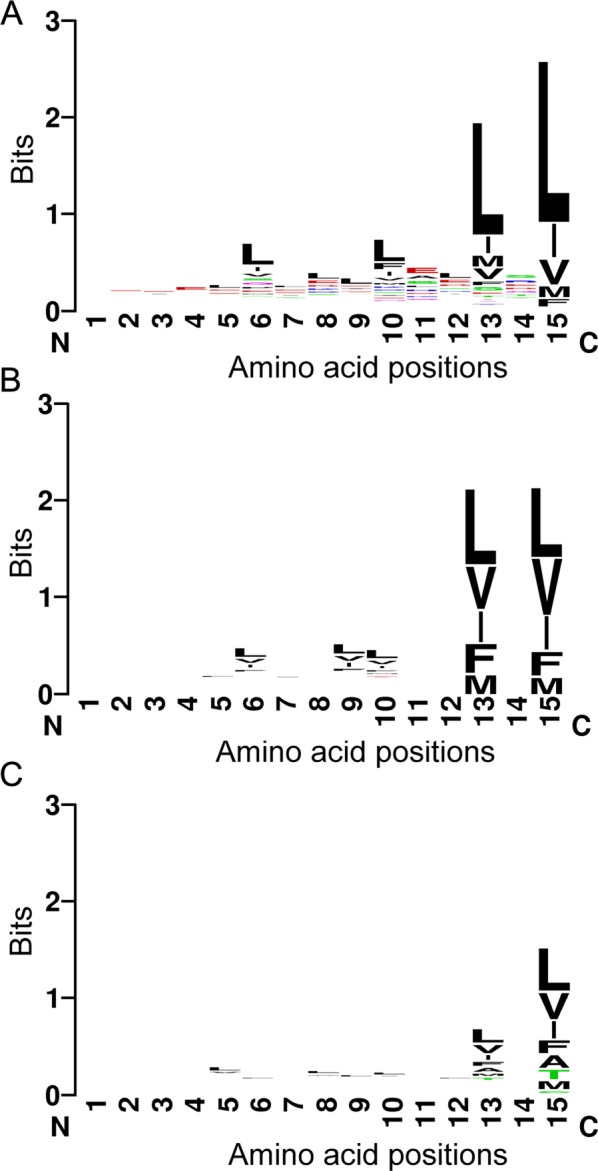
Sequence logos of experimental vs. false-positive NESs. (A) Sequence logo of experimental NESs in NESdb. (B) Sequence logo of negative sequences that fit the traditional NES consensus patterns. (C) Sequence logo of negative sequences that fit the Kosugi NES consensus patterns. Sequence logos were generated by the program WebLogo (http://weblogo.berkeley.edu/), where the x-axis is labeled with amino acid position, with #15 as the last amino acid in the sequence, and the y-axis represents the information content measured in bits. The overall height of each stack of letters indicates the sequence conservation at that position, and the height of a letter within the stack indicates the relative frequency of the amino acid.
As a comparison, we also created sequence logos for negative sequences that fit either the traditional or the Kosugi consensus patterns (Figure 2, B and C, respectively). Figure 2B indicates that only positions 13 and 15 are conserved among the false-positive sequences that match the traditional consensus. The degree of conservation decreases even further for false positives that match the Kosugi consensus (Figure 2C). The reduced functional conservation of hydrophobic positions suggests that false-positive sequences show weaker preference for specific spacing of their hydrophobic residues. When individual hydrophobic positions are examined, we find that Ala, Thr, and Cys are observed at the Φ3 and Φ4 positions of the false positives that fit the Kosugi consensus but not of the experimental NESs (Figure 2C). Furthermore, the false-positive sequences show no increased frequencies of acidic residues at intervening positions between Φs. In summary, we conclude that the strong preference for the Φ1-X3-Φ2-X2-Φ3-X-Φ4 pattern and the abundance of acidic residues may help to distinguish experimental NESs from false-positive sequences.
Position-specific amino acid frequency
To characterize the amino acid composition of NESs in NESdb, we performed frequency analysis of each amino acid at specific positions in the experimental NESs to compare with those of the false positives that match the Kosugi consensus. Consistent with the sequence logos (Figure 2, A–C), Glu residues occur at significantly higher frequency in experimental NESs than in false-positive sequences (0.1 vs. 0.06; Figure 3A). Asp residues are also slightly more prevalent in experimental NESs (Figure 3B). These acidic residues are prevalent in intervening positions between the conserved hydrophobic positions 6, 10, 13, and 15. Overrepresentation of acidic residues suggests that experimental NES sequences are more likely than the false positives to be negatively charged.
FIGURE 3:
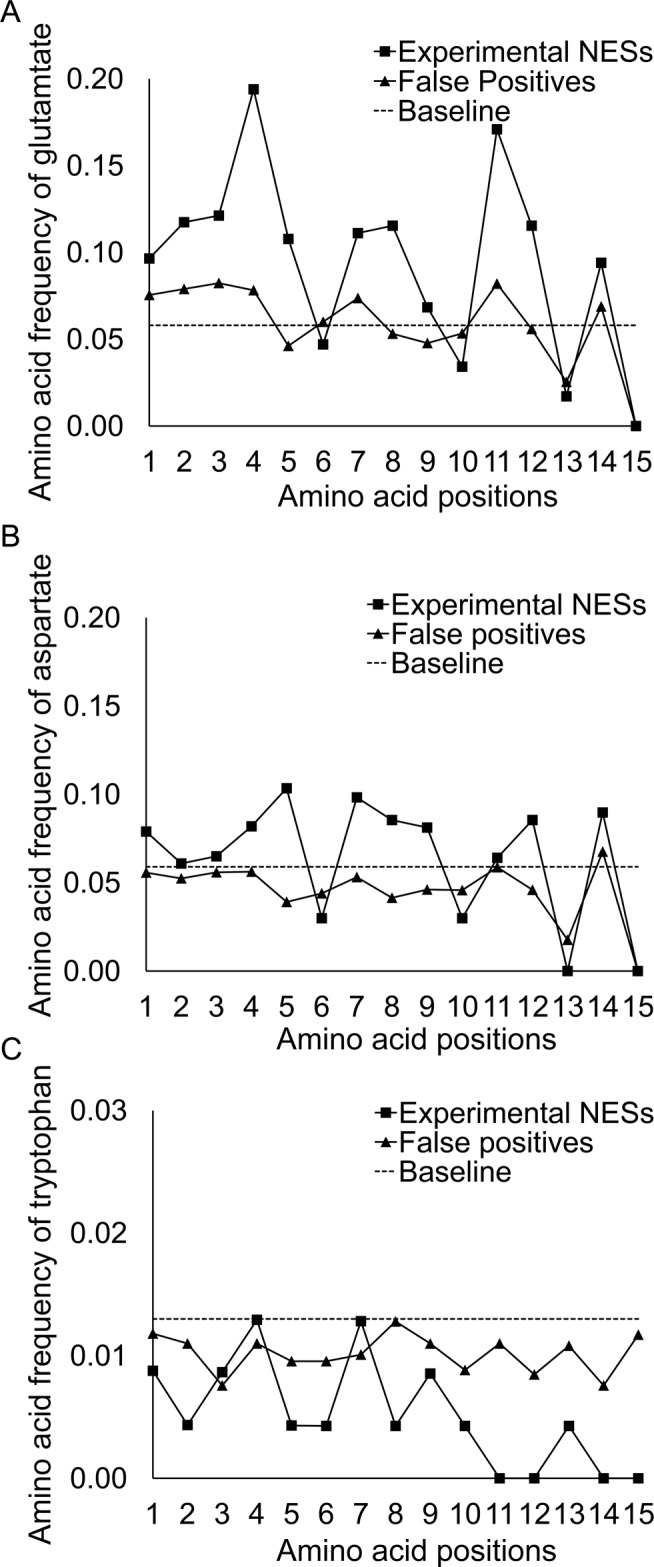
Position-specific amino acids frequencies of experimental vs. false-positive NESs. Amino acid frequency of (A) glutamate, (B) aspartate, and (C) tryptophan. Baseline refers to the background frequency of the amino acid.
We also computed net charges of the NESs by adding up charges from each amino acid. Asp and Glu residues were assigned −1 charge and Lys and Arg residues assigned +1 charge, whereas the other amino acids are assumed to be neutral. Our results showed that ∼62% of the experimental NESs have net negative charge compared with ∼41% of false-positive sequences. This finding is in agreement with previous results by la Cour et al. (2004), who reported that NES segments tend to have lower isoelectric points than full-length protein cargoes. Fu et al. (2011) also incorporated the preference for negatively charged residues into the feature sets of NESsential. Structures of CRM1–NES complexes showed that several positively charged amino acids flank the CRM1 NES-binding groove, poised to engage in electrostatic interactions with acidic residues in NESs (Dong et al., 2009a, b; Güttler et al., 2010).
We also found that tryptophans are mostly absent from positions 10–15 of the experimental NESs but maintain near-normal presence in the false-positive sequences (Figure 3C). Structures of CRM1–NES complexes show that the hydrophobic groove of CRM1 gradually narrows toward the C-terminus of the NES peptide, and modeling of Trp residues into any of the last three residues of the SNUPN NES results in significant steric clashes with CRM1 (unpublished data). Trp side chains may be too bulky to fit into the narrow NES-binding groove.
Coverage of existing NES consensus sequences
Analysis of NES sequences in NESdb showed that only 65% of the experimental NES sequences can be covered by the traditional consensus of Φ1-X2,3-Φ2-X2,3-Φ3-X-Φ4, suggesting that the traditional consensus suffers from low sensitivity (Table 1). The expanded Kosugi consensus sequences increased the recall rate to 89%, with the improvement deriving from two aspects (Table 1). First, two of the six Kosugi sequence patterns—class 2, Φ1-X-Φ2-X2-Φ3-X-Φ4; and class 3, Φ1-X2-Φ2-X3-Φ3-X2-Φ4—are unique and not represented in the traditional consensus and increased the recall rate by 12%, with 13 and 16 additional NES sequences fitting the two patterns, respectively. Second, the Kosugi consensus expanded the amino acid repertoire by allowing unconventional hydrophobic residues Ala, Cys, Thr, and Trp in one and only one of the Φ positions and thus increased recall rate, with 26 additional NESs that fit the consensus.
However, we found that Ala, Cys, Thr, and Trp residues do not appear equally frequently. Among the 26 NESs in NESdb that use these unconventional hydrophobic residues to fit the Kosugi consensus, 16 use Thr in one of their Φ positions, 6 use Ala, 4 use Cys, and none uses Trp. In addition, our analysis showed that the four hydrophobic positions have different tendencies to be occupied by these unconventional hydrophobic residues. There are 13 NESs with unconventional hydrophobic residues in their Φ1 positions, 6 NESs with those residues in the Φ2 positions, 5 NESs with unconventional Φ3 residues, and only 2 NESs with an unconventional Φ4 residue. This result is consistent with the sequence logo in Figure 2A, which shows that Φ3 and Φ4 of experimental NESs are dominated by the conventional hydrophobic residues Leu, Ile, Val, Phe, and Met. Therefore Thr and Ala residues seem more likely to occupy positions Φ1 and Φ2 than Φ3 or Φ4 in experimental NESs.
Güttler et al. (2010; Güttler and Görlich, 2011) recently introduced a structure-based NES consensus that consists of five instead of four key hydrophobic positions. They introduced Φ0 at the N-termini of NESs and divided the signals into two classes: the PKI-class NESs and the Rev-class NESs. The consensus for the PKI-class NES is defined as Φ0-X0,3-Φ1-X3-Φ2-X2,3-Φ3-X-Φ4, where Φ0 can be I, V, M, L, A, Y, F, W, or P; Φ1 can be L, I, V, M, F, A, or W; Φ2 can be F, M, L, I, V, Y, or W; Φ3 can be L, M, I, V, F, W, or A; and Φ4 can be L, I, M, V, or F. The consensus sequence for the Rev-class NES is Φ0-Φ1pro-X1-Φ2-X2-Φ3-X-Φ4, where Pro is strongly preferred in the Φ1 position, and the preferences for the other hydrophobic positions were not described. Analysis of sequences in NESdb showed that 121 of the 234 experimental NESs fit either PKI-class NESs or Rev-class NESs (recall rate of 52%). The structure-based consensus sequences do not include the Φ1-X2-Φ2 spacing, which is covered by the traditional consensus. The exclusion of this specific spacing stems from structural examination of the CRM1-bound NES of the HIV-1 Rev protein (73LQLPPLERLTL83), which suggested that the Φ1-X2-Φ2 NES spacing can accommodate neither helical nor extended peptide conformation (Güttler et al., 2010). On structural analysis, the Φ1-X2-Φ2 spacing of the Rev NES was reassigned to the Φ0-Φ1pro-X1-Φ2 spacing of the structure-based consensus. However, our analysis suggests that the Rev-class NESs are relatively uncommon since only six experimental NESs in NESdb conform to the Φ0-Φ1pro-X1-Φ2 pattern.
The introduction of Φ0 in the structure-based consensus is based on the observation that CRM1 offers five hydrophobic pockets to bind the Φ positions of NESs (Güttler et al., 2010). We found that Φ0 appears at the N-termini of ∼76% of the experimental NESs that fit either Kosugi or traditional consensus. It is not imperative for NESs to have a Φ0, but the common occurrence of Φ0 residues suggests its importance in NES–CRM1 interactions. The absence or presence of Φ0 may explain the large variation of binding affinities between CRM1 and cargoes. In some cases, Φ0 may be necessary to increase affinity when binding energy provided by the rest of the NES is not sufficient for nuclear export. The observation that not all five hydrophobic residues are mandatory, especially when there are four strong hydrophobics, suggests a greater diversity of spacings for the structure-based consensus sequence (Güttler et al., 2010). The relaxed requirement of only four hydrophobic residues increased the recall rate of the structure-based consensus to 83%, but its precision rate dropped to 2%, as many false positives are introduced. The structure-based consensus with 5-Φs appears to be more balanced than the structure-based consensus with 4-Φs (see Supplementary Table S1).
Our performance analysis of the three existing sets of NES consensus sequences showed that each one has its strengths and limitations. The traditional consensus is strict, with the highest precision rate. The Kosugi consensus is more tolerant and thus achieved the best recall rate. The structure-based consensus with 5-Φs is more precise but has a lower recall rate than the Kosugi consensus. Because high-resolution CRM1–NES structures are available for only three different NES peptides, we expect that the structure-based consensus will evolve to improve sensitivity as new structures become available. In fact, each of the three existing versions of NES consensus require further refinements to be used for prediction purposes.
An improved NES consensus
Sequence analyses of NESs in NESdb led us to the following observations: 1) the six hydrophobic patterns of the Kosugi consensus provide sufficient coverage for the diverse conformations that NES peptides might adopt; 2) Φ1 or Φ2 but not Φ3 or Φ4 positions can accommodate Ala or Thr in addition to the conventional hydrophobic residues Leu, Ile, Val, Phe, and Met; and 3) Trp is rarely found at the C-termini of experimental NESs and therefore should be excluded from NES positions 10–15 (Figure 3C). On the basis of these observations, we propose a new NES consensus with three sequence patterns: Φ1-X1,2,3-Φ2-[^W]2-Φ3-[^W]-Φ4 (type 1), Φ1-X2,3-Φ2-[^W]3-Φ3-[^W]-Φ4 (type 2), and Φ1-X2-Φ2-X[^W]2-Φ3-[^W]2-Φ4 (type 3), where [^W] is any of the 20 amino acids except Trp. Furthermore, Ala and Thr residues can be used only once at either position Φ1 or Φ2.
Our new NES consensus has a recall of 84%, which is much higher than the 65% recall rate of the traditional consensus and only slightly lower than the 89% recall of the Kosugi consensus (Table 1). Only 11 experimental NESs that fit the Kosugi consensus are excluded from this new consensus. The slightly reduced sensitivity of our new NES consensus mainly resulted from restricting Φ3 and Φ4 to conventional hydrophobic residues. On the other hand, this new consensus achieves improved precision compared with the Kosugi consensus, reducing the number of false positives by almost half, from ∼5500 to ∼2900 (6 vs. 4% precision). However, the number of false positives remains high, and the precision of the consensus is still low. The intrinsic low precision of this improved NES consensus may be due to the fact that sequences with hydrophobic residues are frequently found in the interior of proteins. Thus, although many false-positive sequences look like NESs and may indeed bind CRM1 as isolated peptides, they are actually not available to bind CRM1 in the context of full-length cargoes. It will be important to consider the context of the NES sequence within its protein to improve the precision of prediction without lowering recall.
Evolutionary conservation of NESs
Although sequence logos identified amino acids and positions that converged to similar patterns among unrelated NESs sequences in NESdb, the analysis provided no information on whether a given NES is preserved through evolution. It is generally believed that homologous proteins share similar fold and functions. Thus one would expect functional units such as NESs to be highly conserved, especially at the key hydrophobic positions, among homologues of the cargo proteins. We analyzed evolutionary conservation patterns of NESs in cargo proteins and their close homologues using the program AL2CO (Pei and Grishin, 2001), which calculates evolutionary conservation index based on multiple sequence alignment of a protein and its homologues. As shown in Figure 4, evolutionary conservation scores gradually increased from the N- to the C-terminus of experimental NESs. In particular, amino acid positions 6, 10, 13, and 15 display higher evolutionary conservation scores than do neighboring positions. The evolutionarily conserved positions match the consensus sequence pattern of Φ1-X3-Φ2-X2-Φ3-X-Φ4, which is the most prevalent pattern describing the experimental NESs. Therefore key hydrophobic positions of experimental NESs are indeed more conserved in homologues than intervening positions, although it is somewhat puzzling to find that position 15 is less conserved than positions 6, 10, and 13. We also examined evolutionary conservation scores of false-positive NESs. Of interest, the average evolutionary conservation score of false-positive sequences is higher than that of the experimental NESs. This may suggest that the likely locations of false positives are in protein hydrophobic cores, which are generally conserved among homologous proteins. Fu et al. (2011) also noticed that NESs may not be necessarily conserved among all orthologues and thus have lower conservation scores, whereas false positives may be highly conserved. However, unlike experimental NESs, in which key hydrophobic positions are more conserved, no specific positions appear more conserved in the false-positive sequences. This subtle feature may be exploited to distinguish real NESs from false positives.
FIGURE 4:

Position-specific evolutionary conservation scores of experimental NESs vs. false positives that fit the newly proposed consensus. The evolutionary conservation scores were computed with the program AL2CO.
NESs in the PDB
To gain information about structural properties of NESs within cargo proteins, we searched the PDB for NESdb entries. We also used the program BLAST (Altschul et al., 1990, 1997) to search for structures of close homologues of CRM1 cargoes. We found 56 structures of NES-containing proteins in the PDB. Of the 56 structures, 27 are monomeric proteins, 9 are homo-oligomers, 16 are part of protein–protein complexes, and 4 are part of protein–nucleic acid complexes. In addition, crystal structures are available for the NESs of SNUPN, HIV-1 Rev, and PKIα bound to CRM1 (Figure 5A). Taking these results together, we obtained structural information for 68 different NESs. Their sequences, consensus patterns, secondary structure assignments, relative surface accessibilities, and CRM1-binding activities are all listed in Table 2. Critical hydrophobic residues in the NESs are shown in boldface and designated based on the following criteria: 1) priority is given to a consensus pattern with L, F, I, M, or V in all four Φ positions; 2) if such pattern is absent, a pattern with T or A in one of the Φ positions is selected; and 3) in cases in which multiple patterns exist, we chose patterns with maximum relative surface accessibility.
FIGURE 5:

Examples of NESs in the PDB. (A) Crystal structures of NESs of snurportin-1 (PDB ID: 3GB8), HIV-1 Rev (3NBZ), and PKIα (3NBY) bound to CRM1. Cargo proteins are drawn as ribbon diagrams and their NESs colored pink, whereas the rest of the cargoes are colored from N- to C-termini in gradients of light to dark blue. CRM1 is shown in gray surface representation. (B) Examples of NESs that are located within 20 amino acids of the termini of protein domains: E7 of HPV16 (2EWL), MAPKK1 (2Y4I), and Gal3 (2XG3). The NESs of E7 and MAPKK1 are surface accessible, but the NES of Gal3 is not. (C) Examples of two NESs that are located far from protein termini: NESs of Yap1p (1SSE) and Tbx5 (2X6U). Both NESs shown here are flanked by long loops.
TABLE 2A:
Consensus patterns, secondary structure element, relative surface accessibility, and direct CRM1-binding results for experimental NESs with available 3D structures. Continued
| Protein name | NESdb ID | UniProt ID | NES sequencea | Consensus fitb | PDB ID | SSEc | RSA-NESd | RSA-Φse | Bind CRM1f |
|---|---|---|---|---|---|---|---|---|---|
| Snurportin | 1 | O95149 | 1MEELSQALASSFSV14 | + | 3GB8 | HHHHHHHHHHCCCC | 0.57 | 0.74 | + |
| HIV-REV | 2 | P69718 | 72PLQLPPLERLTL83 | + | 3NBZ | CCCCCCHHHCCC | 0.77 | 0.81 | NT |
| PKIα | 5 | P61925 | 36NELALKLAGLDI47 | + | 3NBY | HHHHHHHCCCCC | 0.58 | 0.71 | + |
| p53 | 6 | P04637 | 336ERFEMFRELNEALEL350 | + | 1OLG | HHHHHHHHHHHHHHH | 0.31 | 0.12 | NT |
| RanBP1 | 7 | P43487 | 175DHAEKVAEKLEALSV189 | + | 1K5D | --------------- | 1.00 | 1.00 | + |
| BRCA1 | 10 | P38398 | 81QLVEELLKIICAFQL95 | + | 1JM7 | HHHHHHHHHHHHHHH | 0.41 | 0.34 | NT |
| MAPKK1/MEK1 | 11 | Q05116 | 28TNLEALQKKLEELEL42 | + | 2Y4I | ---------CCCCCC | 0.88 | 0.76 | + |
| MK2 | 20 | P49138 | 337DVKEEMTSALATMRV351 | + | 1KWP | HHHHHHHHHHHHHCC | 0.29 | 0.12 | + |
| Stat1 | 27 | P42224 | 395STNGSLAAEFRHLQL409 | + | 1BF5 | CCCCCEEEEEEEEE | 0.24 | 0.01 | + |
| mPER2 | 31 | O54943 | 455PSVQELTEQIHRLLM469 | + | 3GDI | HHHHHHHHHHHHHCC | 0.40 | 0.20 | + |
| PLC-δ1 | 33 | P10688 | 160MNFKELKDFLKELNI174 | + | 1DJX | EEHHHHHHHHHCCCC | 0.39 | 0.20 | + |
| p73 | 39 | O15350 | 363ENFEILMKLKESLEL377 | + | 2WTT | HHHHHCCHHHHHHHC | 0.09 | 0.09 | + |
| Smad1 | 40 | Q15797 | 400FETVYELTKMCTIRM414 | + | 1KHU | HHHHHHHHHHHEEEE | 0.29 | 0.05 | NT |
| λPKC | 42 | Q62074 | 241SGKASSSLGLQDFDL255 | + | 1ZRZ | -------CCCCCEEE | 0.63 | 0.25 | NT |
| Yap1p | 45 | P19880 | 609PKYSDIDVDGLCSEL623 | - | 1SSE | CCCCCCCHHHHHHHH | 0.46 | 0.23 | + |
| Cyclin D1 | 54 | P24385 | 281VDLACTPTDVRDVDI295 | + | 3G33 | --------------- | 1.00 | 1.00 | NT |
| p38 | 56 | P0C796 | 119YGEKTTQRDLTELEI133 | + | 1N93 | CCCCCCCCCCCHHHH | 0.50 | 0.38 | NT |
| E2F4 | 65 | Q16254 | 56RRIYDITNVLEGIGL70 | + | 1CF7 | HHHHHHHHHHHHHCC | 0.37 | 0.41 | + |
| RSV M | 79 | P03419 | 192AKIIPYSGLLLVITV206 | – | 2VQP | CEEECCCCCEEEEEC | 0.11 | 0.03 | NT |
| FAK1 | 83 | Q05397 | 90LRSEEVHWLHVDMGV104 | + | 2AEH | CCCCCEEEECCCCEH | 0.32 | 0.18 | NT |
| Q00944 | 90LQSEEVHWLHLDMGV104 | ||||||||
| FAK1 | 83 | Q05397 | 511LQVRKYSLDLASLIL525 | + | 1MP8 | HHHCCCCCCHHHHHH | 0.34 | 0.14 | NT |
| Vpr | 86 | P05928 | 55AGVEAIIRILQQLLF69 | + | 1ESX | CHHHHHHHHHHHHHH | 0.43 | 0.54 | NT |
| Q73369 | 55TGVEALIRILQQLLF69 | ||||||||
| nsP2 | 92 | P36328 | 514IVLNQLCVRFFGLDL528 | + | 2HWK | HHHHHHHHHHHCCCH | 0.03 | 0.04 | NT |
| NPM | 93 | P06748 | 88SLGGFEITPPVVLRL102 | + | 2P1B | EEEEEEECCCEEEEE | 0.19 | 0.16 | NT |
| NPM mutA | 94 | P06748 | 282EAIQDLCLAVEEVSL296 | + | 2VXD | --------------- | 1.00 | 1.00 | + |
| CHP1 | 96 | Q99653 | 133DELLQVLRMMVGVNI147 | + | 2CT9 | HHHHHHHHHHCCCCC | 0.38 | 0.49 | + |
| E1A | 99 | P03255 | 69SVMLAVQEGIDLLTF83 | + | 2KJE | CCCHHHCCCCCHHHC | 0.44 | 0.49 | + |
| ADAR1 | 110 | P55265 | 126LSSHFQELSI135 | + | 1QGP | CHHHHCCCHH | 0.66 | 0.71 | + |
| E7 | 114 | P03129 | 70QSTHVDIRTLEDLLM84 | + | 2EWL | ECCHHHHHHHHHHHH | 0.48 | 0.45 | + |
| P21736 | 78ESSAEDLRTLQQLFL91 | ||||||||
| N protein | 118 | Q88935 | 103EFSLPTHHTVRLIRV117 | + | 1P65 | CCCCCHHHHHHHHHH | 0.52 | 0.60 | NT |
| Hxk2 | 120 | P04807 | 304MSSGYYLGEILRLAL318 | + | 1IG8 | HHCHHHHHHHHHHHH | 0.06 | 0.05 | NT |
| Topo IIα | 121 | P11388 | 1013DTVLDILRDFFELRL1027 | + | 1BGW | CCHHHHHHHHHHHHH | 0.21 | 0.09 | + |
| P06786 | 988NSVNEILSEFYYVRL1002 | ||||||||
| Survivin | 123 | O15392 | 84CAFLSVKKQFEELTL98 | + | 2QFA | CHHHHCCCCHHHCCH | 0.43 | 0.33 | NT |
| NC2β | 126 | Q01658 | 66ISPEHVIQALESLGF80 | + | 1JFI | CCHHHHHHHHHHHCC | 0.34 | 0.23 | + |
| Tbx5 | 127 | Q9PWE8 | 146AHWMRQLVSFQKLKL160 | + | 2X6U | HHHHHCCEECCCCEE | 0.28 | 0.10 | NT |
| Paxillin | 136 | P49023 | 262ATRELDELMASLSDF276 | + | 2VZI | CHHHHHHHHHHC--- | 0.76 | 0.70 | + |
| BPV E1 | 139 | P03116 | 404YQNIELITFINALKL418 | + | 2V9P | HCCCCHHHHHHHHHH | 0.29 | 0.18 | NT |
| CaMKIα | 143 | Q63450 | 307FNATAVVRHMRKLQL321 | + | 1A06 | ECHHHHHHHC----- | 0.58 | 0.79 | + |
| STRADα | 154 | Q7RTN6 | 412SGIFGLVTNLEELEV426 | + | 3GNI | -------------CC | 1.04 | 0.83 | + |
| LEI | 155 | P30740 | 279EESYTLNSDLARLGV293 | + | 1HLE | EEEEECHHHHHHHCC | 0.28 | 0.06 | + |
| P05619 | 279EESYDLTSHLARLGV293 | ||||||||
| MK5 | 165 | O54992 | 328GIQQAHAEQLANMRI342 | + | 2OZA | HHHHHHHHHHHHHCC | 0.33 | 0.20 | NT |
| P49137 | 351DVKEEMTSALATMRV365 | ||||||||
| Dab1 | 168 | P97318 | 146AAEPVILDLRDLFQL160 | + | 1OQN | CCHHHHHHHHHHHHH | 0.26 | 0.08 | NT |
| p28GANK | 173 | O75832 | 1MEGCVSNLMV10 | + | 1QYM | --CCCCCHH | 0.61 | 0.41 | NT |
| 2EGCVSNLMV10 | |||||||||
| SIRT2 | 199 | Q8IXJ6 | 37DMDFLRNLFSQTLSL51 | + | 1J8F | HHHHHHHHC------ | 0.61 | 0.58 | + |
| Hst2 | 212 | P53686 | 303EQLLEIVHDLENLSL317 | + | 1Q14 | CHHHHHHHHHHC--- | 0.46 | 0.60 | NT |
| PP2ACα | 213 | P67775 | 144NVMKYFTDLFDYLPL155 | + | 2IAE | CHHHHHHHHHHHCCC | 0.10 | 0.00 | NT |
| ORF-9b | 214 | P59636 | 41KVYPIILRLGSNLSL55 | + | 2CME | EEEEEEECCCCCCEE | 0.28 | 0.19 | NT |
CRM1–NES interactions
We chose 40 of the 68 NESs found in the PDB to test for direct CRM1 binding in pull-down binding assays with recombinant GST-NESs, CRM1, and RanGTP. Although direct CRM1-NES interaction is an absolute prerequisite in CRM1-mediated nuclear export, most studies reporting NES identification did not include experimental evidence for direct CRM1–NESs interactions. Only 30 of the 221 proteins in NESdb have been shown to bind CRM1 directly in in vitro assays using recombinant proteins. Another 22 have been shown to interact with CRM1 in immunoprecipitation assays that do not necessarily demonstrate direct interactions with CRM1.
Each NES peptide tested is ∼20 amino acids long and contains the NES sequence that was annotated in NESdb. Results for the binding assays are shown in Figure 6 and Supplemental Figure S2 and summarized in Table 2. Among the 40 NESs that were tested, 24 NES peptides bound CRM1 in stoichiometric manner and are referred to as CRM1 binders, and 16 failed to bind CRM1. We examined the 16 negative binders for proteolytic degradation using mass spectrometry and found that 14 NESs have the expected masses (Supplemental Table S2). We ascertained that these 14 sequences are negative NESs and moved them to the “NESs in doubt” list. We noticed that 3 of the 14 negative NES do not fit any of the consensus patterns, whereas only 1 of 24 CRM1-binders does not fit any consensus pattern. These data suggest that sequences that match consensus patterns are more likely to bind, further supporting the validity of NES consensus sequences.
FIGURE 6:

CRM1–NESs interactions. Direct interactions between recombinant purified GST-NESs and CRM1 are shown by pull-down binding assays. Immobilized GST-NESs of 40 CRM1 cargoes were incubated with CRM1 in the presence and absence of RanGTP. Bound proteins were resolved with SDS–PAGE and visualized by Coomassie staining. The 40 NESs were chosen because their 3D structures are available in the PDB. (A–C) CRM1 binders (except CHP1-2 and Topo2α-2). (D) A subset of the NESs that do not bind CRM1.
It is surprising to find that approximately one-third of the experimental NESs tested by pull-down binding assays did not bind CRM1 directly. This finding underscores the difficulty in accurately identifying NESs and the caution one must take in interpreting experimental results before designating NESs. Although nuclear export of most NESdb entries is CRM1 dependent, some proteins might not contain NESs but bind CRM1 indirectly through adaptor proteins. Alternatively, a cargo protein might in fact bind CRM1 directly but not have its NES identified correctly. Further complications arise as CRM1 may bind to an isolated NES peptide but may not bind the NES in the context of a protein domain or the full-length protein, as in the case of Stat1 and c-Abl (McBride and Reich, 2003; Hantschel et al., 2005). Binding assays using recombinant full-length cargoes and CRM1 combined with mutagenesis and cellular localization studies will be necessary to accurately identify potential NESs.
We classified the 14 negative binders with expected masses identified earlier as misidentified NESs, which reside in the “NES in doubt” list of NESdb. Other than these 14 negative binders, 3D structures are available for 5 other misidentified NESs in that list. We examined structural properties of the remaining 47 experimental NESs and the 19 misidentified NESs found in the PDB and compared them with >200 false-positive NESs found in structures of CRM1 cargoes.
NES locations in CRM1 cargoes
Three-dimensional structures of NES-containing domains or proteins show that NESs reside at various locations within individual protein domains. Of the 47 experimental NESs, 24 reside within 20 residues of the termini of protein domains. Such terminal NESs may be more accessible for CRM1 binding since only one end of the signals is tethered to the cargo polypeptide chains. E7 of HPV16 (PDB ID: 2EWL) and MAPKK 1 (2Y4I) are two examples of proteins with terminal NESs (Figure 5B). We note that NESs at protein or domain termini do not guarantee accessibility if NES residues contribute to the hydrophobic core of the protein or are part of a β-sheet. For example, the NES of Gal3 (2XG3), one of the five misidentified NESs located at protein domain termini, is in the middle of a β-sheet. Interaction of the Gal3 NES with CRM1 would require unraveling of the β-sheet (Figure 5B). Internal NESs that are located adjacent to long loops might also be accessible to CRM1. Fourteen experimental NESs in the PDB precede and/or are preceded by loops longer than five amino acids. Two examples of NESs flanked by long loops are found in the AP-1 transcription factor (1SSE) and T-box protein 5 (2X6U; Figure 5C). Seven experimental NESs in the PDB are located in internal sites of proteins that are not flanked by loops. By comparison, we found that only 35 false positives (17%) are located at protein domain termini, and 53 false positives are located in internal sites of proteins that are not flanked by loops.
Secondary structures of the NESs
Structures of CRM1–NES complexes showed that the C-terminal portions (residues 13–15) of CRM1-bound NESs adopt extended or loop conformations, whereas the rest of NESs are either short α-helices or loops (Figure 5A). In the PDB, 22 of the 44 experimental NESs that are not bound to CRM1 adopt similar secondary structure elements (SSEs) as NESs in the CRM1–NES complexes (Table 2A and Figure 7A). Sixteen of these NESs adopt helix-loop conformations and 6 adopt entirely loop conformations. Four experimental NESs were not modeled due to disorder or flexibility. Thus SSEs of 59% of experimental NESs appear suitable for CRM1 binding. Of the remaining 18 NESs, 10 are entirely helical and part of long helices, and 8 are in β-strands that are part of β-sheets (Table 2A). In contrast, a smaller fraction of the false positives (36%, or 73 of 202) adopt helix-loop or entirely loop conformation, and many more are entirely helical (54) or part of β-sheets (75). Misidentified NESs showed similar distribution of SSEs as false positives, as 31% of misidentified NESs (6 of 19) adopt helix-loop or entirely loop conformations, five are entirely helical, and eight are part of β-sheets (Table 2B and Figure 7, B and C).
FIGURE 7:

Examples of secondary structural elements adopted by NESs. (A) The NES of PLC-δ1 (1DJX) adopts a combined α-helix-loop conformation, similar to the secondary structure of the CRM1-bound SNUPN NES. (B) The NES of Stat1 (298KQVLWDRTFSLFQQL312; 1BF5) is entirely α-helical. (C) The NES of CDK5 (64VRLHDVLHSDKKLTL78; 1UNG) is part of a β-sheet. The color schemes used here are the same as in Figure 5.
TABLE 2B:
Consensus patterns, secondary structure element, relative surface accessibility, and direct CRM1-binding results for NESs in doubt, with available 3D structures.
| Protein name | NESdb ID | UniProt ID | NES sequencea | Consensus fitb | PDB ID | SSEc | RSA-NESd | RSA-Φse | Bind CRM1f |
|---|---|---|---|---|---|---|---|---|---|
| Actin | 14 | P60010 | 166YAGFSLPHAILRIDL180 | + | 1YAG | ECCEECHHHCEEECC | 0.27 | 0.08 | NT |
| Actin | 14 | P60010 | 207EIVRDIKEKLCYVAL221 | + | 1YAG | HHHHHHHHHHCCCCC | 0.22 | 0.17 | NT |
| c-Abl | 21 | P00520 | 1078EAINKLESNLRELQI1092 | + | 1ZZP | HHHHHHHHHHHHHCC | 0.33 | 0.22 | + |
| P00519 | 1085EAINKLENNLRELQI1099 | ||||||||
| Stat1 | 27 | P42224 | 190SDQKQEQLLLKKMYL204 | + | 1BF5 | -------CHHHHHHH | 0.72 | 0.39 | NT |
| Stat1 | 27 | P42224 | 298KQVLWDRTFSLFQQL312 | – | 1BF5 | HHHHHHHHHHHHHHH | 0.30 | 0.10 | NT |
| APC protein | 47 | P25054 | 163AQLQNLTKRIDSLPL174 | + | 1M5I | HHHHHHHHHHCCCC- | 0.53 | 0.38 | – |
| NPM | 93 | P06748 | 35NDENEHQLSLRTVSL49 | + | 2P1B | ----CCEEEEEEEEE | 0.37 | 0.00 | – |
| CHP1g | 96 | Q99653 | 171ISFTEFVKVLEKVDV185 | + | 2CT9 | EEHHHHHHCCCCCCH | 0.34 | 0.25 | – |
| Net | 98 | P41970 | 1MESAITLWQFLLQL14 | + | 1HBX | CCCCHHHHCCCCC | 0.32 | 0.23 | – |
| Hxk2 | 120 | P04807 | 19VPKELMQQIENFEKI33 | – | 1IG8 | CCHHHHHHHHHHHHH | 0.39 | 0.22 | – |
| Topo IIαg | 121 | P11388 | 1051QARFILEKIDGKIII1065 | + | 1BGW | HHHHHHHHHCCCCCC | 0.31 | 0.20 | – |
| P06786 | 1026QVKFIKMIIEKELTV1040 | ||||||||
| Gal3 | 128 | P16110 | 236NHRMKNLREISQLGI250 | + | 2XG3 | ECCCCCHHHCCEEEE | 0.33 | 0.04 | – |
| P17931 | 222NHRVKKLNEISKLGI236 | + | 1XML | CHHHHHHHCCCCEEE | 0.51 | 0.41 | – | ||
| DcpS | 146 | Q96C86 | 136TEKHLQKYLRQDLRL150 | ||||||
| FLIP-L | 147 | O15519 | 432RQERKRPLLDLHIEL446 | – | 3H11 | HHCCCCCHHHHHHHH | 0.34 | 0.29 | – |
| MK5 | 165 | O54992 | 338ANMRIQDLKVSLKPL352 | ||||||
| P49137 | 361ATMRVDYEQIKIKKI375 | + | 2OZA | HHHCCCCCCCCCCCH | 0.59 | 0.65 | – | ||
| ERα | 166 | P03372 | 440LQGEEFVCLKSIILL454 | + | 2OCF | CCHHHHHHHHHHHHH | 0.11 | 0.02 | – |
| FGF-1 | 170 | P05230 | 138THYGQKAILFLPLPV152 | + | 1JQZ | CCCCCCCCCEEEEEC | 0.30 | 0.24 | – |
| Oct-6 | 182 | P21952 | 362PSAHEITGLADSLQL376 | + | 2XSD | CCHHHHHHHHHHHCC | 0.43 | 0.17 | – |
| Cdk5-p27 | 194 | Q00535 | 64VRLHDVLHSDKKLTL78 | – | 1UNG | CCEEEEEEECCEEEE | 0.31 | 0.21 | – |
| Cdk5-p27 | 194 | Q00535 | 133LINRNGELKLANFGL147 | + | 1UNG | EECCCCCEEECCHHH | 0.24 | 0.12 | – |
| APRIL | 196 | Q92688 | 106LEPLKKLECLKSLDL120 | + | 2ELL | HHHHCCCCCCCEEEC | 0.24 | 0.01 | – |
We also performed position-specific SSE analysis of the 15 NES positions (Figure 8). We found that 60–70% of all experimental NESs in the PDB adopt α-helical conformation in the N-terminal region spanning positions 3–10. Only ∼30% of the NESs form loops, and only ∼10% adopt β-strand conformations, in the N-terminal one-third of the signals. However, the trend changes at the C-termini of the NESs, with the last two hydrophobic positions equally likely to be α-helical or coiled (Figure 8A). In contrast, position-specific SSE analysis of the false-positive NESs showed persistent tendency to form α-helix throughout the entire length of the false positive NESs (Figure 8B). Although the curves are rough due to the smaller sample size, position-specific SSE analysis of misidentified NESs displayed similar trends as those of the false positive NESs (Supplementary Figure S1A).
FIGURE 8:

Secondary structure elements (SSEs) of the NESs. (A) SSE composition of experimental NESs based on SSEs extracted from the 3D structures. (B) SSE composition of the false positives based on SSEs extracted from the 3D structures. (C) SSE composition of experimental NESs based on predicted SSEs. (D) SSE composition of the false positives based on predicted SSE. SSEs from 3D structures were extracted using the program DSSP (Kabsch and Sander, 1983), and SSE prediction was carried out using the program PSIPRED (Jones, 1999; Buchan et al. 2010).
Because 3D structures are not available for many NESdb entries, we used the program PSIPRED (Jones, 1999; Buchan et al., 2010) to predict SSEs of all 234 experimental NESs and all the false-positive sequences that fit the newly proposed consensus. The results showed that the predicted SSEs of 63 experimental NESs are helix-loop conformations, 81 are mostly loop conformations, 73 are part of long helices, and 17 are part of β strands. The predicted SSEs demonstrated similar features as the structural data, with a strong preference for α-helices and a bias against β-strands. The C-termini of experimental NESs tend to progress from helical to coiled, whereas false-positive sequences tend to remain entirely helical (Figure 8, C and D). Surprisingly, although SSEs observed in 3D structures of misidentified NESs resembled those in the false positives, predicted SSEs of misidentified NESs showed similar trends as the experimental NESs (Supplementary Figure S1B). Such inconsistency may present a challenge in NES prediction efforts that incorporate predicted SSEs as a feature.
Based on the SSEs of NESs in 3D structures, the α-helix that transitions to loop conformation appears to be the most favored conformation for NESs. This combined helix-loop conformation may require the least rearrangement to fit into the NES groove of CRM1. The all-loop conformations may exert less restraint on the backbone of NESs when the hydrophobic residues dock into the Φ pockets of the CRM1 groove. In contrast, the all-helical conformation will likely require some degree of unfolding at the C-terminal end to occupy the narrow portion of the CRM1-binding groove. The β-strand conformation requires the most significant conformational change due to inappropriate periodicity of hydrophobic residues or inaccessibility of the NES peptide in a β-sheet. Indeed, we observed that 8 of the 19 misidentified NESs occur in β-strands in the PDB, and all but one failed to bind CRM1 in pull-down binding experiments (Figure 6 and Table 2B). We need to the keep in mind that the NESs in the PDB may adopt SSEs in different states of the cargo proteins due to intrinsic protein flexibility, protein modifications, or changes of binding partners.
Crystallographic B-factors and disorder propensity of the NESs
Of the 47 experimental NESs in the PDB, the NESs of RanBP1 (1K5D), cyclin D1 (3G33), STRADα (3GNI), and NPM mutant A (2VXD) are part of constructs used in structure determination, but the NES residues were not modeled in the final structures probably due to high mobility or disorder. In addition, only fractions of seven other NESs were modeled, suggesting that these unobserved residues were also mobile or structurally disordered. These observations prompted us to analyze structural flexibility and intrinsic disorder of the NESs and sequences that flank the signals.
Structural flexibility or mobility can be illustrated by large crystallographic B-factors (temperature factors) that measure the degree to which the electron density spreads out in x-ray structures. It was previously observed that several NESs are located at or close to regions with large B-factors (la Cour et al., 2004). We compared the average B-factors of Cα atoms in the NESs with the average Cα B-factors of the entire proteins or domains. Average Cα B-factors were calculated for the 37 experimental NESs in crystal structures. We found that the majority (78%) of experimental NESs have similar B-factors as the whole proteins, whereas 16% of the experimental NESs have B-factors at least one standard deviation (SD) above the average B-factor of the entire protein, and 6% have B-factors at least one SD below those of the entire protein. Similar analysis of the false-positive NESs in crystal structures showed that 92% have similar B-factors as the whole proteins, and 2 and 6% have higher and lower B-factors, respectively. Therefore B-factors of the false-positive NESs showed similar distribution to those of the experimental NESs. This analysis suggests that experimental NESs are not usually found in protein regions with high B-factors, and this crystallographic parameter is not useful to distinguish experimental from false-positive NESs.
We also examined the propensity for intrinsic disorder of the NESs. Disordered regions of proteins often contain functional sites, and numerous computational tools have been developed to analyze protein sequences for potential intrinsic disorder. Most of these prediction tools are quite successful since there is a clear association between disorder propensity and sequence features such as low complexity and high aromatic composition. We used the program DISOPRED2 to predict disorder propensity of NES-containing proteins in NESdb (Ward et al., 2004). We calculated average disorder scores at each position of the experimental NESs to compare with the disorder scores of the false positives. Disorder propensity plots show that experimental NESs have much higher disorder scores than false positives (Figure 9). These findings are consistent with previous observations that NES sites display more disorder than false-positive NESs (Fu et al., 2011). Disorder scores for misidentified NESs are comparable to those of experimental NESs.
FIGURE 9:
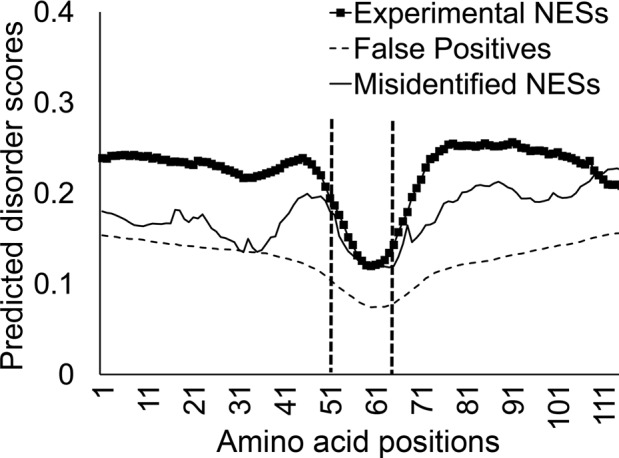
Disorder propensities of the NESs. Predicted disorder scores of experimental NESs compared with those of the misidentified NESs and the false positives. Disorder scores were obtained for 50 residues preceding the NESs, the NESs, and 50 residues following the NESs. NES residues are found within the two vertical dotted lines. Disorder prediction was carried out by DISOPRED2 (Ward et al., 2004).
We observed that N-terminal portions of NESs have higher disorder scores than their C-termini (Figure 9). Of interest, the drop in predicted disorder at the C-termini of NESs correlates well with the increase in evolutionary conservation scores of experimental NESs among homologous cargoes (Figure 4). Although we do not fully understand the basis of such correlation, this relationship may be further investigated and perhaps exploited to improve NES prediction. We also found that sequences that flank experimental NESs (50 amino acids on either side of the NESs) have significantly higher disorder scores than sequences that surround misidentified NESs or false-positive NESs (Figure 9). Disordered regions surrounding experimental NESs may increase their access to CRM1 or may also enhance the ability of NESs to adapt to optimal conformations for CRM1 binding.
Surface accessibility of the NESs
Three-dimensional structures of CRM1-NES complexes revealed that NESs bind to a hydrophobic groove of CRM1 with their Φ0–Φ4 hydrophobic residues docking into hydrophobic pockets within the groove (Dong et al., 2009a, b; Monecke et al., 2009; Güttler et al., 2010). Structural comparison of the binary CRM1–NES, the ternary RanGTP-CRM1-NES, and the binary CRM1–RanGTP structures revealed that conformations of NES-binding grooves are all similar, suggesting that diverse NESs adapt structurally to fit into a structurally invariant binding site (Güttler et al., 2010). A critical requirement for NES association with CRM1 is the exposure of key hydrophobic residues of the NES before CRM1 binding. However, hydrophobic residues, especially those on hydrophobic surfaces of amphipathic helices, tend to be buried in proteins. To investigate the accessibility of NESs within their protein cargoes, we computed their relative surface accessibility (RSA) from the available 3D structures.
The RSA of a residue refers to the ratio of its solvent accessible surface area (ASA) in the 3D structure to its ASA in an extended tripeptide conformation (Ala-X-Ala or Gly-X-Gly; Rost and Sander, 1994). RSA (values range from 0 to 1) rather than ASA values are usually used to compare accessibilities of amino acids of different sizes. A residue is usually classified as exposed or buried based on an arbitrary, user-defined threshold. In this work, we computed RSA values of NES residues using the program NACCESS (Hubbard and Thornton, 1993), and a residue is considered exposed if its RSA is greater than the widely adopted cutoff value of 0.25. The RSA of an NES is calculated by averaging the RSAs of all its residues. The same cutoff value is used to describe exposure of the NES. RSAs of NESs (RSA-NES) are listed in Table 2 along with average RSAs of the key hydrophobic NES residues (RSA-Φs). We grouped the 47 experimental NESs in the PDB into three categories according to their RSA-NES and RSA-Φs values (Table 3). Twenty-three NESs are categorized in the exposed group, where both RSA-NESs and RSA-Φs are >0.25. Another 16 NESs are categorized as partially exposed, with RSA-NESs of >0.25 and RSA-Φs of ≤0.25. Finally, eight NESs are deemed buried, with both RSA-NESs and RSA-Φs values of ≤0.25. Note that for NESs that are located at protein–protein or protein–nucleic acid interfaces, RSA values were computed without their binding partners.
TABLE 3:
Surface accessibility of experimental NESs compared with that of the misidentified NESs and false positives.
| RSA-NESa | RSA-Φ1b | RSA-Φ2 | RSA-Φ3 | RSA-Φ4 | Exposedc (%) | Partially exposedd (%) | Buriede (%) | ||
|---|---|---|---|---|---|---|---|---|---|
| 3D structuresf | Experimental NESs | 0.45 ± 0.25 | 0.31 ± 0.30 | 0.25 ± 0.27 | 0.36 ± 0.34 | 0.36 ± 0.35 | 49 | 34 | 17 |
| Misidentified NESs | 0.36 ± 0.14 | 0.18 ± 0.21 | 0.17 ± 0.23 | 0.17 ± 0.24 | 0.32 ± 0.33 | 26 | 58 | 16 | |
| False positives | 0.27 ± 0.12 | 0.17 ± 0.21 | 0.15 ± 0.20 | 0.12 ± 0.17 | 0.16 ± 0.21 | 17 | 42 | 42 | |
| Predictedg | Experimental NESs | 0.24 ± 0.08 | 0.16 ± 0.14 | 0.12 ± 0.13 | 0.12 ± 0.11 | 0.17 ± 0.13 | 10 | 32 | 58 |
| Misidentified NESs | 0.22 ± 0.11 | 0.13 ± 0.18 | 0.12 ± 0.18 | 0.10 ± 0.14 | 0.12 ± 0.14 | 17 | 13 | 67 | |
| False positives | 0.21 ± 0.10 | 0.16 ± 0.15 | 0.14 ± 0.14 | 0.10 ± 0.11 | 0.12 ± 0.12 | 13 | 22 | 65 |
aAverage relative surface accessibility of all residues in the NES.
bAverage relative surface accessibility of key hydrophobic residues in the NES.
cA sequence is defined as exposed if both its RSA-NES and RSA-Φs are >0.25.
dA sequence is defined as partially exposed if its RSA-NES is >0.25 and RSA-Φs is ≤0.25.
eA sequence is defined as buried if both its RSA-NES and RSA-Φs are ≤0.25.
fCalculated surface accessibility of 47 experimental NESs, 19 misidentified NESs, and 202 false positives in 3D structures.
gPredicted surface accessibility of 234 experimental NESs, 24 misidentified NESs, and 2903 false positives using the program SABLE.
We also studied differences in surface accessibilities between experimental NESs, false-positive NESs, and misidentified NESs, which were all measured in the same manner. The average RSA-NES values for experimental NESs are larger than those of the misidentified NESs or the false positives (0.45 vs. 0.36 and 0.27; Table 3). All four key hydrophobic residues in experimental NESs are also more exposed than the corresponding residues in misidentified or false-positive NESs. This difference is especially striking for the Φ4 position, where the Φ4 RSA of experimental NESs is more than double that of the false positives (0.36 vs. 0.16). Forty-nine percent of experimental NESs are considered exposed, in contrast to only 5 misidentified NESs (26%) and 34 false positives (17%), which are classified as exposed. Therefore it is evident from surface accessibility analysis of cargo structures that experimental NESs are more exposed than false-positive NESs.
We predicted RSA values for all 234 experimental NESs (including NESs with 3D structures), all 24 misidentified NESs, and 2903 false-positive sequences using the program SABLE to gain insight into their surface accessibility features (Adamczak et al., 2004, 2005). Table 3 shows that the predicted RSA-NES and RSA-Φs values for experimental NESs are comparable to those of the misidentified NESs and the false positives. Nevertheless, the trend for predicted RSA is similar to the measured accessibility of NESs in the PDB. More misidentified NESs and the false positives are classified as buried than experimental NESs (67 and 65% vs. 58%).
It is intriguing to find that a substantial fraction of experimental NESs are not accessible, with their hydrophobic residues buried in the cargo proteins. How do these NESs gain access to and bind CRM1? Of interest, among the eight NESs in the PDB that are not accessible, only four are monomers. Three others form oligomers, and one is part of a protein–nucleic acid complex. It is possible that these proteins and their NESs may undergo conformational changes upon removal or changes of the binding partners, thus exposing previously buried NESs. Furthermore, 14 experimental NESs in the PDB are connected to the rest of the cargoes by loops longer than five amino acids, which may allow equilibration between multiple local conformations, some of which may entail more-accessible NESs. Finally, protein modifications like mutations or posttranslational modifications such as phosphorylation, acetylation, methylation, or ubiquitination might unmask previously buried NESs. For example, the normally nucleolar protein nucleophosmin (NPM) is mutated in acute myeloid leukemia to create NESs at the C-terminus of its C-terminal domain, which then mislocalize mutant NPM to the cytoplasm. The new NESs are expected to slightly extend a mostly buried C-terminal helix, but, of interest, the mutations involve loss of two Trp residues in the helix that leads to domain unfolding and NES exposure (Chen et al., 2006; Falini et al., 2006, 2007; Bolli et al., 2007). Therefore, although a real NES should ideally be exposed rather than buried in the cargo protein, we cannot yet simplistically reject a particular sequence as a NES based on its lack of solvent accessibility in a 3D structure.
DISCUSSION
The leucine-rich or classical NES is the only characterized class of NES. Three sets of NES consensus sequences were previously proposed to describe the hydrophobic patterns of the NESs. However, since large portions of all three consensus patterns essentially describe the amphipathic helix, which is found in most proteins, there exist many false-positive sequences that merely conform to NES consensus but do not have nuclear export capability. Therefore the NES appears to be a complex and diverse signal that is captured not just by its consensus sequences but also by other physical properties. In most cases, NESs were experimentally identified and reported on an individual basis. Although several attempts have been made to characterize NES features beyond sequence patterns, these efforts were either carried out with a limited number of NESs or focused on a limited set of NES properties.
We compiled a database named NESdb with >200 experimentally identified NES-containing CRM1 cargoes and conducted a comprehensive analysis of both the sequence and structural features of 234 experimentally defined NESs in the database. Our analysis focused on identifying differences between experimental and the false-positive NESs. We found that despite the shared sequence patterns between experimental NESs and false positives, the C-termini of experimental NESs are less likely to accommodate bulky amino acids than those of the false positives, and substitution of traditional hydrophobic residues by amino acids with weaker hydrophobicity is better tolerated at the N-termini of experimental NESs than at their C-termini. In addition, experimental NESs are more likely to be negatively charged than false positives. On the basis of these subtle differences, we improved the NES consensus to achieve a balanced recall rate and precision.
Experimental NESs are further distinguished from the false positives by their structural features. The secondary structural elements of experimental NESs are usually entirely loops or start with an α-helix that transition to a short loop. In contrast, false positives tend to be α-helical throughout the entire length of the NESs. Presumably, the α-helix-loop or all-loop NES conformations are more complementary to the rigid NES-binding groove of CRM1, as seen in the crystal structures of CRM1–NES complexes. Experimental NESs also show higher propensity for intrinsic structural disorder in the NES and its surrounding sequences than the false positives. Disordered regions in unbound cargoes can provide both the accessibility and flexibility needed for interactions with CRM1. Hydrophobic residues tend to be buried in protein structures, but hydrophobic residues in experimental NESs show remarkably greater accessibilities than those in false positives.
The NES represents another example of complex signals that must be described by a set of structural and sequence properties. Accurate prediction of these complex signals from the genome requires such physical and chemical knowledge. The features revealed here that distinguish experimental from false-positive NESs will be important for future accurate NES prediction.
MATERIALS AND METHODS
NES data sets
NES sequences are available in the NESdb database (http://prodata.swmed.edu/LRNes), which contains 221 experimentally identified CRM1 cargoes that were published up to December 2011 and was compiled by manually curating the published literature. Sequence similarity among the 221 proteins was identified using the program CD-HIT (Li and Godzik, 2006). Two hundred proteins with <40% sequence identity to other proteins in NESdb, which contain 234 experimental NES sequences, were used in the analyses. An experimental NES is defined as a short peptide of 15 amino acids (or less if located at the N-terminus) that was demonstrated experimentally to possess nuclear export capabilities. Negative NESs are defined as 15-residue-long protein sequences that are located outside the experimental NESs. More than 100,000 negative sequences from NESdb entries were generated using a sliding window protocol. False-positive NESs are negative NESs that fit NES consensus patterns. There are also 24 misidentified NESs in NESdb. These were sequences that were initially identified to be NESs but about which subsequent experiments raised doubt on whether the sequences are indeed NESs.
Sequence analyses of the NESs
Sequence logos were generated using the program WebLogo (Crooks et al., 2004). Each amino acid position is represented by a stack of letters. The height of the stack (measured in bits) reflects the degree of sequence conservation at the corresponding position, and the height of each letter represents the relative frequency of the amino acids at the corresponding location (Schneider and Stephens, 1990; Crooks et al., 2004). Evaluation of NES consensus and position-specific amino acid frequency analysis were performed using Excel and Python scripts. Evolutionary conservation scores were calculated with the program AL2CO (Pei and Grishin, 2001). A sequence similarity search was carried out by the BLAST program (Altschul et al., 1990, 1997).
Structural analyses of the NESs
High-resolution structures of CRM1 cargoes or their close homologues were found using BLAST search (Altschul et al., 1990, 1997) against the PDB and the structures visualized with PyMOL (PyMOL Molecular Graphics System; Schrödinger, New York, NY). Secondary structure elements (SSEs) of the NESs in these structures were assigned using the DSSP program (Kabsch and Sander, 1983). SSEs of all NES sequences analyzed were predicted using the program PSIPRED (Jones, 1999; Buchan et al., 2010). Disorder scores of NES sequences were calculated using the program DISOPRED2 with the false-positive threshold set to 10% (Ward et al., 2004). The RSA of a residue within an NES was obtained by dividing its ASA in 3D structures with its ASA when in an extend conformation (Ala-X-Ala or Gly-X-Gly tripeptide; Rost and Sander, 1994). A threshold value of 0.25 was chosen to classify its accessibility status of buried versus exposed. If a residue was included in the construct used in structure determination but not modeled in the final structure, its RSA was set to 1. RSA values in NES-containing protein structures were computed using the program NACCESS (Hubbard and Thornton, 1993), and RSA predictions were carried out using the program SABLE (Adamczak et al., 2004, 2005).
In vitro binding assays
Constructs of 40 different NESs were generated by ligation of annealed oligonucleotides into the pGEX-Tev vector and verified by sequencing. GST-NESs were expressed and purified as previously reported.
Supplementary Material
Acknowledgments
We thank Yeeling (June) Lam for help with locating NESdb entries in the PDB and with initial binding assays. We also thank Maria Islam-Meredith for technical help. This work was funded by the National Institutes of Health (F32GM093493 to D.X., R01-GM069909 to Y.M.C., and R01-GM094575 to N.V.G.), the Welch Foundation (I-1532 to Y.M.C. and I-1505 to N.V.G.), the Leukemia and Lymphoma Society (Scholar, to Y.M.C.), and the UT Southwestern Endowed Scholars Program (to Y.M.C).
Abbreviations used:
- ASA
accessible surface area
- CRM1
chromosome region maintenance 1
- GO
Gene Ontology
- HIV
human immunodeficiency virus
- LMB
leptomycin B
- NES
nuclear export signal
- NLS
nuclear localization signal
- PKIα
cAMP-dependent protein kinase inhibitor alpha
- RSA
relative surface accessibility
- SSE
secondary structure element
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-01-0046) on July 25, 2012.
REFERENCES
- Adamczak R, Porollo A, Meller J. Accurate prediction of solvent accessibility using neural networks-based regression. Proteins. 2004;56:753–767. doi: 10.1002/prot.20176. [DOI] [PubMed] [Google Scholar]
- Adamczak R, Porollo A, Meller J. Combining prediction of secondary structure and solvent accessibility in proteins. Proteins. 2005;59:467–475. doi: 10.1002/prot.20441. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, et al. Gene Ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Fridell RA, Benson RE, Hua J, Cullen BR. Protein sequence requirements for function of the human T-cell leukemia virus type 1 Rex nuclear export signal delineated by a novel in vivo randomization-selection assay. Mol Cell Biol. 1996;16:4207–4214. doi: 10.1128/mcb.16.8.4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolli N, et al. Born to be exported: COOH-terminal nuclear export signals of different strength ensure cytoplasmic accumulation of nucleophosmin leukemic mutants. Cancer Res. 2007;67:6230–6237. doi: 10.1158/0008-5472.CAN-07-0273. [DOI] [PubMed] [Google Scholar]
- Buchan DW, Ward SM, Lobley AE, Nugent TC, Bryson K, Jones DT. Protein annotation and modelling servers at University College London. Nucleic Acids Res. 2010;38:W563–W568. doi: 10.1093/nar/gkq427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Rassidakis GZ, Medeiros LJ. Nucleophosmin gene mutations in acute myeloid leukemia. Arch Pathol Lab Med. 2006;130:1687–1692. doi: 10.5858/2006-130-1687-NGMIAM. [DOI] [PubMed] [Google Scholar]
- Chook YM, Blobel G. Karyopherins and nuclear import. Curr Opin Struct Biol. 2001;11:703–715. doi: 10.1016/s0959-440x(01)00264-0. [DOI] [PubMed] [Google Scholar]
- Chook YM, Süel KE. Nuclear import by karyopherin-betas: recognition and inhibition. Biochim Biophys Acta. 2011;1813:1593–1606. doi: 10.1016/j.bbamcr.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti E, Izaurralde E. Nucleocytoplasmic transport enters the atomic age. Curr Opin Cell Biol. 2001;13:310–319. doi: 10.1016/s0955-0674(00)00213-1. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Zhao L, Guo H, Zheng AC. The nucleocytoplasmic transport of viral proteins. Virol Sin. 2010;25:79–85. doi: 10.1007/s12250-010-3099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingwall C, Robbins J, Dilworth SM, Roberts B, Richardson WD. The nucleoplasmin nuclear location sequence is larger and more complex than that of SV-40 large T antigen. J Cell Biol. 1988;107:841–849. doi: 10.1083/jcb.107.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingwall C, Sharnick SV, Laskey RA. A polypeptide domain that specifies migration of nucleoplasmin into the nucleus. Cell. 1982;30:449–458. doi: 10.1016/0092-8674(82)90242-2. [DOI] [PubMed] [Google Scholar]
- Dong X, Biswas A, Chook YM. Nat Struct Mol Biol. 2009a;16:558–560. doi: 10.1038/nsmb.1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Biswas A, Süel KE, Jackson LK, Martinez R, Gu H, Chook YM. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature. 2009b;458:1136–1141. doi: 10.1038/nature07975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emami S. Interplay between p53-family, their regulators, and PARPs in DNA repair. Clin Res Hepatol Gastroenterol. 2011;35:98–104. doi: 10.1016/j.gcb.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Engelsma D, Bernad R, Calafat J, Fornerod M. Supraphysiological nuclear export signals bind CRM1 independently of RanGTP and arrest at Nup358. EMBO J. 2004;23:3643–3652. doi: 10.1038/sj.emboj.7600370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falini B, Albiero E, Bolli N, De Marco MF, Madeo D, Martelli M, Nicoletti I, Rodeghiero F. Aberrant cytoplasmic expression of C-terminal-truncated NPM leukaemic mutant is dictated by tryptophans loss and a new NES motif. Leukemia. 2007;21:2052–2054. doi: 10.1038/sj.leu.2404839. [DOI] [PubMed] [Google Scholar]
- Falini B, et al. Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML. Blood. 2006;107:4514–4523. doi: 10.1182/blood-2005-11-4745. [DOI] [PubMed] [Google Scholar]
- Fischer U, Huber J, Boelens WC, Mattaj IW, Lührmann R. The HIV-1 Rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell. 1995;82:475–483. doi: 10.1016/0092-8674(95)90436-0. [DOI] [PubMed] [Google Scholar]
- Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–1060. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- Fu SC, Imai K, Horton P. Prediction of leucine-rich nuclear export signal containing proteins with NESsential. Nucleic Acids Res. 2011;39:e111. doi: 10.1093/nar/gkr493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M, Nishida E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature. 1997;390:308–311. doi: 10.1038/36894. [DOI] [PubMed] [Google Scholar]
- Görlich D, Kutay U. Transport between the cell nucleus and the cytoplasm. Annu Rev Cell Dev Biol. 1999;15:607–660. doi: 10.1146/annurev.cellbio.15.1.607. [DOI] [PubMed] [Google Scholar]
- Güttler T, Görlich D. Ran-dependent nuclear export mediators: a structural perspective. EMBO J. 2011;30:3457–3474. doi: 10.1038/emboj.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güttler T, Madl T, Neumann P, Deichsel D, Corsini L, Monecke T, Ficner R, Sattler M, Görlich D. NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nat Struct Mol Biol. 2010;17:1367–1376. doi: 10.1038/nsmb.1931. [DOI] [PubMed] [Google Scholar]
- Hantschel O, Wiesner S, Güttler T, Mackereth CD, Rix LL, Mikes Z, Dehne J, Görlich D, Sattler M, Superti-Furga G. Structural basis for the cytoskeletal association of Bcr-Abl/c-Abl. Mol Cell. 2005;19:461–473. doi: 10.1016/j.molcel.2005.06.030. [DOI] [PubMed] [Google Scholar]
- Henderson BR, Eleftheriou A. A comparison of the activity, sequence specificity, and CRM1-dependence of different nuclear export signals. Exp Cell Res. 2000;256:213–224. doi: 10.1006/excr.2000.4825. [DOI] [PubMed] [Google Scholar]
- Hubbard SJ, Thornton JM. NACCESS. Department of Biochemistry and Molecular Biology. University College London; 1993. [Google Scholar]
- Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Kalderon D, Richardson WD, Markham AF, Smith AE. Sequence requirements for nuclear location of simian virus 40 large-T antigen. Nature. 1984;311:33–38. doi: 10.1038/311033a0. [DOI] [PubMed] [Google Scholar]
- Kosugi S, Hasebe M, Tomita M, Yanagawa H. Nuclear export signal consensus sequences defined using a localization-based yeast selection system. Traffic. 2008;9:2053–2062. doi: 10.1111/j.1600-0854.2008.00825.x. [DOI] [PubMed] [Google Scholar]
- Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, Yoshida M, Horinouchi S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci USA. 1999;96:9112–9117. doi: 10.1073/pnas.96.16.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo N, Wolff B, Sekimoto T, Schreiner EP, Yoneda Y, Yanagida M, Horinouchi S, Yoshida M. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp Cell Res. 1998;242:540–547. doi: 10.1006/excr.1998.4136. [DOI] [PubMed] [Google Scholar]
- Kutay U, Güttinger S. Leucine-rich nuclear-export signals: born to be weak. Trends Cell Biol. 2005;15:121–124. doi: 10.1016/j.tcb.2005.01.005. [DOI] [PubMed] [Google Scholar]
- la Cour T, Gupta R, Rapacki K, Skriver K, Poulsen FM, Brunak S. NESbase version 1.0: a database of nuclear export signals. Nucleic Acids Res. 2003;31:393–396. doi: 10.1093/nar/gkg101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- la Cour T, Kiemer L, Mølgaard A, Gupta R, Skriver K, Brunak S. Analysis and prediction of leucine-rich nuclear export signals. Protein Eng Des Sel. 2004;17:527–536. doi: 10.1093/protein/gzh062. [DOI] [PubMed] [Google Scholar]
- Lanford RE, Butel JS. Construction and characterization of an SV40 mutant defective in nuclear transport of T antigen. Cell. 1984;37:801–813. doi: 10.1016/0092-8674(84)90415-x. [DOI] [PubMed] [Google Scholar]
- Lange A, Mills RE, Lange CJ, Stewart M, Devine SE, Corbett AH. Classical nuclear localization signals: definition, function, and interaction with importin alpha. J Biol Chem. 2007;282:5101–5105. doi: 10.1074/jbc.R600026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BJ, Cansizoglu AE, Süel KE, Louis TH, Zhang Z, Chook YM. Rules for nuclear localization sequence recognition by karyopherin beta 2. Cell. 2006;126:543–558. doi: 10.1016/j.cell.2006.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Lim MJ, Wang XW. Nucleophosmin and human cancer. Cancer Detect Prev. 2006;30:481–490. doi: 10.1016/j.cdp.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marfori M, Mynott A, Ellis JJ, Mehdi AM, Saunders NF, Curmi PM, Forwood JK, Bodén M, Kobe B. Molecular basis for specificity of nuclear import and prediction of nuclear localization. Biochim Biophys Acta. 2011;1813:1562–1577. doi: 10.1016/j.bbamcr.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Matsuyama A, et al. ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol. 2006;24:841–847. doi: 10.1038/nbt1222. [DOI] [PubMed] [Google Scholar]
- McBride KM, Reich NC. The ins and outs of STAT1 nuclear transport. Sci STKE. 2003;2003:RE13. doi: 10.1126/stke.2003.195.re13. [DOI] [PubMed] [Google Scholar]
- Monecke T, Güttler T, Neumann P, Dickmanns A, Görlich D, Ficner R. Crystal structure of the nuclear export receptor CRM1 in complex with Snurportin1 and RanGTP. Science. 2009;324:1087–1091. doi: 10.1126/science.1173388. [DOI] [PubMed] [Google Scholar]
- Neville M, Stutz F, Lee L, Davis LI, Rosbash M. The importin-beta family member Crm1p bridges the interaction between Rev and the nuclear pore complex during nuclear export. Curr Biol. 1997;7:767–775. doi: 10.1016/s0960-9822(06)00335-6. [DOI] [PubMed] [Google Scholar]
- Ossareh-Nazari B, Bachelerie F, Dargemont C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science. 1997;278:141–144. doi: 10.1126/science.278.5335.141. [DOI] [PubMed] [Google Scholar]
- Pei J, Grishin NV. AL2CO: calculation of positional conservation in a protein sequence alignment. Bioinformatics. 2001;17:700–712. doi: 10.1093/bioinformatics/17.8.700. [DOI] [PubMed] [Google Scholar]
- Richards SA, Carey KL, Macara IG. Requirement of guanosine triphosphate-bound ran for signal-mediated nuclear protein export. Science. 1997;276:1842–1844. doi: 10.1126/science.276.5320.1842. [DOI] [PubMed] [Google Scholar]
- Rost B, Sander C. Conservation and prediction of solvent accessibility in protein families. Proteins. 1994;20:216–226. doi: 10.1002/prot.340200303. [DOI] [PubMed] [Google Scholar]
- Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stade K, Ford CS, Guthrie C, Weis K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell. 1997;90:1041–1050. doi: 10.1016/s0092-8674(00)80370-0. [DOI] [PubMed] [Google Scholar]
- Stauber RH, Mann W, Knauer SK. Nuclear and cytoplasmic survivin: molecular mechanism, prognostic, and therapeutic potential. Cancer Res. 2007;67:5999–6002. doi: 10.1158/0008-5472.CAN-07-0494. [DOI] [PubMed] [Google Scholar]
- Suhasini M, Reddy TR. Cellular proteins and HIV-1 Rev function. Curr HIV Res. 2009;7:91–100. doi: 10.2174/157016209787048474. [DOI] [PubMed] [Google Scholar]
- Tran EJ, Bolger TA, Wente SR. SnapShot: nuclear transport. Cell. 2007;131:420. doi: 10.1016/j.cell.2007.10.015. [DOI] [PubMed] [Google Scholar]
- Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol. 2012;83:1021–1032. doi: 10.1016/j.bcp.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Weis K. Regulating access to the genome: nucleocytoplasmic transport throughout the cell cycle. Cell. 2003;112:441–451. doi: 10.1016/s0092-8674(03)00082-5. [DOI] [PubMed] [Google Scholar]
- Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signal for rapid export of proteins from the nucleus. Cell. 1995;82:463–473. doi: 10.1016/0092-8674(95)90435-2. [DOI] [PubMed] [Google Scholar]
- Xu D, Farmer A, Chook YM. Recognition of nuclear targeting signals by karyopherin-β proteins. Curr Opin Struct Biol. 2010;20:782–790. doi: 10.1016/j.sbi.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Grishin NV, Chook YM. NESdb: a database of NES-containing CRM1 cargoes. Mol Biol Cell. 2012;23:3673–3676. doi: 10.1091/mbc.E12-01-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemp I, Kutay U. Nuclear export and cytoplasmic maturation of ribosomal subunits. FEBS Lett. 2007;581:2783–2793. doi: 10.1016/j.febslet.2007.05.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.