

PKCα phosphorylates cofilin at Ser-23 and/or Ser-24 during degranulation, and the novel phosphorylation contributes to F-actin remodeling by regulating the ability of cofilin to bind to 14-3-3ζ and to depolymerize and/or sever F-actin. This highly regulated mechanism is necessary for the proper termination of degranulation.
Abstract
Using specific inhibitors, kinase-negative mutants, and small interfering RNA against protein kinase Cα (PKCα) or PKCβI, we find that PKCβI positively regulates degranulation in rat basophilic leukemia–2H3 cells, whereas PKCα negatively regulates degranulation. Mass spectrometric and mutagenic analyses reveal that PKCα phosphorylates cofilin at Ser-23 and/or Ser-24 during degranulation. Overexpression of a nonphosphorylatable form (S23,24A), but not that of a mutant-mimicking phosphorylated form (S23,24E), increases degranulation. Furthermore, the S23,24A mutant binds to F-actin and retains its depolymerizing and/or cleavage activity; conversely, the S23,24E mutant is unable to sever actin filaments, resulting in F-actin polymerization. In addition, the S23,24E mutant preferentially binds to the 14-3-3ζ protein. Fluorescence-activated cell sorting analysis with fluorescein isothiocyanate–phalloidin and simultaneous observation of degranulation, PKC translocation, and actin polymerization reveals that during degranulation, actin polymerization is dependent on PKCα activity. These results indicate that a novel PKCα-mediated phosphorylation event regulates cofilin by inhibiting its ability to depolymerize F-actin and bind to 14-3-3ζ, thereby promoting F-actin polymerization, which is necessary for cessation of degranulation.
INTRODUCTION
Histamine was identified in the early 1900s as a mediator of biological events, and drugs targeting its receptors have been in clinical use for more than 60 years. Histamine exerts a range of effects on many physiological and pathological processes, and new roles are still being elucidated. The best-characterized property of histamine is its role in inflammation, during which it is released from both mast cells and basophils (Galli, 2000).
Connective tissue mast cells have been used for many years to investigate the mechanisms of histamine release because they are easy to obtain, particularly from the peritoneal cavity. Conversely, mucosal mast cells (MMCs) and basophils are more difficult to isolate and have therefore been disregarded (Befus et al., 1982). However, the rat basophilic leukemia (RBL)-2H3 mast cell line has been developed and used in many studies (Barsumian et al., 1981) because the cells exhibit some of the typical characteristics of both MMCs and basophils (Siraganian et al., 1982).
Through experiments using RBL-2H3 cells, the signaling pathways involved in histamine release have been revealed. Antigen-driven cross-linking of cell-surface FcεRI receptors initiates a signaling cascade via the engagement of protein tyrosine kinases of the Src and Syk families (Turner and Kinet, 1999). Lyn, a Src family kinase, phosphorylates the immunoreceptor tyrosine-based activation motifs present in the FcεRIβ and γ subunits (Pribluda et al., 1994). This phosphorylation leads to the recruitment and subsequent activation of Syk kinase, which phosphorylates several downstream signaling molecules, including the linker for activation of T-cells (LAT; Saitoh et al., 2000). Phosphorylated LAT recruits a number of signaling molecules containing Src homology 2 domains, such as the adaptor protein Grb2 and phospholipase Cγ (PLCγ; Kinet, 1999). Activated PLCγ hydrolyzes phosphatidylinositol-4,5-bisphosphate, resulting in the production of inositol-1,4,5-triphosphate (IP3) and diacylglycerol (DAG). DAG activates protein kinase C (PKC), and IP3 mediates the release of intracellular calcium from the endoplasmic reticulum, which is required to open calcium channels in the plasma membrane. Increases in calcium and PKC activation are required for mast cell degranulation (Ozawa et al. 1993b). The calcium ionophores A23185 and ionomycin can induce mast cell degranulation without additional stimuli (Lo et al., 1987) because the pharmacological agents bypass FcεRI-proximal signaling events and directly stimulate cells by mobilizing free calcium ions and activating PKC (Hanson and Ziegler, 2002).
The FcεRI-mediated increase in cytoplasmic calcium triggers degranulation via reorganization of actin filaments and microtubules (Smith et al., 2003; Blank and Rivera, 2004). Studies with actin-specific drugs have established that cortical actin filaments function as a barrier to prevent granules from reaching the plasma membrane for exocytosis. Calcium-induced cortical F-actin disassembly is particularly important for secretory cells (Burgoyne and Cheek, 1985). Thus actin remodeling is intimately linked to the extensively regulated degranulation process.
PKC, the other essential factor for degranulation, consists of 10 isoforms that are characterized according to their molecular structure and activation requirements. All PKCs require phosphatidylserine (PS) for activation. The conventional PKCs (cPKC), including α, βI, βII, and γ, require both DAG and calcium ions, whereas novel PKCs (δ, ε, η, and θ) depend only on DAG. The atypical PKCs (ζ and ι/λ) are activated by PS but not by DAG or calcium (Nishizuka, 1988, 1992). RBL-2H3 cells contain PKCα, βI, ε, δ, and ζ, and different roles in degranulation have been reported for the particular PKC isoforms. For example, PKCα and ε have been shown to inhibit PLCγ, which suppresses degranulation (Ozawa et al., 1993a). An analysis of bone marrow–derived mast cells (BMMCs) from PKCβ-deficient mice revealed the positive function of PKCβ (Nechushtan et al., 2000); in contrast, PKCα has been reported to perform a negative function (Ozawa et al., 1993a). Conversely, a positive role for PKCα in degranulation has been suggested (Powner et al., 2002), and PKCδ has been shown to negatively regulate degranulation in PKCδ-deficient BMMCs (Leitges et al., 2002). These controversial reports demonstrate the need to determine individual PKC functions in degranulation. In addition, the molecules downstream of PKCs during degranulation have not been reported.
Therefore we examined the distinct functions of PKCs and identified PKC substrates during degranulation in RBL-2H3 cells. Our results revealed a positive role for PKCβI and a negative role for PKCα in degranulation. Furthermore, we identified the actin-remodeling protein cofilin as a substrate of PKCα. Specifically, PKCα phosphorylated cofilin at Ser-23 and/or Ser-24 upon stimulation with FcεRI or ionomycin. Phosphorylation of cofilin inhibited its actin-depolymerizing and actin-severing activities, suppressing excess histamine release from RBL-2H3 cells. The PKCα phosphorylation site is conserved in mammals and birds, suggesting that the well-controlled termination mechanism developed in higher animals for the rapid termination of histamine release.
RESULTS
Distinct roles of PKCα and PKCβI in degranulation
The membrane translocation of PKC is a good marker for its activation (Kraft et al., 1982; Sakai et al., 1997; Shirai et al., 1998), and RBL-2H3 cells possess several PKC isoforms, including the PKCα, βI, ε, δ, and ζ isoforms (unpublished data). To determine which PKC isoform(s) are activated during degranulation, we monitored the translocation of green fluorescent protein (GFP)–fused PKCα, βI, ε, δ, and ζ. Stimulation with 1 μM ionomycin induced significant translocation of PKCα and βI, as well as a slight but significant degree of PKCε translocation to the plasma membrane (Figure 1, A and C). In contrast, no translocation of PKCδ and ζ was detected (Figure 1, A and C). The application of dinitrophenyl–bovine serum albumin (DNP-BSA) to immunoglobulin E (IgE)–treated cells (so-called antigen stimulation) evoked membrane translocation of all PKCs, except for the ζ isoform (Figure 1, B and D); however, statistical analysis indicated that translocation of PKCδ was insignificant. These results suggest that PKCα, βI, and ε are involved in the degranulation of RBL-2H3 cells in response to both ionomycin and antigen stimulation. In addition, a selective inhibitor of cPKCs, Gö6976, inhibited both ionomycin- and antigen-induced degranulation in a dose-dependent manner (Figure 1E); this result confirms the involvement of PKCα and/or βI in the degranulation of RBL-2H3 cells.
FIGURE 1:

The translocation of PKCs during degranulation. Translocation of GFP-PKCs in RBL-2H3 cells induced by ionomycin (A) or antigen (B). RBL-2H3 cells overexpressing GFP-tagged PKCα, βI, δ, ε, or ζ were stimulated with 1 μM ionomycin or 50 ng/ml DNP-BSA (antigen). The translocation of GFP-PKCs was monitored using confocal microscopy. Bars, 10 μm. (C, D) Statistical analysis of membrane translocation. The average percentage of the membrane fluorescence of total fluorescence is represented as the relative membrane fluorescence with SD. Open and closed columns show the results before and after ionomycin (C) or DNP-BSA (D) stimulation, respectively (**p < 0.1, *p < 0.05). (E) The effect of the cPKC inhibitor Gö6976 on antigen- or ionomycin-induced degranulation. RBL-2H3 cells were incubated for 15 min with different concentrations of Gö6976 and then stimulated with either 50 ng/ml DNP-BSA or 1 μM ionomycin for 15 min. The percentage of β-hexosaminidase release is plotted.
To further examine the cPKC isoform(s) involved in the degranulation of RBL-2H3 cells, we observed the movement of PKCs during degranulation. To visualize the granules, we used a DsRed-fused signal peptide of β-hexosaminidase (DsRed-signal peptide), which is included in the same granules and released with histamine. The DsRed-signal peptide colocalized with histamine granules (Figure 2A) and disappeared upon ionomycin-induced (Figure 2B) or antigen-induced degranulation (Supplemental Figure S1, A and B), suggesting that this peptide was effective for visualizing granules during the degranulation process. Careful observation of the GFP-PKCs and granules revealed that after membrane translocation, PKCα accumulated on the membrane at exocytotic sites just after ionomycin-induced secretion (Figure 2B, left). In contrast, PKCβI accumulation around the granules was observed just before secretion (Figure 2B, right). Similar results were obtained upon antigen stimulation (Supplemental Figure S1, A and B), suggesting that PKCα and PKCβI have temporally different individual roles during degranulation.
FIGURE 2:

The distinct roles of PKCα and PKCβI in ionomycin-induced degranulation. (A) The colocalization of signal peptide-DsRed2 and histamine in RBL-2H3 cells. RBL-2H3 cells transfected with signal peptide-DsRed2 were fixed and stained with anti-histamine and Alexa Fluor 488-conjugated secondary antibodies. Bar, 10 μm. (B) Representative images of ionomycin-induced degranulation and the translocation of GFP-tagged PKCα and βI. RBL-2H3 cells were cotransfected with the DsRed-fused signal peptide of β-hexosaminidase and GFP-PKCα (left) or βI (right). After 24 h, the cells were treated with 1 μM ionomycin and monitored using confocal microscopy. White arrows indicate secreting granules. Bars, 10 μm. (C) Effect of overexpression of wild-type (WT) or kinase-negative (KN) forms of PKCα (left) and PKCβI (right). RBL-2H3 cells were infected with adenovirus encoding GFP-tagged WT or KN PKCα and with those of WT or KN PKCβI. The cells were stimulated with 1 μM ionomycin for 15 min, and the percentage of β-hexosaminidase release was plotted. The actual release corresponding to percentage of total β-hexosaminidase from control cells was 34 ± 2%. (D) Effect of specific inhibitors for PKCα and βI. RBL-2H3 cells were pretreated with azelastine or LY333531 for 15 min and stimulated with 1 μM ionomycin in the presence of the inhibitor. Data analysis was performed as described in C. (E) Effect of siRNA PKCα or βI on degranulation. RBL-2H3 cells were transfected with siRNA specific for PKCα or βI, and then GFP-PKCα or βI was overexpressed in some of them. Lysates from the siRNA-treated RBL-2H3 cells were prepared for Western blot analyses and probed for PKCα, PKCβI, and actin as a loading control. The RBL-2H3 cells treated with siRNA targeting PKCα or βI and rescued by GFP-PKCα or βI overexpression were stimulated as described in C. Experiments in C–F were performed in triplicate, and error bars represent ± SD (*p < 0.05, **p < 0.1).
To determine the individual roles of PKCα and βI in degranulation, we examined the effects of selective PKC inhibitors and PKCα or βI overexpression. Overexpression of the PKCα kinase-negative (KN) mutant enhanced ionomycin-induced degranulation, but overexpression of wild-type (WT) PKCα did not significantly alter degranulation (Figure 2C). In contrast, overexpression of GFP-PKCβI WT increased ionomycin-induced degranulation, whereas overexpression of GFP-PKCβI KN suppressed degranulation (Figure 1C). Similar results were obtained upon antigen stimulation (Supplemental Figure S1C). Moreover, azelastine, a PKCα inhibitor, promoted ionomycin-induced degranulation in a dose-dependent manner (Figure 2D). In contrast, LY333531, a specific inhibitor of PKCβI, prevented degranulation (Figure 2D). Similar effects were observed in the case of antigen stimulation (Supplemental Figure S1D). Finally, the distinct roles of PKCα and βI in degranulation were confirmed via small interfering RNA (siRNA)–mediated knockdown. After transfection into RBL-2H3 cells, siRNA against either PKCα or βI successfully reduced PKCα or βI expression, respectively (Figure 2E). Treatment of siRNA against PKCα enhanced degranulation, but PKCβI knockdown inhibited ionomycin-induced degranulation (Figure 2E). Similar results were obtained in the case of antigen stimulation (Supplemental Figure S1E). More important, overexpression of GFP-PKCα or PKCβI rescued the respective effect of siRNA of PKCα or PKCβI (Figure 2E). These results clearly indicate that PKCβI plays a stimulatory role in degranulation, whereas PKCα inhibits the process.
Identification of PKC substrates during degranulation
To identify the substrate(s) of PKCα and/or βI in the degranulation process, we treated 32P-loaded RBL-2H3 cells with ionomycin, and analyzed phosphoproteins via two-dimensional electrophoresis and autoradiography. Stimulation with ionomycin induced phosphorylation of a protein of ∼19 kDa, but this phosphorylation was abolished by treatment with Gö6976 (Figure 3A), indicating that this protein was phosphorylated by PKCα and/or βI during degranulation. Mass spectrometric analysis of the two-dimensional gel spot revealed that the protein was cofilin-1 (cofilin). To confirm that cofilin was indeed phosphorylated by cPKC during degranulation of RBL-2H3 cells, we loaded cells expressing FLAG-tagged cofilin (cofilin-FLAG) with [γ-32P] and stimulated them with ionomycin or antigen; we then performed immunoprecipitation and autoradiography. Phosphorylation of cofilin-FLAG was observed at 5 min after ionomycin stimulation, and Gö6976 significantly inhibited this phosphorylation (Figure 3B, left). A similar result was obtained with antigen stimulation (Figure 3B, right), confirming that cofilin was phosphorylated in a cPKC-dependent manner during degranulation.
FIGURE 3:

Identification of the PKCα substrate during the degranulation process. (A) Detection of proteins phosphorylated by cPKC during degranulation. The 32P-loaded RBL-2H3 cells were pretreated with or without 5 μM Gö6976 for 15 min and then stimulated with 5 μM ionomycin. The lysates were subjected to two-dimensional electrophoresis and autoradiography. The circles indicate proteins that were phosphorylated by cPKC during degranulation. (B) In vivo phosphorylation of cofilin-FLAG by cPKC. RBL-2H3 cells were transfected with cofilin-FLAG. Twenty-four hours later, the cells were stimulated with 1 μM ionomycin or 50 ng/ml DNP-BSA for 5 min in the presence or absence of 5 μM Gö6976. Soluble proteins were immunoprecipitated via anti-FLAG affinity gel. The immunoprecipitated cofilin-FLAG was then detected by immunoblotting with an anti-FLAG antibody (bottom). The level of radioactivity was analyzed with a BAS-2500 bio-imaging analyzer (top), and the average phosphorylation level from three independent experiments is shown in the bottom graphs. Error bars and asterisks show the SD and probability relative to the control (i.e., no stimulation). *p < 0.05. (C) Effect of azelastine on ionomycin- and antigen-induced cofilin phosphorylation in vivo. RBL-2H3 cells were transfected with cofilin-FLAG. Twenty-four hours later, after preincubation with 10 μM or 50 μM azelastine for 15 min, the cells were stimulated with 1 μM ionomycin or 50 ng/ml DNP-BSA for 5 min and lysed. The phosphorylation level of the immunoprecipitated cofilin-FLAG was analyzed as described in B. (D) Effect of siRNA targeting PKCα on ionomycin- and antigen-induced cofilin phosphorylation in vivo. RBL-2H3 cells treated with siRNA for PKCα were transfected with cofilin-FLAG. Twenty-four hours later, the cells were stimulated with 1 μM ionomycin or 50 ng/ml DNP-BSA for 5 min and lysed. The phosphorylation level of the immunoprecipitated cofilin-FLAG was analyzed as described in B.
Cofilin is an actin-binding protein. Although it shows complex behavior toward actin, cofilin phosphorylation generally promotes actin polymerization (Bamburg, 1999), which has an inhibitory effect on degranulation (Frigeri and Apgar, 1999). In light of these reports, we hypothesized that PKCα phosphorylates cofilin. Therefore the effects of a PKCα inhibitor on the in vivo phosphorylation of cofilin were examined. As expected, 10 μM azelastine abolished both ionomycin- and antigen-induced cofilin phosphorylation (Figure 3C). In addition, the same concentration of azelastine inhibited ionomycin-induced activation of PKCα but not that of PKCβI (Supplemental Figure S2B), confirming the specificity of azelastine. The involvement of PKCα in cofilin phosphorylation was also confirmed by PKCα siRNA treatment (Figure 3D), suggesting that cofilin is a substrate of PKCα.
Phosphorylation of cofilin at Ser-3 in the actin-binding domain by the Lin-11/Isl-1/Mec-3 (LIM) and testicular protein (TES) kinases results in cofilin inactivation (Bamburg, 1999). However, Ser-3 is not a consensus PKC phosphorylation site. In addition, cofilin was directly phosphorylated by PKCα in vitro, but a substitution of Ser-3 to Ala (S3A) did not alter the phosphorylation level (Supplemental Figure S3A), indicating that Ser-3 is not the phosphorylation site(s) by PKCα. To identify the residue(s) phosphorylated by PKCα, we performed an in vitro phosphorylation assay with purified PKCα and maltose-binding protein (MBP)–cofilin in tandem with mass spectrometric analysis. Comparing the mass spectra in the absence and presence of ATP showed that at least one serine was phosphorylated in the peptide fragment, VFNDMKVRKSSTPEEVKK, which corresponds to amino acids 14–31 of cofilin (Figure 4A). This peptide contains two serines, Ser-23 and Ser-24, which are present in the consensus phosphorylation sequence recognized by PKC. To examine whether these serines are phosphorylated by PKCα, we performed an in vitro kinase assay with cofilin mutants. PKCα phosphorylated wild-type MBP-cofilin (Figure 4B), confirming that PKCα directly phosphorylates recombinant cofilin in vitro. However, when 24 amino acids, including Ser-23 and Ser-24, were deleted from the N-terminus (Nd) or when both Ser-23 and Ser-24 were converted to alanines (S23,24A), phosphorylation by PKCα was nearly eliminated (Figure 4B). In addition, point mutations of Ser-23 (S23A) or Ser-24 (S24A) substantially attenuated cofilin phosphorylation (Figure 4B); however, phosphorylation was not completely abolished, suggesting that PKCα phosphorylates cofilin at both Ser-23 and Ser-24 in vitro. Of importance, an in vivo kinase assay proved that phosphorylation of Ser-23 and Ser-24 in cofilin occurred during degranulation in RBL-2H3 cells (Figure 4, C and D). Ionomycin- and antigen-induced phosphorylation of cofilin was markedly reduced by the S23,24A substitution. In contrast, the S3A mutant, in which Ser-3 (a phosphorylation site by LIMK) was converted to alanine, exhibited an insignificant reduction in phosphorylation during the degranulation process (Figure 4, C and D). Phosphorylation of cofilin at Ser-23 and Ser-24 during degranulation was also confirmed in mouse BMMCs (Figure 4E and Supplemental Figure S3B). These results indicate that Ser-23 and Ser-24 of cofilin are phosphorylated by PKCα during degranulation.
FIGURE 4:

Determination of the PKCα phosphorylation site(s) in cofilin. (A) Mass spectrometric analysis of cofilin phosphorylated by purified PKCα in vitro. Purified MBP-cofilin was incubated with PKCα in the presence or absence of ATP, Ca2+, and PS/DO at 30°C for 15 min. After separation by SDS–PAGE, MBP-cofilin was subjected to mass spectrometry. The detected mass values for untreated (top) and ATP-treated cofilin (bottom) are shown. Spectra that were dependent on the presence of ATP are indicated by red boxes. The first peak is mono isotopic and the second is isotopic; detailed data are shown in Supplemental Figure S8. (B) In vitro phosphorylation of cofilin at Ser-23 and/or Ser-24 by PKCα. An in vitro kinase assay was performed with purified PKCα and MBP-cofilin wild-type (WT), the N-terminal–deletion mutant (Nd; lacking the N-terminal 24 amino acids), and the serine mutants (S3A, S23A, S24A, S23,24A) in the presence (+) or absence (–) of PKC activators. (C, D) The in vivo phosphorylation of cofilin at Ser-23 and Ser-24 during degranulation in RBL-2H3 cells. Plasmids of FLAG-tagged wild-type cofilin or the S3A, or S23,24A mutants were transfected into RBL-2H3 cells and then stimulated with 1 μM ionomycin (C) or 50 ng/ml DNP-BSA (D). (E) The in vivo phosphorylation of cofilin at Ser-23 and Ser-24 during degranulation in mouse BMMC. Plasmids of FLAG-tagged wild-type cofilin or the S3A or S23,24A mutants were transfected into BMMC cells and then stimulated with 1 μM ionomycin. Quantitative analysis of cofilin phosphorylation from three independent experiments is shown in the bottom graphs of B– E; bars represent SD. Quantification was performed by normalizing the radioactive bands in the kinase assay to the total amount of cofilin (*p < 0.05).
Function of novel cofilin phosphorylation during degranulation
To further examine how the phosphorylation of cofilin at Ser-23 and Ser-24 influences degranulation, we used two cofilin mutants—S23,24A and S23,24E, the latter of which mimics phosphorylation by substituting glutamic acid residues for Ser-23 and Ser-24. After we established cell lines that stably expressed DsRed-tagged WT cofilin or its mutant derivatives, immunoblotting analyses revealed that the expression of DsRed-cofilin and its mutants was comparable in each cell line (Figure 5A). Ionomycin-induced degranulation was enhanced in S23,24A-cofilin–expressing cells but unchanged in S23,24E-cofilin–expressing cells (Figure 5B, left). Similar results were obtained after antigen stimulation, although degranulation in S23,24E-cofilin cells was slightly inhibited (Figure 5B, right). These data demonstrate that reduced cofilin phosphorylation at Ser-23 and Ser-24 promotes degranulation in RBL-2H3 cells. Correspondingly, treatment with azelastine increased degranulation from both native RBL-2H3 cells (Figure 2D, left) and a cell line stably expressing a WT cofilin-DsRed fusion protein (Figures 5, C and D, left). In contrast, the effect of azelastine was abolished by both mutations, as azelastine treatment did not alter ionomycin- or antigen-induced degranulation in S23,24A-cofilin and S23,24E-cofilin cell lines (Figure 5, C and D). These results indicate that phosphorylation of cofilin at Ser-23 and Ser-24 by PKCα plays an important role in the control of excess histamine release.
FIGURE 5:

The effect of expression of cofilin mutants on degranulation. (A) Western blotting analyses of the expression of DsRed-cofilin WT and the S23,24A and S23,24E mutants. Approximately 50 μg of protein from each cell line stably expressing wild-type, S23,24A, or S23,24E was subjected to SDS–PAGE and Western blotting with anti-DsRed (top) and anti-actin (bottom) antibodies. After measuring the density of immunoreactive bands with ImageJ software (National Institutes of Health, Bethesda, MD) and normalizing the cofilin immunoreactive signal to that of actin, an average value ± SD was plotted in the bottom graph. (B) Each stable cell line was stimulated with 1 μM ionomycin or 50 ng/ml DNP-BSA for 15 min, and the percentage of β-hexosaminidase release was plotted. (C, D) After pretreatment with 10 μM azelastine or no treatment, each stable cell line was stimulated with 1 μM ionomycin (C) or 50 ng/ml DNP-BSA (D) for 15 min; the percentage of β-hexosaminidase release was plotted. Asterisk indicates a significant difference relative to control release (WT, –AZ) with p < 0.05; number sign indicates difference between control (–AZ) and test (+AZ) with p < 0.05; AZ, azelastine; N.S., not significant. All experiments were performed in triplicate; error bars indicate SD.
Effect of the novel phosphorylation on cofilin activity
Previous studies showed that cofilin has several complex functions, but we focused on two general biochemical functions: depolymerizing actin filaments into actin monomers (Carlier et al., 1997) and severing actin filaments to create free, barbed ends (Ichetovkin et al., 2000). In addition to its actin-binding ability, these functions of cofilin are attenuated by LIMK-mediated phosphorylation at Ser-3 (Arber et al., 1998). In other words, phosphorylation of cofilin at Ser-3 leads to actin polymerization. Therefore we examined whether phosphorylation at Ser-23 and Ser-24 also affected the actin-depolymerizing and actin-severing activity of cofilin, via an F-actin sedimentation assay with wild-type or mutant forms of cofilin. When G-actin was polymerized to F-actin in the absence of cofilin and subjected to ultra-high-speed centrifugation, a significant amount of F-actin was found in the pellet fraction (P); in contrast, a smaller amount of G-actin was found in the supernatant (S) (Figure 6A, lanes 7 and 8). When WT cofilin was added before centrifugation, the amount of G actin increased and F-actin decreased because of the depolymerizing and/or severing activity of cofilin (Figure 6A, lanes 1 and 2). This distribution was not altered in the case of the S23,24A mutant (Figure 6A, lanes 3 and 4), indicating that it retained its actin-depolymerizing and/or actin-severing activity. In contrast, in the case of the S23,24E mutant (Figure 6A, lanes 5 and 6), which mimics phosphorylated cofilin, an actin distribution was similar to that of control (no cofilin). Moreover, the amount of S23,24E cofilin that cosedimented with F-actin was significantly decreased relative to both wild-type cofilin and the S23,24A mutant (Figure 6A, lanes 1–6, bottom, and see “cofilin(ppt)” in the bottom graph). These results indicate that actin-binding activity of cofilin was weakened when phosphorylated at Ser-23 and Ser-24, which abrogated its depolymerization and/or severing activity.
FIGURE 6:

Cofilin phosphorylation at Ser-23 and Ser-24 influences depolymerization of F-actin and binding to 14-3-3ζ. (A) The F-actin sedimentation assay. Purified wild-type (WT) cofilin, S23,24A (SA), and S23,24E (SE) were incubated with F-actin for 30 min. To serve as a negative control, F-actin was incubated alone (–). F-actin and F-actin–bound cofilin were precipitated via centrifugation, and the amounts of F-actin and cofilin in the supernatant (S) and pellet (P) were analyzed by SDS–PAGE and Coomassie blue staining. Quantitative analysis of amounts of cofilin and F-actin from three independent experiments is shown in the bottom graphs; bars represent SD. Quantification was performed by normalizing the density of bands in the assay to the amount of cofilin or actin in control (*p < 0.05). (B) Microscopic analyses of cofilin-induced F-actin depolymerization and/or severing. Alexa 488– or biotin-labeled actin filaments were incubated with purified MBP or MBP-cofilin wild-type, S23,24A, or S23,24E. Subsequently the filaments were tethered to a glass slide and observed via confocal microscopy. The result of quantitative analyses of the length of actin filaments in three independent experiments is shown in the lower graph (*p < 0.05). The experiments were performed in triplicate, and error bars represent ± SD. (C) Cofilin binding to 14-3-3ζ. Purified MBP-cofilin WT, S3A, S3E, S23,24A, or S23,24E was incubated with FLAG-14-3-3ζ for 60 min. Cofilin-bound 14-3-3ζ was pulled down with an anti-FLAG affinity gel and subjected to SDS–PAGE and Western blotting.
Next we compared effects of WT and mutant cofilin on length of actin filaments by direct observation of Alexa Fluor 488–labeled actin in accordance with the method described in previous reports (Ichetovkin et al., 2000; Ono et al. 2004). When actin was polymerized in the presence of WT cofilin or S23,24A, the length of F-actin filaments was dramatically shortened compared with the control (Figure 6B); this finding confirms that the nonphosphorylated form of cofilin possesses F-actin–depolymerizing and/or F-actin–severing activity. Conversely, S23,24E cofilin was unable to depolymerize and/or sever F-actin (Figure 6B), suggesting that, in addition to Ser-3, the novel phosphorylation sites on cofilin are also important for its F-actin–depolymerizing and/or F-actin–severing activity. Furthermore, these results suggest that PKCα-mediated phosphorylation of cofilin contributes to actin polymerization during degranulation by regulating the F-actin–depolymerizating and/or F-actin–severing activity of cofilin, which may be important for the proper termination of histamine release.
A previous report indicated that cofilin bound to 14-3-3ζ, which has been suggested to play a dynamic role in the regulation of cytoplasmic actin structure via the RKS23S24TP motif (Gohla and Bokoch, 2002). Therefore we tested the interaction between 14-3-3ζ and the cofilin mutants. The phosphorylation-mimic mutant (S23,24E) bound to 14-3-3ζ more strongly than WT, similar to the S3E mutant (Figure 6C). Conversely, the S23,24A mutant did not bind to 14-3-3ζ, indicating that the Ser-23/Ser-24 phosphorylation sites are important for the 14-3-3ζ binding. These results suggest that, in addition to Ser-3 phosphorylation by LIMK, PKCα phosphorylation-dependent binding of cofilin to 14-3-3ζ somehow contributes to actin polymerization during degranulation.
PKCα-dependent actin polymerization during degranulation in vivo
To confirm the role of PKCα-mediated cofilin phosphorylation in the regulation of actin polymerization during degranulation, we visualized actin polymerization in vivo using DsRed-actin. Before stimulation, DsRed-actin showed two distributions, in which either a portion of the DsRed-actin accumulated in the cortical region of the cell or there was equal distribution throughout the cytosol (Supplemental Figure S4A). The former pattern accurately represented cortical F-actin in the resting cells (Supplemental Figure S4A, green; Yanase et al., 2011), and the latter pattern seemed to be due to small portion of DsRed-actin polymerized into cortical F-actin, in addition to a small amount of cortical F-actin itself (Supplemental Figure S4A). Because it was very hard to see the accumulation of DsRed-actin during degranulation in the cells exhibiting the former distribution due to masking of the fluorescence of newly polymerized DeRed-actin by those formerly present in the cortical region, we chose cells exhibiting the latter distribution.
DsRed-actin accumulated at the plasma membrane in response to stimulation with ionomycin or antigen (Supplemental Figure S4B, top). However, this accumulation was abolished by the F-actin–depolymerizing agent latrunculin A (Supplemental Figure S4B, middle), which indicated that the actin that accumulated at the plasma membrane was F-actin. Using this system, we investigated whether PKC could influence actin polymerization during degranulation. Ionomycin- or antigen-induced actin polymerization was inhibited by treatment with Gö6976 (Supplemental Figure S4B, bottom), indicating that cPKCs were involved in the polymerization of actin. In addition, cotransfection of GFP-PKCαKN inhibited the polymerization of DsRed-actin; however, actin polymerization was observed with PKCβI KN (Figure 7A and Supplemental Figure S5A), suggesting that actin polymerization was dependent on PKCα activity. The importance of PKCα in actin polymerization was confirmed by using azelastine, which inhibited actin polymerization in vivo without inhibiting PKCβI activation (translocation; Figure 7B and Supplemental Figure S5B). In addition to the membrane accumulation, a ring-shaped accumulation of F-actin was observed (Supplemental Figure S6A, white arrows). To investigate the ring-shape F-actin accumulation, we simultaneously visualized actin polymerization and degranulation using GFP-actin and the DsRed-signal peptide. In response to ionomycin or antigen stimulation, the ring-shaped F-actin accumulation was found around the granules just after degranulation in a manner similar to PKCα (Supplemental Figure S6B). Therefore we compared the timing of the accumulation of both PKCα and actin polymerization. After ionomycin stimulation, actin was recruited immediately after PKCα accumulation (compare Figure 7C at 114 and 125 s), and both proteins were colocalized near the area previously occupied by the granules (Figure 7C at 125 s). In contrast, the ring-shaped F-actin did not colocalize with accumulated PKCβI (Figure 7D). Similar results were obtained in the case of the antigen stimulation (Supplemental Figure S4, C and D). More important, colocalization of the ring-shaped F-actin and PKCα, but not that of PKCβI, was confirmed by phalloidin staining and after scan analysis (Figure 7, E and F, and Supplemental Figure S5, E and F). The spatiotemporal difference between PKCα and βI strongly supports the importance of PKCα in actin polymerization during degranulation.
FIGURE 7:

Simultaneous observations of actin polymerization and PKCα or PKCβI during ionomycin-induced degranulation. (A) The effect of coexpression of wild-type or either kinase-negative PKCα or PKCβI on actin polymerization in vivo. RBL-2H3 cells were transfected with DsRed-actin and either WT or KN of GFP-PKCα (left) or PKCβI (right). The cells were then treated with 1 μM ionomycin. The relative change in fluorescence of DsRed-actin at the plasma membrane in response to ionomycin is shown in the bottom graphs. *p < 0.05. (B) The effect of a PKCα inhibitor on actin polymerization. RBL-2H3 cells were transfected with DsRed-actin and GFP-PKCβI. After preincubation with 10 μM azelastine, cells were treated with 1 μM ionomycin. The relative change in fluorescence of GFP-PKCβI or DsRed-actin at the plasma membrane is shown in the bottom graph. (C, D) Comparison of F-actin localization with PKCα or PKCβI. RBL-2H3 cells were cotransfected with DsRed-actin and GFP-PKCα (C) or βI (D). After 24 h, the cells were treated with 1 μM ionomycin and were monitored using confocal microscopy. (E, F) Comparison of intact F-actin localization with PKCα or PKCβI. RBL-2H3 cells were transfected with GFP-PKCα (E) or βI (F). After 24 h, the cells were treated with 1 μM ionomycin and fixed. Intact F-actin was visualized using rhodamine-phalloidin and monitored using confocal microscopy. Bottom graph shows profiles of fluorescence of GFP and rhodamine when line scan was performed, along with red arrow in the insets. White arrows indicate areas where PKCα or PKCβI accumulated. Bars, 10 μm.
To further confirm the PKCα-dependent assembly of F-actin, we used quantitative fluorescence-activated cell sorting (FACS) and fluorescein isothiocyanate (FITC)–phalloidin. In control treatments, the fluorescence peak was shifted to the right by ionomycin, indicating that actin polymerization occurred during degranulation (Figure 8A, left). However, in the case of siRNA treatment against PKCα, this rightward shift was not observed; instead, depolymerization occurred within 15 min after stimulation (Figure 8A, right). Statistical analysis of average values for FITC–phalloidin fluorescence, which reflects intracellular F-actin content, confirmed the importance of PKCα for actin polymerization during degranulation. Ionomycin stimulation significantly increased the mean fluorescence in a time-dependent manner (Figure 8B, open column), whereas PKCα siRNA suppressed this increase (Figure 8B, closed column). Azelastine also abolished ionomycin-induced actin polymerization (Figure 8C). Similar results were obtained in the case of antigen stimulation (Supplemental Figure S7). Together with microscopy observations and the inhibitory effect of PKCα-dependent phosphorylation of cofilin on degranulation described earlier, these results implicate a important role for PKCα in actin polymerization via phosphorylation of cofilin at novel sites (the ring-shaped accumulation), which occurs at sites of exocytosis just after degranulation and is necessary for rapid termination of histamine release.
FIGURE 8:

PKCα-dependent actin polymerization during ionomycin-induced degranulation. (A) Histograms of FACS analyses. Cells transfected with PKCα siRNA and control RBL-2H3 were stimulated with ionomycin, and actin polymerization was monitored with FITC-phalloidin via FACS. Red lines indicate the position of the peak at 0 min. (B) Statistical analysis of PKCα-dependent actin polymerization. The same experiments described in A were performed three times, and the mean of the fluorescence intensity for each FITC histogram is shown, together with the SD. Asterisk indicates significant differences relative to the control at 0 min, with p < 0.05, and number sign indicates significant differences between the control (–AZ or –PKCα siRNA) and test (+AZ or +PKCα siRNA) conditions, with p <0.05. (C) Effect of azelastine on actin polymerization. RBL-2H3 cells were pretreated with or without azelastine for 15 min and stimulated with 1 μM ionomycin in the presence of the inhibitor. Data analysis was performed as described in B.
DISCUSSION
Previous studies established that PKC activation is essential for mast cell degranulation (Ozawa et al. 1993b). However, the role of the individual PKC isoforms in degranulation has not been determined. In the present study, experiments with the specific inhibitor azelastine showed that PKCα negatively regulates degranulation. We confirmed that azelastine directly inhibits PKCα but not PKCβI at low concentrations (between 1 and 10 μM) in vitro (Supplemental Figure S2A). However, azelastine has also been shown to inhibit histamine release and is a known antiallergy drug (Hide et al., 1997, Shichijo et al., 1998). These discrepancies may be the result of specific assay conditions, such as the concentration of azelastine, as concentrations of azelastine >10 μM also inhibit PKCβ in vitro (Supplemental Figure S2A). Alternatively, even if the same concentration of azelastine was used, the degree of cell permeability may have been different. Furthermore, it is noteworthy that the increased release of histamine was easily detectable in our system because the maximum release was lower than in previous reports. The maximal release from control cells was 34 ± 2% in this study, whereas it was ∼73% in a previous report (Hide et al., 1997). Of importance, the inhibitory effect of PKCα was confirmed via the adenoviral overexpression of PKCα KN and the PKCα-directed siRNA treatment. The inhibitory role of PKCα was further supported by the recruitment of PKCα immediately after secretion to the area where secretory granules had been located and by F-actin being accumulated with similar timing as PKCα. In contrast, PKCβI, the other conventional PKC isoform expressed in RBL-2H3 cells, enhanced degranulation. In support of this finding, Nechushtan et al. (2000) demonstrated that PKCβ-deficient mice exhibit a decrease in degranulation. The recruitment of PKCβI to the vicinity of granules before degranulation suggests that PKCβI phosphorylates proteins located near secretory granules, such as soluble N-ethylmaleimide–sensitive factor attachment protein receptors and other proteins that promote membrane fusion, including calcium channels. Further experiments are necessary to identify the substrate of PKCβI during degranulation.
We provided the first evidence that cofilin, a major actin-remodeling protein, is a substrate of PKCα during degranulation in RBL-2H3 cells. In addition, we identified Ser-23 and Ser-24 of cofilin as major, novel sites of PKCα phosphorylation, although there may be an additional phosphorylation site(s) because Nd and S23,24A were slightly phosphorylated by PKCα in vitro and in vivo. Similar to phosphorylation at Ser-3 by LIMK, mimicking the phosphorylation of Ser-23 and Ser-24 inhibited the depolymerization and/or severing activity of cofilin and the binding ability to F-actin. Furthermore, the expression of the cofilin S23,24A mutant up-regulated degranulation in RBL-2H3 cells, and actin polymerization in vivo was dependent on PKCα activity. In addition, PKCα but not PKCβI colocalized with polymerized actin around the granules immediately after degranulation. These results indicate that PKCα accumulates at sites of exocytosis, where it inactivates cofilin via Ser-23 and Ser-24 phosphorylation, resulting in F-actin polymerization; this relationship is summarized in Figure 9. We propose that the preferential binding of phospho-cofilin to 14-3-3ζ also plays an important role in F-actin polymerization because 14-3-3ζ regulates actin dynamics by stabilizing phospho-cofilin (Gohla and Bokoch, 2002). Finally, actin polymerization appears to suppress excess degranulation. In fact, previous studies reported that cortical F-actin serves as a barrier to prevent vesicular access to the plasma membrane (Frigeri and Apgar, 1999). Our data raise the possibility that accelerated actin polymerization via the PKCα-mediated phosphorylation of cofilin may serve as a negative feedback mechanism in mast cell degranulation. This hypothesis is in agreement with a previous report that PKC regulates the actin cytoskeleton in a wide range of cell types via the phosphorylation of actin-regulating proteins, including myristoylated alanine-rich C kinase substrate and ezrin/radixin/moesin proteins (Larsson, 2006), which are involved in the regulation of exocytosis (Bretscher et al., 2002).
FIGURE 9:
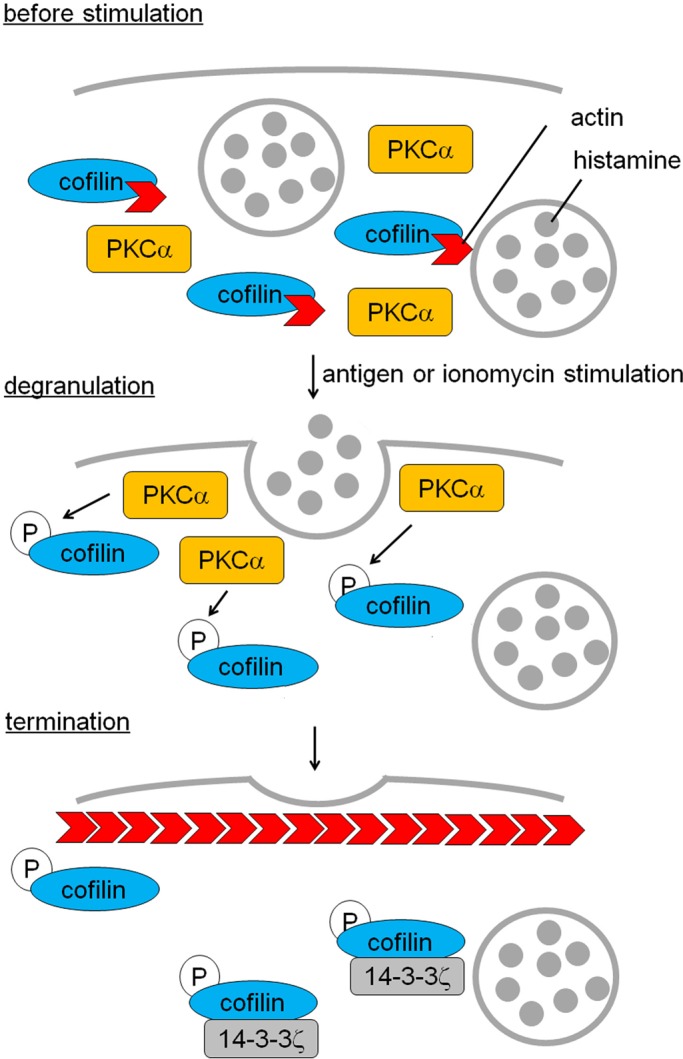
A model for preventing excess degranulation via PKCα-mediated cofilin phosphorylation. The aggregation of FcεRI triggers PKCα activation, resulting in phosphorylation of cofilin at Ser-23 and/or Ser-24. The novel phosphorylation results in inactivation of cofilin, leading to an increase in cortical F-actin polymerization, which is necessary to prevent excess degranulation. Phosphorylation strengthens cofilin binding to 14-3-3ζ, contributing to the system. To simplify, cortical F-actin in the resting state and actin depolymerization at early phase are omitted in the model. Newly polymerized F-actin is indicated with red arrows.
Unfortunately, we could not rule out the possibility that PKCβI also phosphorylates cofilin, because treatments with LY333531 and PKCβI siRNA inhibited degranulation; thus no events, including cofilin phosphorylation and actin polymerization, occurred. However, several findings indicate that PKCα but not PKCβI phosphorylates Ser-23 and/or Ser-24 of cofilin, resulting in actin polymerization to inhibit excess histamine release. 1) Actin polymerization was inhibited by PKCαKN but not PKCβI KN (Figure 4A). 2) The accumulation of PKCα but not PKCβI was temporally and spatially coincident with F-actin accumulation around the area where granules had been located (Figure 4, C–F, and Supplemental Figure S5). 3) The stable cell line expressing the nonphosphorylated form of cofilin (S23,24A) resulted in an increase in histamine release (Figure 5B), similar to the effect of azelastine, the overexpression of PKCαKN, and the siRNA treatment for PKCα (Figure 2, C–E). 4) Furthermore, the effect of azelastine on histamine release was abolished by the overexpression of the phosphorylated form of cofilin (S23,24E; Figure 5, C and D).
We used S23,24A and S23,24E mutants to test our hypothesis that both serines are phosphorylated; this hypothesis was derived from the finding that a single point mutation of Ser-23 or Ser-24 did not completely abolish phosphorylation. Indeed, a recent report of the phospho-proteome of human embryonic stem cells showed that both Ser-23 and Ser-24 of human cofilin were phosphorylated (Rigbolt et al., 2011). However, we still do not know whether both Ser-23 and Ser-24 are simultaneously phosphorylated in one molecule. Of interest, cofilin mutants (S23E and S24E) in which Ser-23 or Ser-24 was replaced with glutamic acid also lost the ability to sever and/or depolymerize F-actin (unpublished data). Furthermore, the degranulation of cell lines stably expressing S23A or S24A was up-regulated (unpublished data). These results suggest that at least a single phosphorylation event at Ser-23 or Ser-24 is enough to regulate cofilin function in histamine release, and only single phosphorylation may occur in some cases. Of interest, Ser-24 corresponds to the phospho-Ser in the prototypical 14-3-3 recognition motif RSXpSP (Fu et al., 2000). Indeed, S24E preferentially bound to 14-3-3ζ, whereas S23E did not (unpublished data). Ser-24 is conserved among cofilin-1, cofilin-2, and actin-depolymerizing factor (Table 1), suggesting that Ser-24 is more important than Ser-23.
TABLE 1:
Sequence alignment of cofilin within vertebrates.
| 19 23 24 28 | ||
|---|---|---|
| Human | Cofilin-1 | K V R K S ST P E E |
| Cofilin-2 | K V R K S ST Q E E | |
| Actin-depolymerizing factor | K V R K C S T P E E | |
| Mouse | Cofilin-1 | K V R K S S T P E E |
| Chicken | Cofilin-1 | K V R K S S T P E E |
| Gecko | Cofilin-1 | K V R K C S T P E E |
| Frog | Cofilin-1 | K V R H P L S P E E |
| Zebrafish | Cofilin-1 | R V R L Q G T D E K |
The alignment of the conserved PKC consensus sites is shown with red letters; those residues are identified as novel phosphorylation sites by PKCα.
How can this novel phosphorylation pattern regulate cofilin function? Ser-3 is located in the N-terminus close to the β4 sheet, which is important for actin binding, in the three-dimensional structure (Pope et al., 2004). The substitution of Ser-3 to Asp affects the spatial relationship between the N-terminus and the β4 sheet, resulting in the inactivation of cofilin. In contrast, Ser-23 and Ser-24 are located between α1 and α2 helices and do not appear to directly interact with the β4 sheet in the 3D structure. Accordingly, phosphorylation at Ser-23 and/or Ser-24 allosterically regulates the positional relationship between the β4 sheet and the N-terminus around Ser-3, resulting in cofilin inactivation.
Our finding raises one more question regarding why other phosphorylation site(s) in addition to Ser-3 are necessary to regulate cofilin function. In platelet degranulation, the dephosphorylation of cofilin at Ser-3 (activation) occurs rapidly in an initial phase, and it is subsequently slowly rephosphorylated by LIMK-1 (inactivation) (Pandey et al., 2006, 2009). Dephosphorylation seems to occur through the calcineurin-dependent activation of slingshot (Wang et al., 2005). Although the involvement of cofilin dephosphorylation in histamine release was not reported, it seems to occur in mast cells, judging from the finding that actin depolymerization is generally necessary for an initial phase of secretion and the report that calcineurin is involved in the FcεRI-mediated exocytosis from RBL mast cells (Hultsch et al., 1998). However, unlike degranulation from platelets, the inactivation of cofilin should occur rapidly in histamine release to avoid a hyperallergic response. In the process, an additional regulation system of cofilin by PKCα may be developed. Indeed, an optimal sequence alignment indicates that Ser-23 and Ser-24 are only conserved for mammals and birds, although Ser-24 is also conserved in reptiles (Table 1), in which the presence of histamine in mast cells has been reported (Mulero et al., 2007). Of importance, these conserved serine residues share the PKC phosphorylation consensus sequence (K/R)-X-(S/T). In contrast, Ser-3, which is phosphorylated by LIMK (but not by PKC), is conserved between fish and mammals. These facts suggest that the PKC-mediated regulation of cofilin may be specific for the release of histamine in higher animals, for which rapid polymerization may be necessary to immediately halt secretion. Indeed, azelastine treatment did not enhance the secretion of catecholamine from chromaffin cells (unpublished data). Conversely, phosphorylation at Ser-3 by LIMK may play some role in fundamental actin remodeling. To answer the question, further experiments to compare the effect of phosphorylation on the kinetics of actin and 14-3-3ζ binding to cofilin and/or its severing activity would be necessary. Furthermore, the regulation of actin by cofilin is more complicated. For example, a positive role of F-actin polymerization in degranulation has been reported (Pendelton and Koffer, 2001). In addition, cofilin is regulated not only by phosphorylation but also other factors, including phosphatidylinositol 4,5-bisphosphate and pH (van Rheenen et al., 2007; Pope et al., 2004). These facts indicate a complex role of actin polymerization at different stages or different areas of degranulation through a well-regulated mechanism. Further experiments to investigate actin polymerization and cofilin phosphorylation temporally and locally would be helpful to understand the comprehensive role of actin and cofilin in degranulation. In future studies, DsRed-Signal peptide and DsRed-actin would be useful, and our finding that cofilin is regulated by additional phosphorylation sites at Ser-23 and Ser-24 is important.
In conclusion, PKCα phosphorylates cofilin at Ser-23 and/or Ser-24 during degranulation. Furthermore, these novel PKC-mediated phosphorylation events regulate F-actin remodeling by modifying the ability of cofilin to bind to 14-3-3ζ and depolymerize and/or serve F-actin. This highly regulated mechanism is necessary for the termination of degranulation and appears to have been developed specifically for histamine release.
MATERIALS AND METHODS
Materials
Ionomycin and Gö6976 were purchased from Calbiochem (La Jolla, CA). DNP-specific IgE (clone SPE7), DNP-BSA, p-nitrophenyl-N-acetyl-β-d-glucosaminide, anti-FLAG M2 monoclonal antibody, and anti-FLAG M2 Affinity Gel were obtained from Sigma-Aldrich (St. Louis, MO). [γ-32P]ATP and monosodium [γ-32P]phosphate were products of MP Biomedicals (Irvine, CA). Azelastine was kindly donated by Eisai (Tokyo, Japan). LY333531 was obtained from Axon Medchem (Groningen, Netherlands). Mouse cofilin-hexahistidine in pUDC2SRα was described previously (Yang et al., 1998). The cDNA encoding GFP, GFP-PKCα and βI and kinase-negative mutants were prepared as described previously (Sakai et al., 1997; Shirai et al., 1998).
Cell culture and protein overexpression and down-regulation
RBL-2H3 cells were grown in RPMI 1640 (Nacalai Tesque, Kyoto, Japan) supplemented with 10% FBS (Sigma-Aldrich), 100 U/ml penicillin, and 100 μg/ml streptomycin (GIBCO, Grand Island, NY) in a 37°C humidified atmosphere containing 5% CO2. Transient transfections of plasmids were performed with Fugene6 (Roche, Basel, Switzerland) or via electroporation (Amaxa; Lonza, Basel, Switzerland) according to the manufacturer's protocol. In the case of adenovirus infection, 2 × 105 RBL-2H3 cells were seeded in a 3.5-cm glass-bottom dish (MatTek, Ashland, MA) with 2 ml of medium 1 d before the infection. After removal of the medium, a high-titer virus solution (multiplicity of infection >5) in 100 μl OPTI-MEM was added to the dish. After 4 h of incubation, the medium was changed to RPMI 1640, and the cells were incubated overnight.
For down-regulation of PKCα and βI, RBL-2H3 cells were transfected with siRNA at a final concentration of 100 nM using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. The siRNA sequences were as follows: 5′-CACGAGGGCAGCCUGUCUUAA-3′ and 5′-UUAAGACAGGCUGCCCUCGUG-3′ were used for rat PKCα and 5′-CGGUGCGCUUCGCCCGCAATT-3′ and 5′-UUGCGGGCGAAGCGCACCGTG-3′ were used for rat PKCβ. Control scramble siRNA oligos were purchased from Qiagen (Valencia, CA) and transfected into cells as a negative control.
Degranulation assay
RBL-2H3 cells (5.0 × 105/ml) were incubated overnight. Without any pretreatment, the cells were treated with ionomycin for 15 min at 37°C. For antigen stimulation, the cultures were incubated with 0.5 μg/ml IgE. The cells were then washed twice and treated with DNP-BSA for 15 min at 37°C. The supernatants were transferred to 96-well plates and incubated with β-hexosaminidase substrate (1 mM p-nitrophenyl-N-acetyl-d-glucosaminide) for 1 h at 37°C. To stop the reaction, 0.2 M Tris was added, and the absorbance at 405 nm was measured.
Construction of plasmids encoding wild-type and cofilin mutants
The cDNA fragments of cofilin with EcoRI site at the N-terminus and the C-terminus were produced via PCR using mouse cofilin-hexahistidine in pUDC2SRα. The primers were 5′-TTGAATTCATGGCCTCTGGTGTG-3′ and 5′-TTGAATTCTCACAAAGGCTTGCCC-3′. The PCR products were first subcloned into a pUC118 vector (Takara, Shiga, Japan). After digestion with EcoRI, the cDNA encoding cofilin was subcloned into the EcoRI site in the pMAL-2c (New England BioLabs, Ipswich, MA), in the p3xFLAG-CMVTM-14 (Sigma-Aldrich), or in the pDsRed monomer vector (Clontech, Palo Alto, CA).
The cDNA encoding the mutants lacking 24 N-terminal amino acids were produced via PCR with the following primers: 5′-CCGAATTCCCAGAAGAAGTGAAGA-3′ and 5′-TTGAATTCTCACAAAGGCTTGCCC-3′. The PCR products were cloned into pMAL-2c as described. In addition, point mutants of cofilin were produced via site-directed mutagenesis according to the manufacturer's instructions with the Quick Change II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA), using mouse cofilin and DsRed monomer in p3xFLAG-CMV-14 or in pMAL-2c as the template. The sense and antisense primers used were as follows: for S23A cofilin, 5′-GGTTCGCAAGGCCTCAACACAAG-3′ and 5′-CTGGTTTGAGGCCTTGCGAACC-3′; for S24A cofilin, 5′-GGTTCGCAAGAGCGCTACACCAGAAG-3′ and 5′-CTTCTGGTGTAGCGCTCTTGCGAACC-3′; for S23,24A cofilin, 5′-GGTTCGCAAGGCAGCAACACCAGAAG-3′ and 5′-CTTCTGGTGTTGCTGCCTTGCGAACC-3′; for S3E cofilin, 5′-ATTTCAGAATTCATGGCCGAGGGTGTGGCTGTCTCTGAT-3′ and 5′-ATCAGAGACAGCCACACCCTCGGCCATGAATTCTGAAAT-3′; and for S23,24E cofilin, 5′-TTCAATGACATGAAGGGTTCGAAAGGAGGAGACACCAGAAGAAGTGAAG-3′ and 5′-CTTCACTTCTTCTGGTGTCTCCTCCTTTCGAACCTTCATGTCATTGAA-3′. All PCR products were verified by sequencing.
RNA isolation and construction of β-hexosaminidase signal peptide-DsRed
Total mRNA was isolated from the cells with TRIzol reagent (Life Technologies, Grand Island, NY) according to the manufacturer's protocol. The mRNA was reverse transcribed to cDNA using random hexamers and Thermoscript thermostable reverse transcriptase (Invitrogen). Primers, 5′-GCTAGCATGGCCGGCTGCAGGCTCTGG-3′ and 5′-GCTAGCGCAGTCTGCTCAAACTCCTGCTC-3′, were used to amplify β-hexosaminidase in the presence of Taq polymerase. The PCR product was ligated into the pGEM-T Easy Vector (Takara). The signal peptide of β-hexosaminidase was amplified via PCR and cloned into AseI and BamHI sites of pWDsRed2-N1 (modified Clontech pDsRed2-N1). All PCR products were verified by sequencing.
Construction of adenovirus vectors encoding GFP-PKCα and βI
After digestion with XbaI and XhoI, the respective cDNA fragments encoding GFP-PKCα or βI were subcloned into the XbaI and XhoI sites of the pShuttle-cytomegalovirus vector. After linearization by Pme I, the pShuttle vector containing cDNA encoding a fusion protein of GFP with PKCα or βI or its mutant was coelectroporated with a pAdEasy backbone vector into BJ5183 bacterial cells. The recombination was verified via a Pac1 digestion, and the plasmid was purified by CsCl banding. Approximately 10 μg of the purified plasmids was digested by Pac1 and transfected with FuGENE 6 (Roche) into 50–70% confluent HEK293 cells plated on a 6-cm dish. The cells were scraped on day 7 posttransfection and resuspended in 1 ml of phosphate-buffered saline(–). After sonication, 50–70% confluent HEK293 cells in a T75 flask were infected with the supernatant and cultured in DMEM supplemented with penicillin (100 U/ml), streptomycin (100 μl/ml), and 10% horse serum (Life Technologies). To further amplify the viruses, we repeated infection using 30–50% of the viral supernatant on 50–70% confluent HEK293 cells in T75 flasks at least five times. Finally, the adenovirus was purified via CsCl banding and titrated.
Confocal microscopy
RBL-2H3 cells expressing signal peptide-DsRed and GFP-PKCα or βI were seeded onto a glass-bottom culture dish (MatTek) and incubated for 24 h before observation. In the case of stimulation of DNP-BSA, 1 μl of 100 μg/ml IgE was added to medium (final 50 ng/ml) 1 d before the examination. The culture medium was replaced with Siraganian buffer (119 mM NaCl, 5 mM KCl, 0.4 mM MgCl2, 5.6 mM glucose, 1 mM CaCl2, 0.1% BSA, and 25 mM 1,4-piperazinediethanesulfonic acid, pH 7.4). Translocation of signal peptide-DsRed and GFP-tagged PKCα or βI was triggered by direct addition of 10 μM ionomycin or 500 ng/ml DNP-BSA into the Siraganian buffer to yield a final concentration of 1 μM or 50 ng/ml. GFP fluorescence was monitored via a confocal laser scanning fluorescence microscope (LSM 510 invert; Carl Zeiss, Jena, Germany) with a 488-nm argon laser excitation and a 515- to 535-nm bandpass barrier filter. Red fluorescence was monitored at 543-nm HeNe excitation using a 560-nm long-pass barrier filter. To qualify the membrane translocation, >10 cells were analyzed via line scanning, and the percentage of membrane fluorescence of that total was estimated.
Two-dimensional electrophoresis
For the first dimension, immobilized pH gradient (IPG) dry strip gels (pH 3–10 and a length of 13 cm or pH 4–7 and a length of 13 cm; GE Healthcare Amersham Biosciences, Tokyo, Japan) were rehydrated with 280 μl of 32P-labeled protein sample containing 1 mg of protein in IPG strip holders (GE Healthcare Amersham Biosciences) for 10 h. The temperature was maintained at 20°C. Isoelectric focusing (IEF) was then conducted to a total of 65,500 Vh. After IEF, IPG gels were equilibrated with buffer containing 6 M urea, 50 mM Tris-HCl (pH 8.8), 30% glycerol, 10 mg/ml dithiothreitol, and 2.0% SDS for 15 min at room temperature. For the second dimension of separation, the equilibrated IPG dry strip gels were applied to 12.5% polyacrylamide gradient gels, and SDS–PAGE was performed.
In-gel digestion and mass spectrometry for protein identification
After electrophoresis, silver-stained spots corresponding to proteins were excised and destained. For reduction-alkylation reactions, excised bands were incubated with 10 mM dithiothreitol in 25 mM ammonium bicarbonate for 45 min at room temperature and then with 0.1 M iodoacetamide in 50 mM ammonium bicarbonate for 60 min at 50ºC. Proteins in gels were digested with porcine trypsin (sequencing grade; Promega, Madison, WI) in 50 mM ammonium bicarbonate for 15 h at 37ºC. The peptide fragments were extracted from gels, concentrated in vacuo, and then subjected to liquid chromatography/mass spectrometry/mass spectrometry (LC/MS/MS) using a high-performance liquid chromatography system (Paradigm MS4; Michrom Bioresources, Auburn, CA) coupled to a linear ion trap mass spectrometer (Finnigan LTQ; Thermo Scientific, San Jose, CA). LC/MS/MS data were interpreted through the MASCOT MS/MS ions search (Matrix Science, London, United Kingdom).
Identification of phosphopeptide by mass spectrometry
In-gel reduction-alkylation and in-gel digestion of the MBP-cofilin bands excised from a Coomassie brilliant blue–stained SDS gel were performed as described. After desalting with ZipTipC18 (Millipore, Billerica, MA), positive-ion mass spectra of peptide fragments were acquired in a Micromass Q-Tof2 mass spectrometer (Waters Corporation, Milford, MA) equipped with a direct infusion nano electrospray ionization source.
Expression and purification of recombinant protein
The cDNAs encoding wild-type cofilin or mutants were digested with EcoRI and subcloned into the EcoRI site of pMal-2c. BL21 (DE3) pLys cells were used to express MBP-fusion proteins, which was induced with 0.3 mM isopropyl 1-thio-d-galactoside at 25°C for 4 h. The cells were then harvested and lysed in column buffer (20 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1 mM dithiothreitol, 200 mM NaCl, 1% Triton X-100, 20 μg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride) with a handy sonic instrument (Tomy Seiko, Tokyo, Japan). After centrifugation at 10,000 × g for 1 h, fusion proteins were purified on an amylose resin (New England BioLabs, Ipswich, MA) affinity column, according to the manufacturer's instructions.
In vitro cofilin phosphorylation assay
Purified MBP-cofilin was incubated with purified PKCα at 30°C in the presence or absence of 8 μg/ml PS, 0.8 μg/ml (±)-1,2-didecanoylglycerol (DO), and 0.5 mM CaCl2. This reaction was initiated by the addition of 0.2 mM ATP with [γ-32P]ATP. After 15 min, the reaction was terminated via addition of sample buffer (186 mM Tris-HCl, pH 6.7, 15% glycerol, 9% SDS, 6% 2-mercaptoethanol, bromophenol blue) and boiling at 95°C for 5 min. The amount and radioactivity of MBP-cofilin were monitored by SDS–PAGE in conjunction with immunoblotting and autoradiography with a BAS-2500 analyzer (Fujifilm, Tokyo, Japan).
In vivo cofilin phosphorylation assay
RBL-2H3 cells were transfected with cofilin-FLAG and its mutants. In the case of DNP-BSA stimulation, 1 μl of 100 μg/ml IgE was added to medium (final concentration of 50 ng/ml) 1 d before the examination. For overnight culture after transfection, the cells were loaded with 32P for 1 h and were stimulated with ionomycin or DNP-BSA for 1 min. The cells were harvested and lysed in lysis buffer (20 mM Tris-HCl, pH 7.4, 10 mM ethylene glycol tetraacetic acid [EGTA], 2 mM EDTA, 150 mM NaCl, 200 μM leupeptin, 1 mM phenylmethylsulfonyl fluoride, 1 mM Na3VO4, 1 μg/ml pepstatin, and 20 mM β-glycerophosphate) containing 1% Triton X-100; subsequently the cells were sonicated. After centrifugation, the supernatant was immunoprecipitated with an anti-FLAG M2 affinity gel and subjected to SDS–PAGE, autoradiography, and immunoblotting with an anti-FLAG antibody.
Generation of stable cell lines
Plasmids encoding mDsRed-cofilin and each mutant were transfected via the Amaxa Nucleofector System in accordance with the manufacturer's instructions. Geneticin (0.5 mg/ml) was added to the medium 24 h posttransfection. After being cultured for more than 24 h, the transfected cells were transferred to 96-well plates at a density of 1 cell/well for cloning. Positive clones were identified by fluorescence detected using confocal microscopy.
F-actin cosedimentation assay
The F-actin sedimentation assay was performed as described by Moriyama et al. (1996). Briefly, 2 mg/ml actin from rabbit muscle (Molecular Probes, Eugene, OR) was polymerized for 60 min at room temperature in 10× polymerization buffer (0.2 M Tris-HCl, pH 7.5, 1 M KCl, 20 mM MgCl2, and 1 mM dithiothreitol). After purification of MBP-cofilin and mutants, MBP was excised by factor Xa (Novagen, Gibbstown, NJ) and removed with amylose resin. Purified cofilin (5 μg of wild type or mutant) was incubated with 10 μg of F-actin in 200 μl of 10 mM Tris-HCl (pH 8.0) for 30 min at room temperature. Then, the mixtures were ultracentrifuged at 100,000 × g for 30 min. The supernatants and pellets were separately subjected to SDS–PAGE and stained with Coomassie blue.
Direct observation of F-actin severing
The F-actin–severing assay was performed as previously described (Ono et al. 2004). Briefly, unlabeled actin (1.4 μM), Alexa Fluor 488–labeled actin (0.4 μM, Molecular Probes), and biotin-labeled actin (0.2 μM; Cytoskeleton, Dover, CO) were copolymerized for 1 h in ISAP buffer (50 mM KCl, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid–KOH, 5 mM EGTA, 2 mM MgCl2, 1 mM ATP, and 1 mM dithiothreitol, pH 7.2). Slide glass (76 × 26 mm; Matsunami, Osaka, Japan) was coated with 0.1% nitrocellulose in methanol. The cover glass (24 × 32 mm; Matsunami) was mounted on the coated glass slide to form a perfusion chamber. The chamber was perfused with 30 μg/ml anti-biotin antibody and washed twice with ISAP buffer containing 0.5 mg/ml BSA. Alexa Fluor 488/biotin–labeled F-actin was diluted 30-fold with anti-bleaching buffer (ISAP buffer containing 36 μg/ml catalase, 20 μg/ml glucose oxidase, 6 mg/ml glucose, and 100 mM dithiothreitol). After preincubation with purified cofilin or its mutants, the mixture was perfused into the chamber and incubated for 5 min. After washing twice with anti-bleaching buffer, fluorescence was monitored via confocal microscopy.
F-actin FACS Assay
In total, 2 × 106 RBL cells were transfected with 100 ng/ml siRNA in a 10-cm dish. After trypsinization and fixation with 70% ethanol, the cells were stained with FITC-phalloidin (Molecular Probes). Appropriate numbers of cells were subjected to analysis with a FACScan (BD Biosciences, San Diego, CA). To compare the cellular F-actin content, a ratio of the mean fluorescence intensity of FITC-phalloidin staining was plotted as “relative to F-actin.”
Supplementary Material
Abbreviations used:
- DAG
diacylglycerol
- KN
kinase negative
- MBP
maltose-binding protein
- PKC
protein kinase C
- PS
phosphatidylserine
- RBL
rat basophilic leukemia
- WT
wild type
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-01-0053) on August 1, 2012.
REFERENCES
- Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- Barsumian EL, Isersky C, Petrino MG, Siraganian RP. IgE-induced histamine release from rat basophilic leukemia cell lines: isolation of releasing and nonreleasing clones. Eur J Immunol. 1981;11:317–323. doi: 10.1002/eji.1830110410. [DOI] [PubMed] [Google Scholar]
- Befus AD, Pearce FL, Gauldie J, Horsewood P, Bienenstock J. Mucosal mast cells. Isolation and functional characteristics of rat intestinal mast cells. J Immunol. 1982;128:2475–2480. [PubMed] [Google Scholar]
- Blank U, Rivera J. The ins and outs of IgE-dependent mast-cell exocytosis. Trends Immunol. 2004;25:266–273. doi: 10.1016/j.it.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD, Cheek TR. Is the transient nature of the secretory response of chromaffin cells due to inactivation of calcium channels? FEBS Lett. 1985;182:115–118. doi: 10.1016/0014-5793(85)81166-2. [DOI] [PubMed] [Google Scholar]
- Bretscher A, Edwards K, Fehon RG. ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol. 2002;3:586–599. doi: 10.1038/nrm882. [DOI] [PubMed] [Google Scholar]
- Carlier MF, Laurent V, Santolini J, Melki R, Didry D, Xia GX, Hong Y, Chua NH, Pantaloni D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigeri L, Apgar JR. The role of actin microfilaments in the down-regulation of the degranulation response in RBL-2H3 mast cells. J Immunol. 1999;162:2243–2250. [PubMed] [Google Scholar]
- Fu H, Subramarian RR, Masters SC. 14-3-3 proteins: structure, function, and regulation. Annu Rev Pharmacol Toxicol. 2000;40:617–647. doi: 10.1146/annurev.pharmtox.40.1.617. [DOI] [PubMed] [Google Scholar]
- Galli SJ. Mast cells and basophils. Curr Opin Hematol. 2000;7:32–39. doi: 10.1097/00062752-200001000-00007. [DOI] [PubMed] [Google Scholar]
- Gohla A, Bokoch G. 14-3-3 regulates actin dynamics by stabilizing phosphorylated cofilin. Cur Biol. 2002;12:1704–1710. doi: 10.1016/s0960-9822(02)01184-3. [DOI] [PubMed] [Google Scholar]
- Hanson DA, Ziegler SF. Regulation of ionomycin-mediated granule release from rat basophil leukemia cells. Mol Immunol. 2002;38:1329–1335. doi: 10.1016/s0161-5890(02)00083-4. [DOI] [PubMed] [Google Scholar]
- Hide I, Toriu N, Nuibe T, Inoue A, Hide M, Yamamoto S, Nakata Y. Suppression of TNF-α secretion by azelastin in a rat mast (RBL-2H3) cell line. J Immunol. 1997;159:2932–2940. [PubMed] [Google Scholar]
- Hultsch T, Brand P, Lohman S, Saloga J, Kincaid RL, Knop J. Direct evidence that FK506 inhibition of FcεRI-mediated exocytosis from RBL cells involves calcineurin. Arch Dermatol Res. 1998;290:258–263. doi: 10.1007/s004030050301. [DOI] [PubMed] [Google Scholar]
- Ichetovkin I, Han J, Pang KM, Knecht DA, Condeelis JS. Actin filaments are severed by both native and recombinant Dictyostelium cofilin but to different extents. Cell Motil Cytoskeleton. 2000;45:293–306. doi: 10.1002/(SICI)1097-0169(200004)45:4<293::AID-CM5>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Kinet JP. The high-affinity IgE receptor (Fc epsilon RI): from physiology to pathology. Annu Rev Immunol. 1999;17:931–972. doi: 10.1146/annurev.immunol.17.1.931. [DOI] [PubMed] [Google Scholar]
- Kraft AS, Anderson WB, Cooper HL, Sando JJ. Decrease in cytosolic calcium/phospholipid-dependent protein kinase activity following phorbol ester treatment of EL4 thymoma cells. J Biol Chem. 1982;257:13193–13196. [PubMed] [Google Scholar]
- Larsson C. Protein kinase C and the regulation of the actin cytoskeleton. Cell Signal. 2006;18:276–284. doi: 10.1016/j.cellsig.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Leitges M, Gimborn K, Elis W, Kalesnikoff J, Hughes MR, Krystal G, Huber M. Protein kinase C-delta is a negative regulator of antigen-induced mast cell degranulation. Mol Cell Biol. 2002;22:3970–3980. doi: 10.1128/MCB.22.12.3970-3980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo TN, Saul W, Beaven MA. The actions of Ca2+ ionophores on rat basophilic (2H3) cells are dependent on cellular ATP and hydrolysis of inositol phospholipids. A comparison with antigen stimulation. J Biol Chem. 1987;262:4141–4145. [PubMed] [Google Scholar]
- Moriyama K, Iida K, Yahara I. Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells. 1996;1:73–86. doi: 10.1046/j.1365-2443.1996.05005.x. [DOI] [PubMed] [Google Scholar]
- Mulero I, Sepulcre MP, Meseguer J, Garcia-Ayala A, Mulero V. Histamine is stored in mast cells of most evolutionarily advanced fish and regulates the fish inflammatory response. Proc Natl Acad Sci USA. 2007;104:19434–10439. doi: 10.1073/pnas.0704535104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechushtan H, Leitges M, Cohen C, Kay G, Razin E. Inhibition of degranulation and interleukin-6 production in mast cells derived from mice deficient in protein kinaseCbeta. Blood. 2000;95:1752–1757. [PubMed] [Google Scholar]
- Nishizuka Y. The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature. 1988;334:661–665. doi: 10.1038/334661a0. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- Ono S, Mohri K, Ono K. Microscopic evidence that actin-interacting protein 1 actively disassembles actin-depolymerizing factor/cofilin-bound actin filaments. J Biol Chem. 2004;279:14207–14212. doi: 10.1074/jbc.M313418200. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Szallasi Z, Kazanietz MG, Blumberg PM, Mischak H, Mushinski JF, Beaven MA. Ca(2+)-dependent and Ca(2+)-independent isozymes of protein kinase C mediate exocytosis in antigen-stimulated rat basophilic RBL-2H3 cells. Reconstitution of secretory responses with Ca2+ and purified isozymes in washed permeabilized cells. J Biol Chem. 1993a;268:1749–1756. [PubMed] [Google Scholar]
- Ozawa K, Yamada K, Kazanietz MG, Blumberg PM, Beaven MA. Different isozymes of protein kinase C mediate feedback inhibition of phospholipase C and stimulatory signals for exocytosis in rat RBL-2H3 cells. J Biol Chem. 1993b;268:2280–2283. [PubMed] [Google Scholar]
- Pendelton A, Koffer A. Effect of latrunculin reveal requirements for the actin cytoskeleton during secretion from mast cells. Cell Motil Cytoskeleton. 2001;48:37–51. doi: 10.1002/1097-0169(200101)48:1<37::AID-CM4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Pandey D, Goyal P, Bamburg JR, Siess W. Regulation of LIM-kinase 1 and cofilin in thrombin-stimulated platelets. Blood. 2006;107:575–583. doi: 10.1182/blood-2004-11-4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey D, Goyal P, Dwivedi S, Siess W. Unraveling a novel Rac1-mediated signaling pathway that regulates cofilin dephosphorylation and secretion in thrombin-stimulated platelets. Blood. 2009;114:415–424. doi: 10.1182/blood-2008-10-183582. [DOI] [PubMed] [Google Scholar]
- Pope BJ, Zierler-Gould KM, Kuhne R, Weeds AG, Ball LJ. Solution structure of human cofilin. Actin binding, pH sensitivity, and relationship to actin-depolymerizing factor. J Biol Chem. 2004;279:4840–4848. doi: 10.1074/jbc.M310148200. [DOI] [PubMed] [Google Scholar]
- Powner DJ, Hodgkin MN, Wakelam MJ. Antigen-stimulated activation of phospholipase D1b by Rac1, ARF6, and PKCalpha in RBL-2H3 cells. Mol Biol Cell. 2002;13:1252–1262. doi: 10.1091/mbc.01-05-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribluda VS, Pribluda C, Metzger H. Transphosphorylation as the mechanism by which the high-affinity receptor for IgE is phosphorylated upon aggregation. Proc Natl Acad Sci USA. 1994;91:11246–11250. doi: 10.1073/pnas.91.23.11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigbolt KTG, Prokhorova TA, Akimov V, Henningsen J, Johansen PT, Kratchmarova I, Kassem M, Mann M, Olsen JV, Blagoev B. System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci Signal. 2011;4:rs3. doi: 10.1126/scisignal.2001570. [DOI] [PubMed] [Google Scholar]
- Saitoh S, Arudchandran R, Manetz TS, Zhang W, Sommers CL, Love PE, Rivera J, Samelson LE. LAT is essential for FcεRI-mediated mast cell activation. Immunity. 2000;12:525–535. doi: 10.1016/s1074-7613(00)80204-6. [DOI] [PubMed] [Google Scholar]
- Sakai N, Sasaki N, Sasaki K, Ikegaki N, Shirai Y, Ono Y, Saito N. Direct visualization of the translocation of the gamma-subspecies of protein kinase C in living cells using fusion proteins with green fluorescent protein. J Cell Biol. 1997;139:1465–1476. doi: 10.1083/jcb.139.6.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shichijo M, Inagaki N, Nakai N, Kimata M, Nakata T, Serizawa I, Ikura Y, Saito H, Nagai H. The effect of anti-asthma drugs on mediator release from cultured human mast cells. Clin Exp Allergy. 1998;28:1228–1236. doi: 10.1046/j.1365-2222.1998.00394.x. [DOI] [PubMed] [Google Scholar]
- Shirai Y, Kashiwagi K, Yagi K, Sakai N, Saito N. Distinct effect of fatty acids on translocation ofγ and ε-subspecies of protein kinase C. J Cell Biol. 1998;143:511–521. doi: 10.1083/jcb.143.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siraganian RP, McGivney A, Barsumian EL, Crews FT, Hirata F, Axelrod J. Variants of the rat basophilic leukemia cell line for the study of histamine release. Fed Proc. 1982;1:30–34. [PubMed] [Google Scholar]
- Smith AJ, Pfeiffer JR, Zhang J, Martinez AM, Griffiths GM, Wilson BS. Microtubule-dependent transport of secretory vesicles in RBL-2H3 cells. Traffic. 2003;4:302–312. doi: 10.1034/j.1600-0854.2003.00084.x. [DOI] [PubMed] [Google Scholar]
- Turner H, Kinet JP. Signaling through the high-affinity IgE receptor FcεRI. Nature. 1999;402:B24-B30. doi: 10.1038/35037021. [DOI] [PubMed] [Google Scholar]
- van Rheenen J, Song X, van Roosmalen W, Commer M, Chen X, DesMarais V, Yip SC, Backer JM, Eddy RJ, Condeelis JS. EGF-induced PIP2 hydorsis releases and activates cofilin locally in carcinoma cells. J Cell Biol. 2007;179:1247–1259. doi: 10.1083/jcb.200706206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Shibasaki F, Mizuno K. Calcium signal-induced cofilin dephosphorylation is mediated by slingshot via calcineurin. J Biol Chem. 2005;280:12683–12689. doi: 10.1074/jbc.M411494200. [DOI] [PubMed] [Google Scholar]
- Yanase Y, Hide I, Mihara S, Shirai Y, Saito N, Nakata Y, Hide M, Sakai N. A critical role of conventional protein kinase C in morphological changes of rodent mast cells. Immunol Cell Biol. 2011;89:149–159. doi: 10.1038/icb.2010.67. [DOI] [PubMed] [Google Scholar]
- Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393:809–812. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.