

Abstract
Glaucoma, a neurodegenerative disease, is currently being treated by modulation of one of its primary risk factors, the elevated intraocular pressure. Newer therapies that can provide direct neuroprotection to retinal ganglion cells are being extensively investigated. Tumor necrosis factor-α, a cytokine, has been recognized to play an important role in pro and antiapoptotic cellular events. In this paper we review the relevant literature to understand (1) The association of increased expression of tumor necrosis factor-α with glaucomatous neurodegeneraion, (2) Modulation of tumor necrosis factor-α expression by exposure to various risk factors of glaucoma, (3) Downstream cellular signaling mechanisms following interaction of tumor necrosis factor-α with its receptors and (4) Role of tumor necrosis factor-α as a possible target for therapeutic intervention in glaucoma. Literature was reviewed using PubMed search engine with relevant key words and a total of 82 English language papers published from 1990 to 2010 are included in this review.
Keywords: Apoptosis, glaucoma, intraocular pressure, retinal ganglion cells, retinal ischemia, tumor necrosis factor-alpha
Glaucoma is a heterogeneous group of disorders affecting more than 70 million people world-wide and is characterized by cupping of the optic disc due to loss of retinal ganglion cells (RGCs) and their axons leading to progressive visual loss.[1–3] Currently available drugs primarily aim to lower the intraocular pressure (IOP). However, it has been observed that disease progression may continue despite significant IOP reduction. Clearly, modulation of the primary risk factor of glaucoma such as elevated IOP alone is not enough to completely prevent the RGC loss. Hence, only a targeted therapeutic intervention that can prolong the survival of RGCs and possibly regenerate the lost RGCs, will successfully preserve the vision in glaucoma patients. For the development of such therapeutic interventions, the key is to recognize the underlying pathogenic mechanisms leading to RGC loss. In the recent years involvement of several cytokines such as interleukins, interferon-γ and tumor necrosis factor-α (TNF-α) has extensively been investigated. This review paper is an effort to understand the role of TNF-α in the pathogenesis of glaucoma. Literature was reviewed using Pubmed search engine with relevant key words like TNF-α, glaucoma, retinal ganglion cell apoptosis, and IOP. A total of 82 English language papers published from 1990 to 2010 are included in this review. All publications included in this review are from journals listed in science citation index. The review paper attempts to summarize the large body of evidence from genetic linkage analyses, cell culture studies, in vivo experimental and human studies showing involvement of TNF-α in glaucomatous neurodegeneration.
TNF-α: Neuroprotective and Neurodegenerative Actions
TNF-α is a cytokine that belongs to TNF super family of 19 different protein ligands. These cytokines mediate their cellular responses through 29 receptors of TNF-receptor (TNF-R) super family.[4] This polypeptide cytokine serves a variety of cellular functions ranging from growth stimulating to growth inhibitory processes. Low levels help in maintaining the homeostasis, regulating the circadian rhythm and promoting the replacement of injured tissue.[5] TNF-α plays a significant role in local immune response to invading organisms by initiating a cascade of reactions that culminates into inflammatory response in an attempt to eliminate the invading organisms.[6] TNF-α also plays an important role in diseases of noninflammatory origin. It promotes necrosis of some tumor cell types but is also known to promote growth of other tumor cells.[7,8] TNF-α expression was found to be upregulated in the brain of patients with neurodegenerative diseases such as Alzheimer's and Parkinson's disease suggesting a causative role of TNF-α in neurodegenerative disorders.[9–11] Recent studies have shown that TNF-α plays a significant role both in neuroprotection and neurodegeneration and under physiological conditions a balance exists between the two opposing actions.[12,13] Exposure to risk factors might initiate a shift in balance so as to favor progression of neurodegenerative process.
TNF-α in Glaucoma
The correlation of TNF-α with glaucomatous changes has been established in human and animal in vivo studies that have either shown that serum or intraocular TNF-α levels are elevated in patients with glaucoma or the exposure of normal eyes to TNF-α induces RGC apoptosis. Sawada et al., have shown that the level of TNF-α in aqueous humor of glaucoma patients are significantly elevated as compared to patients with nonglaucomatous eyes.[14] In this study the glaucoma group which included 29 cases of primary open-angle glaucoma (POAG), 28 cases of normal-tension glaucoma (NTG) and 27 cases of exfoliation glaucoma showed significantly high aqueous humor levels of TNF-α as compared to the group of 79 patients with cataract which served as control group. Significantly elevated TNF-α levels have also been demonstrated in the serum of patients with severe optic neuropathy as compared to those with mild optic neuropathy.[15] Madigan et al., showed that intravitreal injection of TNF-α in rabbits induces optic nerve axonal damage similar to that observed in glaucomatous neuropathy.[16] Intravitreal injection of TNF-α in rats was found to induce axonal degeneration from 2 weeks to 2 months after injection, whereas significant RGC loss was noted at 2 months after injection.[17] Intravitreal injection of TNF-α in mice has also been shown to induce degenerative changes in RGCs. However, similar changes were not induced in mice with the TNF-α gene deleted or by immune depletion of TNF-α in wild-type mice.[18] In one of the studies, 4 postmortem eyes from patients with POAG, 7 eyes from patients with normal-pressure glaucoma, and 4 eyes from age-matched normal donors were studied by immunohistochemistry. Increased immunostaining for TNF-α and TNF-R1 was observed in the optic nerve heads of postmortem eyes from glaucoma patients, particularly those with NTG as compared to normal controls.[19] Yuan and Neufeld studied the expression of TNF-α and TNF- R1 in human glaucomatous optic nerve heads from patients with different stages of disease using double-labeling fluorescence immunohistochemistry. They showed that not only the expression of TNF-α and TNF-R1 in glaucomatous optic nerve head is upregulated but it also parallels the progression of optic nerve damage.[20] Thus the coexistence of increased TNF-α expression and glaucomatous changes clearly implicates TNF-α in the pathogenesis of RGC loss in glaucoma.
Risk factor of Glaucoma induced TNF-α Expression
Normal retinal tissue shows constitutive expression of TNF-R1 in the vasculature of the optic nerve heads but weak or no positive labeling for TNF-α. Production and release of TNF-α occurs very early on following exposure to stresses such as elevated IOP or ischemia. In one of the studies, rat retinal gene expression profiling was done in two models of RGC injury- laser photocoagulation-induced ocular hypertension and optic nerve transection. Whole genome microarray analysis was used as initial screening and this was followed by real time PCR to detect significant changes. It was observed that there was an 8-fold increase in the expression of TNF-R1 gene in rats with elevated IOP after laser photocoagulation. Furthermore, there was upregulation of lipopolysaccharide-induced TNF Factor, a transcription factor that positively regulates TNF-α expression.[21] Retinal cell culture under ischemic conditions leads to massive RGC death; however, addition of TNF-α or TNF-R1 antibody to culture medium provides significant protection from cell death.[22] Tezel and Wax also showed that RGC apoptosis was attenuated ~66% by a neutralizing antibody against TNF-α.[23] Under normoxic conditions, TNF-α is not toxic to RGCs when they are surrounded by other retinal cells. However, when purified RGCs are exposed to TNF-α, apoptosis is induced even under normoxic conditions. These results indicate absence of a survival pathway in purified cells and recovery of this pathway in presence of other retinal cells under normoxic conditions.[22] Apparently, the survival signals fail to prevent RGC apoptosis, in the presence of risk factors of glaucoma, due to excessive production of TNF-α.
The glial cells, are of extreme importance in providing the survival signals, as they are known to make external environment favorable for the survival of RGCs. Transition of these normally functioning glial cells to stimulated glial cells in response to stress factors seems to be the key event leading to RGC apoptosis [Fig. 1]. In a mouse model with laser-induced ocular hypertension it was observed that TNF-α mediates the cytotoxic effects of elevated IOP on RGCs via an indirect route that involves microglial activation and the loss of oligodendrocytes.[18] In vitro experiments have revealed that glial cells are stimulated under simulated ischemia or elevated hydrostatic pressure. Stimulated glial cells secrete TNF-α along with other neurotoxic substances such as nitric oxide.[23] It has been suggested that activation of microglia early on in the disease process attempts to stabilize the tissue; however, with increasing severity, intense microglial activation leads to detrimental consequences.[24] Furthermore, it has been observed that exposure to stress factors such as elevated IOP causes upregulation of both the proapoptotic and prosurvival genes. In one of the studies, rats with laser photocoagulation-induced glaucoma showed significantly upregulated expression of proapototic gene, Gadd45a, 1 week after induction of increased IOP which stayed upregulated for 2 months and long after IOP returned to baseline. The prosurvival gene, Inhibitor of Apoptosis Protein 1 (IAP-1), was simultaneously upregulated but returned to baseline earlier than the proapoptotic gene. The expression of corresponding proteins was also upregulated as detected by Western blot and immunohistochemistry localized these changes to the retinal ganglion cell layer.[25,26] IAPs are known to interact with caspases 3, 7 and 9 and thereby inhibit apoptosis.[27–29] The upregulated expression of proapoptotic genes for periods longer than the upregulated expression of pro survival genes is possibly the reason for continued degeneration of RGCs even after the IOP returns to normal.[25]
Figure 1.
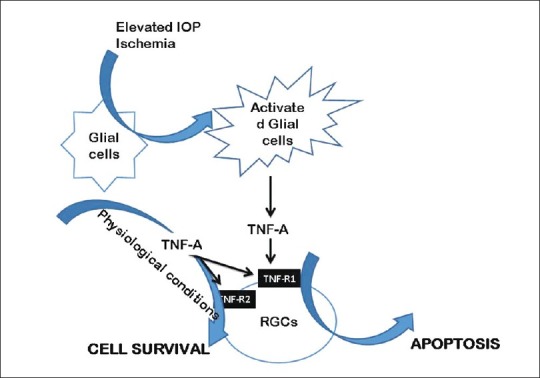
Role of TNF-A in retinal ganglion cell apoptosis induced by elevated intraocular pressure and retinal ischemia
Differential Expression of TNF-α and TNF-R1
In NTG, as compared to age-matched controls, increased immunostaining for TNF-α and TNF-R1 has been observed in the glial cells and their processes around axons and blood vessels in all regions of optic nerve head, especially in the postlaminar region.[19] Evidences show that the normal tissue constitutively expresses TNF-R1 in the vasculature of the optic nerve heads but not the TNF-α. In glaucomatous eyes, expression of both the TNF-α and TNF-R1 increases in astrocytes and microglia. Bai et al., have shown that upregulation of TNF-α in glial cells is controlled by TrkC.T1, which is a neurotrophin receptor truncated isoform lacking the kinase domain and has been shown to be upregulated in the retina of glaucomatous rat eyes.[30] In severely damaged optic nerve heads the axons of the RGCs express TNF-R1 and may be the direct targets for TNF-α-mediated neurodegeneration.[20] Using immunohistochemistry and in situ hybridization, Tezel et al., studied the expression and localization of TNF-α and TNF-R1 in the retinal sections from 20 eyes of donors with glaucoma, and 20 eyes of age-matched normal donors. The intensity as well as the number of TNF-α and TNF-R1 positive cells were significantly high in glaucomatous eyes. Both the mRNA and protein expression for TNF and TNF-R1 was also significantly high in the inner retinal layers. The double-immunofluorescence labeling revealed that the immunostaining for TNF-α was predominantly positive in the glial cells, whereas for TNF-R1, it was mainly positive in the RGCs. The increased TNF-α protein and gene expression in glial cells was observed especially in the retinal nerve fiber layer, ganglion cell layers and in glial cells located close to a retinal blood vessel. Further, the expression of both TNF-α and TNF-R1 was found to parallel the progression of optic nerve degeneration.[31] The pattern of increased expression of these proteins in glaucomatous eyes explains the selective sensitivity of RGCs to TNF-α-induced apoptosis. Moreover, it also explains that the TNF-α-induced RGCs degeneration is a glial-initiated pathogenic mechanism in glaucomatous optic nerve heads[19Fig. 1].
Cell Signaling Mechanisms in TNF-α Mediated Effects on RGCs
Cellular mechanisms involved in TNF-α mediated RGC apoptosis have been demonstrated by morphology, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling, and caspase activity.[23] TNF-α signaling involves multiple second messenger pathways that can function independently or in coordination with each other to transduce its cellular functions.
The diverse activities of TNF-α are mediated via two distinct receptors: a 55-kDa receptor 1 (TNF-R1) and a 75 kDa receptor (TNF-R2). An upregulation of TNF-α, TNF-R1 and TNF-R2 has been observed in response to retinal ischemia preceding the neuronal death. Among the two, TNF-R1 appears to be the key mediator of proapoptotic functions of TNF-α in vast majority of cells whereas TNF-R2 promotes neuroprotection. Animals deficient of TNF-R1 were found to have better neuronal cell survival in response to retinal ischemia-reperfusion injury. TNF-R2 dependent neuroprotection has been correlated with the presence of Akt/protein kinase B and involves phosphatidylinositol 3- kinase (PI3-kinase) signaling pathways.[32] PI3-kinase causes phosphorylation of Akt/protein kinase B. Activated Akt/protein kinase B has the ability to phosphorylate and inhibit the proapoptotic proteins.[33,34] Studies have shown that 24 hour TNF-α treatment can rescue neurons from glutamate-induced excitotoxic death by increasing the neuronal phosphorylated Akt/protein kinase B.[35,36] The neuroprotective actions of TNF- α through TNF-R2 also involve recruitment of TNF-R Associated Factor 2 (TRAF-2) and subsequent activation of nuclear factor-kappa B (NF-κB).[37] The relevant downstream mechanisms are discussed in the following paragraphs.
Binding of TNF-α to TNF-R1 triggers the trimerization of the receptor and exposure of its intracellular domain subsequent to the release of an inhibitory protein known as Silencer of Death Domain (SODD).[38] The exposed intracellular domain of TNF-R1 is recognized by a death domain-containing adaptor protein, TNF Receptor-Associated Death Domain (TRADD).[39] TRADD acts as a platform to recruit Fas-Associated Death Domain (FADD), TRAF2 and Receptor Interacting Protein (RIP), which act on downstream signaling components.[40–42]
Interaction of TRAF2 and RIP with downstream signaling mechanisms leads to activation of transcription factors such as NF-κB. These transcription factors induce expression of genes that are involved in a variety of cellular functions such as cell proliferation, inflammation and suppression of apoptosis.[43,44] NF-κB plays a key role in regulating the antiapoptotic effects of TNF-α and in particular its RelA (p65) subunit is required for induction of TNF-α-dependent genes.[45] NF-κB is a heterodimer consisting of 50- and 65-kDa subunits. The two subunits are present in the cytoplasm complexed with an inhibitory protein, IκB. Following interaction of the receptor with the ligand, IκB dissociates from NF-κB, which now translocates to nucleus. The activation and subsequent nuclear translocation of NF-κB involves an atypical isoform of the enzyme, protein kinase C (PKC), PKCζ, which regulates nuclear events essential for the initiation of the apoptotic pathway.[46] An overexpression of PKCζ protects neurons by facilitating the nuclear translocation of NF-κB but inhibition of PKCζ predisposes to neuronal death.[47] In one of the studies, intravitreal injection of TNF-α alone in rat eyes could not produce significant RGC apoptosis. But when TNF-α was injected in combination with a specific inhibitor of PKCζ, RGCs were exposed to TNF-α toxicity and significant RGC apoptosis was observed in the inner nuclear and ganglion cell layers.[48]
FADD is the proapoptotic mediator engaged by TNF-R1.[40,49] Interaction of FADD with downstream signaling pathways involves activation of caspase-8. TNF-α-activated caspase 8 subsequently activates caspase 3, which in turn activates executioner proteolysis cascade, finally resulting in apoptosis.[50] Fas death pathway may be inhibited by PKCζ, by phosphorylating the FADD.[51] The expressions of apoptosis-related genes, caspase-8, Fas, FADD and the activity of caspase-3 were found to be increased in the iris/ciliary body of DBA/2J mice, an animal model of human pigmentary glaucoma.[52] According to some studies, caspase-independent pathways may also be involved in RGC apoptosis as the cultured RGCs were found to survive only temporarily when exposed to TNF-α in the presence of a caspase inhibitor.[53] Caspase -3 has also been shown to cause cleavage of Akt/protein kinase B. As discussed earlier, Akt/protein kinase B plays an important role in promoting the cell survival, its negative regulation by TNF- promotes apoptotic signaling.[54]
Mutation of a gene, OPTN, encoding a protein optineurin has been found to be associated with normal tension and POAG. OPTN is located on chromosome 10p14. The OPTN gene codes for a conserved 66-kDa protein known as optineurin which is preferentially expressed in RGCs and has been implicated in the TNF-α signaling pathway. At present, there are more than 20 mutations found in the OPTN coding region, four of which have close correlation with POAG. These mutations are respectively E50K of exon 4, M98K of exon 5, discontinuous protein coding of exon 6 and R545Q of exon 16. Among all, E50K has been found to be the most common disease causing mutant in which glutamic acid in position 50 is replaced by lysine in the coded protein.[55] This mutant E50K has been shown to cause RGC death by overexpression of the intracellular domain of 55-kDa TNF-α receptor in HeLa cells.[56] Optineurin shares homology with NF-κB essential modulator which is essential for NF-κB activation. Normally expressed optineurin does not compete with NF-κB essential modulator but overexpression of optineurin causes its competitive inhibition and prevention of TNF-α-induced NF-κB activation thus abolishing the prosurvival signaling.[57] Chalasani et al., also showed that optineurin E50K mutation potentiates the TNF-α-induced RGC death in culture.[58] Moreover, the occurrence of single nucleotide polymorphism of TNF-α gene was found to be significantly higher in high-tension glaucoma patients as compared to control group.[59] While some of the researchers have shown a strong association of POAG and pseudoexfoliative glaucoma with TNF-α polymorphism G-308A[60,61] others could not detect such association.[62,63]
Cellular signaling mechanisms described above indicate that TNF-α mediates both pro- and antiapoptotic effects. Although our understanding of TNF-α is not complete, TNF-α seems to play a unique role in regulating the critical balance between both pro- and antiapoptotic cellular responses. As stated above, some of the experiments have demonstrated the TNF-α-mediated RGC protection after axotomy and elevated IOP.[32,64] Lack of TNF-α induced toxicity in these studies can be explained by activation of NF-κB following exposure to TNF-α, as addition of a suppressor of NF-κB activation led to significant increase in RGC death. Chronic and excessive exposure to stress factors (such as elevated IOP or optic nerve head ischemia) results in dysregulation of TNF-α expression and/or signaling, triggering the shift toward proapoptotic responses in RGCs.
TNF-α may indirectly induce RGC apoptosis
Although, TNF-α produced by activated glial cells has been shown to directly induce apoptosis of RGCs, there is evidence that activated retinal glial cells induce production of other cytotoxic substances such as nitric oxide (NO) and endothelin-1 (ET1) that lead to neuronal damage. Retinal astrocytes in vitro express TNF-R1 and their stimulation by TNF-α was found to induce nitric oxide synthase-2 (iNOS).[20] Shafer and Murphy showed that activated cerebral astrocytes released TNF-α which then induces iNOS expression in endothelial cells.[65] Muller glial cells have also been demonstrated to produce NO in response to endogenous TNF-α.[66] Increased expression of iNOS causes exposure of optic nerve head to excess quantities of nitric oxide (NO) which causes neurodestructive changes in RGCs. Aminoguanidine, a relatively specific inhibitor of NOS-2, has been shown to protect RGCs from apoptosis.[67,68] Tezel and Wax also showed that a selective inhibitor of iNOS can reduce RGC apoptosis by about 50%.[23] NO has been shown to induce apoptosis by activating caspase 3.[69]
Treatment with TNF-α was shown to cause disruption and subsequent breakdown of the tight junction barrier in retinal pigment epithelium (RPE). The RPE, which helps maintain the outer blood-retinal barrier, is a local source for ET1. Following the breakdown of the tight junction barrier in RPE subsequent to TNF-α exposure, there is significant increase in the expression of preproendothelin-1 and secretion of ET-1 by retinal pigment epithelial cells in culture.[70] Increased TNF-α production in response to hypoxia has been shown to increase synthesis and release of ET1 by 5-fold which then causes astrocyte activation and proliferation in the optic nerve head.[71] ET1 is able to induce NOS-2 expression, both at mRNA and protein levels, via ETA/B receptor activation.[72] Both ET-1 and NOS (specially NOS-1 and -2) levels are elevated in glaucoma and are putatively involved in the progression of the disease.[68,73–75] TNF-α and ET-1 have been shown to increase the expression and activity of matrix metalloproteinase -2 (MMP-2) and tissue inhibitors of matrix metalloproteinases-1 and -2 (TIMP-1 and -2). Modulation of extracellular matrix remodeling may further contribute to neuronal cell death.[76]
TNF-α can also act as a downstream mediator of the proapoptotic factors like nerve growth factor (NGF). Pro-NGF has been shown to induce expression of TNF-α in Muller cells. Genetic or biochemical ablation of TNF-α blocks pro-NGF-induced neuronal cell death in retina.[77] Interaction of NGF and pro-NGF with its p75 neurotropin receptor (NTR) mediates the apoptotic signaling. P75(NTR) is upregulated in glaucomatous retina and mediates RGC apoptosis by a paracrine mechanism involving TNF-α.[78]
TNF-α, therefore, not only acts as a direct mediator of RGC apoptosis but can also be an upstream or downstream mediator of other proapoptotic factors. Fig. 2 explains the possible pathways and molecular mediators involved in TNF-α-induced prosurvival and apoptotic signaling.
Figure 2.
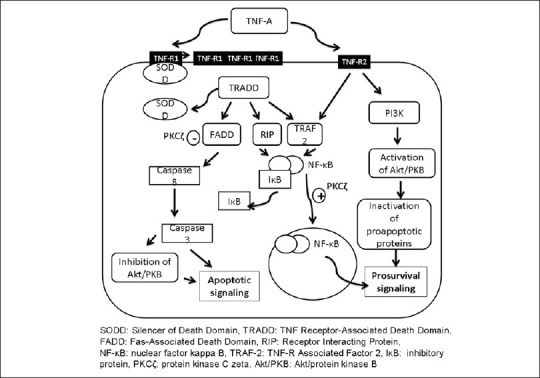
Molecular mechanisms in TNF-induced prosurvival and apoptotic signaling
Therapeutic Interventions Targeting TNF-α
Currently, the search for pharmacological agents as therapeutic interventions in the treatment of glaucoma has greater emphasis on providing direct neuroprotection to RGCs. Simple modulation of risk factors such as elevated IOP is not enough to prevent RGC loss. TNF-α has widely been recognized as an attractive therapeutic target and in several diseases involving TNF-α as a key mediator, anti-TNF therapy has been found to be effective. Agmatine, an aminoguanidine, has been shown to protect RGCs against TNF-α-induced apoptosis.[79] Aminoguianidine also inhibits iNOS expression and prevents RGC loss without affecting IOP.[67] Thus besides directly affecting the TNF-α levels, it can also attenuate TNF-α-induced iNOS expression. Although such effects of agmatine have been demonstrated, its precise mechanism of RGC protection may involve other pathways as it is also known as an inhibitor of the N-methyl-D-aspartate receptor and an agonist for the α2-adrenergic and imidazoline receptors.[80–82]
In spite of the large body of evidence indicating TNF-α as a possible therapeutic target in glaucoma, so far, none of the anti-TNF-α therapy has been shown to be significantly effective in human or in vivo animal models. The utility of anti-TNF-α therapy in glaucoma will depend upon its ability to selectively block excessive TNF-α and TNF-R1 expression without significantly affecting its physiological functions such as local immunity. Moreover, the outcome of anti-TNF-α therapy may be influenced by several other patient-related factors. Introducing anti-TNF-α therapy after significant damage to RGCs has already happened may not be able to rescue remaining RGCs due to involvement of several other cytotoxic substances and highly amplified apoptosis cascade at this stage. Special patient populations such as those with mutant OPTN gene may show higher responsiveness to anti-TNF-α therapy. Designing formulations that can effectively deliver adequate amounts of active substance close to cellular target is another challenge. Therefore, further studies that can incorporate several factors such as those related to the potential drug and those related to patient characteristics are of vital importance in introducing anti-TNF-α therapy as an effective therapeutic intervention in glaucoma.
Conclusions
TNF-α is a pleiotropic cytokine and is involved in a wide range of physiological functions. Several studies have shown that the risk factors of glaucoma activate retinal glial cells which produce cytotoxins like TNF-α. Increased expression of TNF-α and its receptor TNF-R1 causes RGC apoptosis and involves caspases. The utility of the concept of neuroprotection heavily relies on the knowledge of pathophysiological mechanisms involved in the onset and progression of glaucomatous neurodegeneration. Activation of glial cells has been recognized as an early event following exposure to risk factors. Activated glial cells produce cytotoxic substances of which TNF-α has been documented in several studies. Since this cytokine has been shown to have direct as well as an indirect toxicity to RGCs by acting as upstream regulator, it is an attractive target for future antiglaucoma medications. Efforts are also being made to target downstream mechanisms of TNF-α signaling pathway. Anti-TNF-α therapy has been shown to be effective in several diseases that involve TNF-α as a key mediator. However, in spite of large body of evidences to show involvement of TNF-α in the pathogenesis of glaucoma, none of the in vivo studies has so far demonstrated the efficacy of anti-TNF therapy in glaucoma models. Moreover, as the TNF-α has multiple physiological functions, it is important to device the pharmacological means that do not affect its normal homeostatic signaling.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Quigley HA. Number of people with glaucoma worldwide. Br J Ophthalmol. 1996;80:389–93. doi: 10.1136/bjo.80.5.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Resnikoff S, Pascolini D, Etya’ale D, Kocur I, Pararajasegaram R, Pokharel GP, et al. Global data on visual impairment in the year 2002. Bull World Health Organ. 2004;82:844–51. [PMC free article] [PubMed] [Google Scholar]
- 3.Weinreb RN, Khaw PT. Primary open-angle glaucoma. Lancet. 2004;363:1711–20. doi: 10.1016/S0140-6736(04)16257-0. [DOI] [PubMed] [Google Scholar]
- 4.Aggarwal BB. Signaling pathways of the TNF superfamily: A double-edged sword. Nat Rev Immunol. 2003;3:745–56. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 5.Tracey K, Cerami A. Metabolic Response to Cachectin/TNF. In: Boland B, Cullinan J, Kimball C, editors. Annals of the New York Academy of Sciences. New York: New York Academy of Sciences; 1990. pp. 325–30. [Google Scholar]
- 6.Janeway C, Travers P, Walport M, Capra J. New York: Garland Publishers; 1999. Immunobiology : The Immune System in Health and Disease. [Google Scholar]
- 7.Strieter R, Kunkel S, Bone R. Role of Tumor Necrosis Factor-Alpha in Disease States and Inflammation. Crit Care Med. 1993;21:S447–63. doi: 10.1097/00003246-199310001-00006. [DOI] [PubMed] [Google Scholar]
- 8.Rink L, Kirchner H. Recent Progress in the Tumor Necrosis Factor-Alpha Field. Int Arch Allergy Immunol. 1996;111:199–209. doi: 10.1159/000237369. [DOI] [PubMed] [Google Scholar]
- 9.Perry RT, Collins JS, Wiener H, Acton R, Go RC. The role of TNF and its receptors in Alzheimer's disease. Neurobiol Aging. 2001;22:873–83. doi: 10.1016/s0197-4580(01)00291-3. [DOI] [PubMed] [Google Scholar]
- 10.Nagatsu T, Sawada M. Inammatory process in Parkinson's disease: Role for cytokines. Curr Pharm Des. 2005;11:999–1016. doi: 10.2174/1381612053381620. [DOI] [PubMed] [Google Scholar]
- 11.Comabella M, Romera C, Camina M, Perkal H, Moro MA, Leza JC, et al. TNF-alpha converting enzyme (TACE) protein expression in different clinical subtypes of multiple sclerosis. J Neurol. 2006;253:701–6. doi: 10.1007/s00415-006-0090-6. [DOI] [PubMed] [Google Scholar]
- 12.Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–53. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- 13.Hermann GE, Rogers RC, Bresnahan JC, Beattie MS. Tumor necrosis factor-alpha induces cFOS and strongly potentiates glutamate-mediated cell death in the rat spinal cord. Neurobiol Dis. 2001;8:590–9. doi: 10.1006/nbdi.2001.0414. [DOI] [PubMed] [Google Scholar]
- 14.Sawada H, Fukuchi T, Tanaka T, Abe H. Tumor necrosis factor-alpha concentrations in the aqueous humor of patients with glaucoma. Invest Ophthalmol Vis Sci. 2010;51:903–6. doi: 10.1167/iovs.09-4247. [DOI] [PubMed] [Google Scholar]
- 15.Huang P, Qi Y, Xu YS, Liu J, Liao D, Zhang SSM, et al. Serum Cytokine Alteration is Associated with Optic Neuropathy in Human Primary Open Angle Glaucoma. J Glaucoma. 2010;19:324–30. doi: 10.1097/IJG.0b013e3181b4cac7. [DOI] [PubMed] [Google Scholar]
- 16.Madigan MC, Sadun AA, Rao NS, Dugel PU, Tenhula WN, Gill PS. Tumor necrosis factor-alpha (TNF-alpha)-induced optic neuropathy in rabbits. Neurol Res. 1996;18:176–84. doi: 10.1080/01616412.1996.11740399. [DOI] [PubMed] [Google Scholar]
- 17.Kitaoka Y, Kitaoka Y, Kwong JM, Ross-Cisneros FN, Wang J, Tsai RK, et al. TNF-α-Induced Optic Nerve Degeneration and Nuclear Factor-κB p65. Invest Ophthalmol Vis Sci. 2006;47:1448–57. doi: 10.1167/iovs.05-0299. [DOI] [PubMed] [Google Scholar]
- 18.Nakazawa T, Nakazawa C, Matsubara A, Noda K, Hisatomi T, She H, et al. Tumor Necrosis Factor-α Mediates Oligodendrocyte Death and Delayed Retinal Ganglion Cell Loss in a Mouse Model of Glaucoma. J Neurosci. 2006;26:12633–41. doi: 10.1523/JNEUROSCI.2801-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan X, Tezel G, Wax MB, Edward DP. Matrix metalloproteinases and tumor necrosis factor alpha in glaucomatous optic nerve head. Arch Ophthalmol. 2000;118:666–73. doi: 10.1001/archopht.118.5.666. [DOI] [PubMed] [Google Scholar]
- 20.Yuan L, Neufeld AH. Tumor necrosis factor-alpha: A potentially neurodestructive cytokine produced by glia in the human glaucomatous optic nerve head. Glia. 2000;32:42–50. [PubMed] [Google Scholar]
- 21.Yang Z, Quigley HA, Pease ME, Yang Y, Qian J, Valenta D, et al. Changes in Gene Expression in Experimental Glaucoma and Optic Nerve Transection: The Equilibrium between Protective and Detrimental Mechanisms. Invest Ophthalmol Vis Sci. 2007;48:5539–48. doi: 10.1167/iovs.07-0542. [DOI] [PubMed] [Google Scholar]
- 22.Fuchs C, Forster V, Balse E, Sahel JA, Picaud S, Tessier LH. Retinal-Cell-Conditioned Medium Prevents TNF-{alpha}-Induced Apoptosis of Purified Ganglion Cells. Invest Ophthalmol Vis Sci. 2005;46:2983–91. doi: 10.1167/iovs.04-1177. [DOI] [PubMed] [Google Scholar]
- 23.Tezel G, Wax MB. Increased production of tumor necrosis factor-alpha by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J Neurosci. 2000;20:8693–700. doi: 10.1523/JNEUROSCI.20-23-08693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yuan L, Neufeld AH. Activated microglia in the human glaumatous optic nerve head. J Neurosci Res. 2001;64:523–32. doi: 10.1002/jnr.1104. [DOI] [PubMed] [Google Scholar]
- 25.Levkovitch VH, Dardik R, Vander S, Nisgav Y, Kalev-Landoy M, Melamed S. Experimental glaucoma and optic nerve transaction induce simultaneous upregulation of pro apoptotic and prosurvival genes. Invest Ophthalmol Vis Sci. 2006;47:2491–7. doi: 10.1167/iovs.05-0996. [DOI] [PubMed] [Google Scholar]
- 26.Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, et al. IAPs block apoptotic events induced by caspase-8 and cytochrome C by direct inhibition of distinct caspases. EMBO J. 1998;17:2215–23. doi: 10.1093/emboj/17.8.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deveraux QL, Stennicke HR, Salvesen GS, Reed JC. Endogenous inhibitors of caspases. J Clin Immunol. 1999;19:388–98. doi: 10.1023/a:1020502800208. [DOI] [PubMed] [Google Scholar]
- 28.Perrelet D, Ferri A, MacKenzie AE, Smith GM, Korneluk RG, Liston P, et al. IAP family proteins delay motoneuron cell death in vivo. Eur J Neurosci. 2000;12:2059–67. doi: 10.1046/j.1460-9568.2000.00098.x. [DOI] [PubMed] [Google Scholar]
- 29.McPhail LT, Vanderluit JL, McBride CB, Oschipok LW, Crocker SJ, Xu D, et al. Endogenous expression of inhibitor of apoptosis proteins in facial motoneurons of neonatal and adult rats following axotomy. Neurosci. 2003;117:567–75. doi: 10.1016/s0306-4522(02)00742-x. [DOI] [PubMed] [Google Scholar]
- 30.Bai Y, Shi Z, Zhuo Y, Liu J, Malakhov A, Ko E, et al. In glaucoma the upregulated truncated TrkC.T1 receptor isoform in glia causes increased TNF-alpha production, leading to retinal ganglion cell death. Invest Ophthalmol Vis Sci. 2010;51:6639–51. doi: 10.1167/iovs.10-5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tezel G, Li LY, Patil RV, Wax MB. TNF-α and TNF-α Receptor-1 in the Retina of Normal and Glaucomatous Eyes. Invest Ophthalmol Vis Sci. 2001;42:1787–94. [PubMed] [Google Scholar]
- 32.Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: Opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci. 2002;22:1–7. doi: 10.1523/JNEUROSCI.22-07-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–9. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 34.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 35.Marchetti L, Klein M, Schlett KP, Zenmaier K, Eisel UL. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation.Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J Biol Chem. 2004;279:32869–81. doi: 10.1074/jbc.M311766200. [DOI] [PubMed] [Google Scholar]
- 36.Lawlor MA, Alessi DR. PKB/Akt: A key mediator of cell proliferation, survival and insulin responses? J Cell Sci. 2001;114:2903–10. doi: 10.1242/jcs.114.16.2903. [DOI] [PubMed] [Google Scholar]
- 37.Rothe M, Sharma V, Dixit VM, Goeddel DV. TRAF2-mediated activation of NF-kappa B by TNF receptor 2 and CD40. Science. 1995;269:1424–7. doi: 10.1126/science.7544915. [DOI] [PubMed] [Google Scholar]
- 38.Jiang Y, Woronicz JD, Liu W, Goeddel DV. Prevention of constitutive TNF receptor 1 signaling by silencer of death domains. Science. 1999;283:543–6. doi: 10.1126/science.283.5401.543. [DOI] [PubMed] [Google Scholar]
- 39.Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 40.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD–TRAF2 and TRADD–FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 41.Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1996;85:803–15. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 42.Ting AT, Pimentel-Muinos FX, Seed B. RIP mediates tumor necrosis factor receptor 1 activation of NF-kappaB but not Fas/APO-1-initiated apoptosis. EMBO J. 1996;15:6189–96. [PMC free article] [PubMed] [Google Scholar]
- 43.Barnes PJ, Karin M. Nuclear factor-κB: A pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–71. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 44.Karin M, Liu ZG, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–6. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 45.Beg AA, Baltimore D. An Essential Role for NF-κB in Preventing TNF-α-Induced Cell Death. Science. 1996;274:782–4. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 46.Crisanti P, Leon A, Lim DM, Omri B. Aspirin prevention of NMDA-induced neuronal death by direct protein kinase Cæ inhibition. J Neurochem. 2005;93:1587–93. doi: 10.1111/j.1471-4159.2005.03157.x. [DOI] [PubMed] [Google Scholar]
- 47.Diaz-Meco MT, Municio MM, Sanchez P, Lozano J, Moscat J. Lambda-interacting protein, a novel protein that specifically interacts with the zinc finger domain of the atypical protein kinase C isotype lambda/iota and stimulates its kinase activity in vitro and in vivo. Mol Cell Biol. 1996;16:105–14. doi: 10.1128/mcb.16.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang H, Baudouin C, Behar-Cohen F, Cristani P, Omri B. Protein kinase C-zeta mediates retinal degeneration in response to TNF. J Neuroimmunol. 2007;183:104–10. doi: 10.1016/j.jneuroim.2006.11.028. [DOI] [PubMed] [Google Scholar]
- 49.Yeh WC, de la Pompa L, Elia AJ, Shahinian A, Ng M, Mitchell K, et al. FADD: Essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 1998;279:1954–8. doi: 10.1126/science.279.5358.1954. [DOI] [PubMed] [Google Scholar]
- 50.Rath PC, Aggarwal BB. TNF-induced signaling in apoptosis. J Clin Immunol. 1999;19:350–64. doi: 10.1023/a:1020546615229. [DOI] [PubMed] [Google Scholar]
- 51.Smith L, Smith JB. Lack of constitutive activity of the free kinase domain of protein kinase C zeta. Dependence on transphosphorylation of the activation loop. J Biol Chem. 2002;277:45866–73. doi: 10.1074/jbc.M206420200. [DOI] [PubMed] [Google Scholar]
- 52.Xiaohong Z, Feng L, Li K, Hiroshi T, Chao L, Wei C. Involvement of Inflammation, Degradation, and Apoptosis in a Mouse Model of Glaucoma. J Biol Chem. 2005;280:31240–48. doi: 10.1074/jbc.M502641200. [DOI] [PubMed] [Google Scholar]
- 53.Tezel G, Yang X. Caspase-independent component of retinal ganglion cell death, in vitro. Invest Ophthalmol Vis Sci. 2004;45:4049–59. doi: 10.1167/iovs.04-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bachelder RE, Wendt MA, Fujita N, Tsuruo T, Mercuriob AM. The Cleavage of Akt/Protein Kinase B by Death Receptor Signaling Is an Important Event in Detachment-induced Apoptosis. J Biol Chem. 2001;276:34702–7. doi: 10.1074/jbc.M102806200. [DOI] [PubMed] [Google Scholar]
- 55.Rezaie T, Child A, Hitchings R, Brice G, Miller L, Coca-Prados M, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002;295:1077–9. doi: 10.1126/science.1066901. [DOI] [PubMed] [Google Scholar]
- 56.Li Y, Kang J, Horwitz MS. Interaction of an adenovirus E3 14.7-kilodalton protein with a novel tumor necrosis factor alpha-inducible cellular protein containing leucine zipper domains. Mol Cell Biol. 1998;18:1601–10. doi: 10.1128/mcb.18.3.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu G, Wu CJ, Zhao Y, Ashwell JD. Optineurin negatively regulates TNFalpha- induced NF-kappaB activation by competing with NEMO for ubiquitinated RIP. Curr Biol. 2007;17:1438–43. doi: 10.1016/j.cub.2007.07.041. [DOI] [PubMed] [Google Scholar]
- 58.Chalasani ML, Gupta RV, Agarwal N, Balasubramanian D, Swarup G. A Glaucoma-associated mutant of optineurin selectively induces death of retinal ganglion cells which is inhibited by antioxidants. Invest Ophthalmol Vis Sci. 2007;48:1607–14. doi: 10.1167/iovs.06-0834. [DOI] [PubMed] [Google Scholar]
- 59.Fan BJ, Liu K, Wang DY, Tham CC, Tam PO, Lam DS, et al. Association of polymorphisms of tumor necrosis factor and tumor protein p53 with primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2010;51:4110–6. doi: 10.1167/iovs.09-4974. [DOI] [PubMed] [Google Scholar]
- 60.Khan MI, Micheal S, Rana N, Akhtar F, den Hollander AI, Ahmed A, et al. Association of tumor necrosis factor alpha gene polymorphism G-308A with pseudoexfoliative glaucoma in the Pakistani population. Mol Vis. 2009;15:2861–7. [PMC free article] [PubMed] [Google Scholar]
- 61.Razeghinejad MR, Rahat F, Kamali-Sarvestani E. Association of TNFA -308 G/A and TNFR1 +36 A/G gene polymorphisms with glaucoma. Ophthalmic Res. 2009;42:118–24. doi: 10.1159/000226108. [DOI] [PubMed] [Google Scholar]
- 62.Tekeli O, Turacli ME, Egin Y, Akar N, Elhan AH. Tumor necrosis factor alpha-308 gene polymorphism and pseudoexfoliation glaucoma. Mol Vis. 2008;14:1815–8. [PMC free article] [PubMed] [Google Scholar]
- 63.Mossböck G, Renner W, El-Shabrawi Y, Faschinger C, Schmut O, Wedrich A, et al. TNF-α –308 G>A and –238 G>A polymorphisms are not major risk factors in Caucasian patients with exfoliation glaucoma. Mol Vis. 2009;15:518–22. [PMC free article] [PubMed] [Google Scholar]
- 64.Diem R, Meyer R, Weishaupt JH, Bahr M. Reduction of potassium currents and phosphatidylinositol 3-kinase-dependent AKT phosphorylation by tumor necrosis factor-(alpha) rescues axotomized retinal ganglion cells from retrograde cell death in vivo. J Neurosci. 2001;21:2058–66. doi: 10.1523/JNEUROSCI.21-06-02058.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shafer RA, Murphy S. Activated astrocytes induce nitric oxide synthase-2 in cerebral endothelium via tumor necrosis factor α. Glia. 1998;21:370–9. doi: 10.1002/(sici)1098-1136(199712)21:4<370::aid-glia4>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 66.Goureau , O , Amiot F, Dautry F, Courtois Y. Control of nitric oxide production by endogenous TNF-alpha in mouse retinal pigmented epithelial and Muller glial cells. Biochem Biophys Res Commun. 1997;240:132–5. doi: 10.1006/bbrc.1997.7581. [DOI] [PubMed] [Google Scholar]
- 67.Toda N, Toda MN. Nitric oxide: Ocular blood flow, glaucoma, and diabetic retinopathy. Prog Retin Eye Res. 2007;26:205–38. doi: 10.1016/j.preteyeres.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 68.Neufeld AH. Nitric oxide: A potential mediator of retinal ganglion damage in glaucoma. Surv Ophthalmol. 1999;43:S129–35. doi: 10.1016/s0039-6257(99)00010-7. [DOI] [PubMed] [Google Scholar]
- 69.Chae IH, Park KW, Kim HS, Oh BH. Nitric oxide-induced apoptosis is mediated by Bax/Bcl-2 gene expression, transition of cytochrome c, and activation of caspase-3 in rat vascular smooth muscle cells. Clin Chim Acta. 2004;341:83–91. doi: 10.1016/j.cccn.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 70.Narayan S, Prasanna G, Krishnamoorthy RR, Zhang X, Yorio T. Endothelin-1 Synthesis and Secretion in Human Retinal Pigment Epithelial Cells (ARPE-19): Differential Regulation by Cholinergics and TNF-α. Invest Ophthalmol Vis Sci. 2003;44:4885–94. doi: 10.1167/iovs.03-0387. [DOI] [PubMed] [Google Scholar]
- 71.Desai D, Shaoqing H, Thomas Y, Krishnamoorthy RR, Ganesh P. Hypoxia augments TNF-alpha-mediated endothelin-1 release and cell proliferation in human optic nerve head astrocytes. Biochem Biophys Res Commun. 2004;4(318):642–8. doi: 10.1016/j.bbrc.2004.04.073. [DOI] [PubMed] [Google Scholar]
- 72.Prasanna G, Krishnamoorthy R, Hulet C, Zhang H, Zhang X, Yorio T. Endothelin-1 induces nitric oxide synthase-2 expression in human non-pigmented ciliary epithelial cells. Exp Eye Res. 2000;71:535–9. doi: 10.1006/exer.2000.0908. [DOI] [PubMed] [Google Scholar]
- 73.Neufeld AH, Hernandez MR, Gonzalez M. Nitric oxide synthase in the human glaucomatous optic nerve head. Arch Ophthalmol. 1997;115:497–503. doi: 10.1001/archopht.1997.01100150499009. [DOI] [PubMed] [Google Scholar]
- 74.Noske W, Hensen J, Wiederholt M. Endothelin-like immunoreactivity in aqueous humor of primary open-angle glaucoma and cataract patients. Graefes Arch Clin Exp Ophthalmol. 1997;235:551–2. doi: 10.1007/BF00947082. [DOI] [PubMed] [Google Scholar]
- 75.Sugiyama T, Moriya S, Oku H, Azuma I. Association of endothelin-1 with normal tension glaucoma: Clinical and fundamental studies. Surv Ophthalmol. 1995;39:S49–56. doi: 10.1016/s0039-6257(05)80073-6. [DOI] [PubMed] [Google Scholar]
- 76.Shaoqing H, Ganesh P, Thomas Y. Endothelin-1-mediated signaling in the expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in astrocytes. Invest Ophthalmol Vis Sci. 2007;48:3737–45. doi: 10.1167/iovs.06-1138. [DOI] [PubMed] [Google Scholar]
- 77.Frédéric LJ, Mathieu JB, Olivier DB, David S, Carlos RM, Adriana DP, et al. ProNGF induces TNFα-dependent death of retinal ganglion cells through a p75NTR non-cell autonomous signaling pathway. Proc Natl Acad Sci U S A. 2010;107:3817–22. doi: 10.1073/pnas.0909276107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bai Y, Dergham P, Nedev H, Xu J, Galan A, Rivera JC, et al. Chronic and acute models of retinal neurodegeneration TrkA activity are neuroprotective whereas p75NTR activity is neurotoxic through a paracrine mechanism. J Biol Chem. 2010;285:39392–400. doi: 10.1074/jbc.M110.147801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hong S, Kim CY, Lee JE, Seong GJ. Agmatine protects cultured retinal ganglion cells from tumor necrosis factor-alpha-induced apoptosis. Life Sci. 2009;84:28–32. doi: 10.1016/j.lfs.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 80.Wang WP, Iyo AH, Miguel-Hidalgo J, Regunathan S, Zhu MY. Agmatine protects against cell damage induced by NMDA and glutamate in cultured hippocampal neurons. Brain Res. 2006;1084:210–6. doi: 10.1016/j.brainres.2006.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li G, Regunathan S, Barrow CJ, Eshraghi J, Cooper R, Reis DJ. Agmatine: An endogenous clonidine-displacing substance in the brain. Science. 1994;263:966–9. doi: 10.1126/science.7906055. [DOI] [PubMed] [Google Scholar]
- 82.Reis DJ, Regunathan S. Agmatine: An endogenous ligand at imidazoline receptors is a novel neurotransmitter. Ann N Y Acad Sci. 1999;881:65–80. doi: 10.1111/j.1749-6632.1999.tb09343.x. [DOI] [PubMed] [Google Scholar]