

Background: The structure of fibrils formed in Huntington disease remains unknown.
Results: Local structure and domain organization of huntingtin exon 1 fibrils were determined by EPR spectroscopy.
Conclusion: In contrast to the C terminus, the N terminus, and polyglutamine core regions become closely packed, without forming a parallel, in-register structure.
Significance:The results provide structural constraints and insight into how adjacent domains affect polyglutamine structure.
Keywords: Amyloid, Electron Paramagnetic Resonance (EPR), Huntington Disease, Polyglutamine, Structural Biology
Abstract
Misfolding and aggregation of huntingtin is one of the hallmarks of Huntington disease, but the overall structure of these aggregates and the mechanisms by which huntingtin misfolds remain poorly understood. Here we used site-directed spin labeling and electron paramagnetic resonance (EPR) spectroscopy to study the structural features of huntingtin exon 1 (HDx1) containing 46 glutamine residues in its polyglutamine (polyQ) region. Despite some residual structuring in the N terminus, we find that soluble HDx1 is highly dynamic. Upon aggregation, the polyQ domain becomes strongly immobilized indicating significant tertiary or quaternary packing interactions. Analysis of spin-spin interactions does not show the close contact between same residues that is characteristic of the parallel, in-register structure commonly found in amyloids. Nevertheless, the same residues are still within 20 Å of each other, suggesting that polyQ domains from different molecules come into proximity in the fibrils. The N terminus has previously been found to take up a helical structure in fibrils. We find that this domain not only becomes structured, but that it also engages in tertiary or quaternary packing interactions. The existence of spin-spin interactions in this region suggests that such contacts could be made between N-terminal domains from different molecules. In contrast, the C-terminal domain is dynamic, contains polyproline II structure, and lacks pronounced packing interactions. This region must be facing away from the core of the fibrils. Collectively, these data provide new constraints for building structural models of HDx1 fibrils.
Introduction
Huntington disease (HD)4 is a progressive, fatal neurodegenerative disorder caused by a polyglutamine (polyQ) expansion in the first exon of the huntingtin (Htt) (1). Like other polyQ diseases (2), there is a threshold of about 40 Gln beyond which patients get the disease, and the age of onset of the disease is inversely correlated with the length of the expansion (3). HD is associated with neurodegeneration, especially of the caudate nucleus of the striatum (4), and the severity of neurodegeneration correlates with polyQ length (5). Huntingtin exon 1 (HDx1), with a polyQ expansion beyond a threshold of about 40 Gln, is sufficient to cause disease in mice (6).
A hallmark of HD in mice and humans is the formation of intracellular inclusions. In patients, these inclusions consist largely of N-terminal fragments of mutant Htt that are often ubiquitinated (7, 8). Aggregates purified from HD patient brains bind Congo Red with green birefringence, indicating that they have an amyloid-like structure (9). In vitro, mutant HDx1 spontaneously forms fibrils that similarly bind Congo Red (9). Fourier transform infrared spectrometry demonstrated that these fibrils have a β-sheet signature similar to amyloid fibrils (10). Furthermore, HDx1 fibrils have an x-ray diffraction pattern consistent with the cross-β structure characteristic of amyloid fibrils (11, 12). In the cross-β structure, β-strands separated by 4.8 Å run normal to the fibril axis and form a sheet that is parallel to the fibril axis.
HDx1 consists of three domains, an N-terminal 17 amino acids, a polyQ region of variable length, and a C terminus that is rich in prolines. There are several lines of evidence indicating that the polyQ region of HDx1 forms the amyloid core. For example, studies of synthetic polyQ peptides indicate that aggregation propensity increases with polyQ length (13), and that amyloid-like aggregates are formed (14). The polyQ flanking regions in HDx1 further modulate its aggregation and toxicity (15–20). Interestingly, a recent solid-state NMR study using a fragment of HDx1 indicates that the N terminus can adopt an α-helical structure in the fibril (21). The proline-rich C terminus is thought to retard fibril formation by preventing the transition of the polyQ region to an amyloid-like structure (22). Aside from the assignment of helical secondary structure to the N terminus and a cross-β structure to the polyQ region, little is known about the molecular organization of HDx1 fibrils. Moreover, previous studies employed the smaller fragments of HDx1 rather than the entire HDx1 protein. As a consequence, the structural features are even less understood in the context of the entire HDx1 protein.
Here we present electron paramagnetic resonance (EPR) results characterizing the domain organization of recombinantly made, full-length HDx1 fibrils. We show that although much of the polyQ region is immobilized in the fibril, it does not have the parallel, in-register signature characteristic of many other disease-causing amyloid structures. The C terminus exhibits the greatest mobility, takes up polyproline II structure, and loosens the packing of adjacent polyQ residues. Finally, the N terminus is ordered and, in contrast to the C terminus, makes tertiary or quaternary contact with other N termini, leading to stabilization of the fibril core.
EXPERIMENTAL PROCEDURES
Protein Expression, Labeling, and Purification
Using site-directed mutagenesis, both thioredoxin cysteines were mutated to serines in pET32a-HD46Q to create a parent construct for making cysteine mutants of Htt. Cysteine mutations were introduced by site-directed mutagenesis into the parent construct, which expresses a thioredoxin fused to the N terminus of Htt that has 46 glutamines and a C-terminal His tag (NTRX-Q46). Overnight cultures of BL21(DE3) were diluted 50-fold into LB medium and grown at 37 °C to 0.6 A600. Isopropyl 1-thio-β-d-galactopyranoside was added to 1 mm, and the temperature was reduced to 30 °C for 4 h. Pellets were collected by centrifugation at 3500 × g, resuspended in 20 mm Tris-HCl, pH 8.0, 300 mm NaCl, and 10 mm imidazole containing 1× CelLytic B Cell Lysis reagent (Sigma-Aldrich), and incubated for 20 min at room temperature on a rocker. Lysates were clarified by centrifugation at 21,000 × g for 10 min and incubated with nickel-nitrilotriacetic acid-agarose beads (Qiagen) for 1 h at 4 °C on a rocker. Beads were decanted into an Econo-Pac chromatography column (Bio-Rad) and washed with several column volumes of 20 mm Tris-HCl, pH 8.0, 300 mm NaCl, 20 mm imidazole. Purified proteins were eluted with 20 mm Tris-HCl, pH 8.0, 300 mm NaCl, 250 mm imidazole. Volumes were reduced to ∼250 μl using Amicon Ultra-4 or Ultra-15 3000 MWCO centrifugal filters (Millipore), after which the protein was incubated with an equal volume of immobilized TCEP disulfide reducing gel (Pierce) for 1 h at room temperature. Following reduction, the disulfides were spin-labeled by incubation with a 5–15-fold excess of MTSL spin label (Toronto Research Chemicals, Inc., North York, ON, Canada) for 1 h at room temperature. Labeled protein was FPLC-purified on a Superdex-75 gel-filtration column (GE Healthcare) using phosphate-buffered saline (137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 1.76 mm NaH2PO4), adjusted to pH 6.8 with phosphoric acid, containing 1 mm EDTA. For dilution spectra, unlabeled fusion protein without cysteines was purified in the same manner, except no MTSL was added. Unlabeled fusion protein without cysteines used to make seeds was purified similarly, except during FPLC, 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA were used.
Preparation of Seeds
Unlabeled fusion protein without cysteines in 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA was diluted to 5 μm (225 μg/ml). To cleave the thioredoxin tag and initiate fibril formation, EKMax (Invitrogen) was added to 1 unit/10 μg of NTRX-Q46. The reaction was incubated without agitation at 4 °C for 3 days. Fibril formation appeared to be complete by electron microscopy, as judged by the absence globular species. Fibrils were collected by ultracentrifugation at 150,000 × g for 20 min and resuspended in one tenth of the original volume. To fragment the fibrils, this suspension was then sonicated on maximum power for 10 min at 30-s intervals. Sonicated seeds were stored at −80 °C.
Fibril Formation
All Htt protein concentrations were measured by BCA assay and adjusted to 225 μg/ml (5 μm) for fibril formation. For mobility measurements, reactions were set up using 10% labeled mutant protein and 90% unlabeled protein without cysteines. To measure spin-spin interactions, reactions containing 100% labeled protein were used. To minimize batch-to-batch variation, all reactions were seeded with 10% preaggregated seed fibrils, and fibril formation was initiated by the addition of 1 unit of EKMax/10 μg of protein. Reactions were incubated overnight at 4 °C without agitation. Fibrils were collected by ultracentrifugation at 150,000 × g for 20 min and resuspended in 7 μl of PBS, pH 6.8, with 1 mm EDTA.
Electron Microscopy
Prior to ultracentrifugation, 6 μl of fibrils was removed and adsorbed onto copper mesh electron microscopy grids (Electron Microscopy Sciences, Hatfield, PA) for 2 min. These grids were negatively stained with 2% uranyl acetate for 2 min. Subsequently, the grids were examined with a JEOL JEM-1400 electron microscope (JEOL, Peabody, MA) at 100 kV and photographed using a Gatan digital camera.
Continuous Wave EPR Spectra
After ultracentrifugation, the resuspended fibrils were loaded into quartz capillaries (0.6-mm inner diameter × 0.84-mm outer diameter, VitroCom, Mt. Lakes, NJ), and EPR spectra were recorded on an X-band Bruker EMX spectrometer (Bruker Biospin Corporation) at room temperature. The scan width was 150 gauss using a HS cavity at an incident microwave power of 12.60 milliwatts. EPR spectra of fusion proteins were also collected. The spectra were normalized by double integration.
Circular Dichroism (CD)
CD measurements of monomeric fusion protein were performed at a concentration of 5 μm in buffer (10 mm phosphate with no salt, pH 7.4). CD spectra of fibrils were obtained at 30 μm in the same phosphate buffer. The CD spectra were obtained using a Jasco 815 spectropolarimeter (Jasco, Inc., Easton, MD). Measurements were taken every 0.5 nm at a scan rate of 50 nm/min with average time of 1 s. Approximately 20–30 scans were averaged between 190 and 260 nm, and buffer backgrounds were subtracted. Spectra were analyzed using the DichroWeb suite of programs (23) including CDSSTR (24–26), SELCON3 (27), K2D (28, 29), and CONTINLL (30, 31).
Temperature dependence of the CD spectra was measured using a Jasco PFD-425s temperature controller. HDx1 fibrils were incubated at a given target temperature for at least 20 min prior to recording the CD spectra. All other experimental details remained the same as described above. In the temperature range between 0 and 50 ºC, spectral changes were 95–100% reversible. The difference spectrum for 50 ºC and 0 ºC was smoothed using the Savitzky-Golay method available on the Jasco spectrometer.
RESULTS
To investigate the structural changes that occur in 46Q-containing HDx1 upon aggregation, we generated 17 different derivatives (Fig. 1A) that contained the spin-labeled side chain R1 (Fig. 1B). We examined the derivatives by EPR in their soluble and fibrillar forms. Due to its better solubility, the HDx1-thioredoxin fusion protein was used to study the soluble form whereas fibrils were formed from HDx1 in which the thioredoxin partner had been cleaved off. In agreement with previous studies (10, 32, 33), we find that HDx1 forms predominantly relatively short fibrils (100–500-nm length, 10–12-nm diameter) that have a tendency to associate laterally and form bundles (Fig. 1C).
FIGURE 1.

Spin labeling and fibril formation of HDx1 with 46Q. A, spin labels introduced at the indicated sites in different HDx1 domains. Colors highlight the indicated domains. B, structure of spin-labeled side chain R1. C, representative negative stain electron micrograph of spin-labeled HDx1, labeled at position 64. Scale bar is 100 nm.
Soluble HDx1 as a Fusion Protein Is Highly Dynamic
To ensure that the native protein structure was not perturbed by the addition of the spin label, we compared CD spectra of the soluble fusion protein with and without label and found that they did not differ significantly (supplemental Fig. S1).
As illustrated in Fig. 2, the EPR spectra of all spin-labeled, soluble HDx1-thioredoxin fusion proteins consist of three sharp and narrowly spaced lines typical of highly mobile sites. Given the relatively large size of the fusion protein (28 kDa), this high mobility cannot solely be caused by rapid tumbling of the protein. Rather, the data indicate a highly dynamic structure consistent with a previous study using CD and NMR that found unpolymerized HDx1-thioredoxin fusion protein to be largely disordered (34). Although all of our spectra indicate that soluble HDx1 is highly dynamic, there are subtle differences in the sharpness (i.e. spin label motion) of the spectra. The widths of the central resonance line for sites within the first 30 amino acids are consistently larger (1.8–2.0 gauss) than those of subsequent sites (1.6–1.75 gauss). These data indicate that the first 30 amino acids are less mobile than the latter amino acids. Tethering of the HDx1 to the fusion partner could cause some of this reduction in mobility. Inasmuch as tethering typically affects the mobility of just the first 5–10 amino acids (35, 36), residual secondary is likely to be present. This notion is consistent with computational work suggesting that the N terminus of HDx1 is likely to be partially ordered (17).
FIGURE 2.

EPR spectra of the thioredoxin-HDx1 fusion protein indicate a highly dynamic structure prior to aggregation. X-band EPR spectra were collected at 150-gauss scan width at room temperature. Numbers indicate locations of the spin label in the amino acid sequences of HDx1 containing 46 Gln. For better visualization, all spectra are shown at the same amplitude.
HDx1 Fibril Formation and CD Analysis
To obtain uniform fibrils for the different spin-labeled derivatives, we seeded each fibril reaction with the same batch of native HDx1 seeds. Such seeding has previously been shown to faithfully propagate the biophysical properties of HDx1 through multiple generations (37). Spin-labeled HDx1 grew readily from native fibril seeds, indicating that the derivatives took up native structure. In addition, CD spectra of fibrils formed from several derivatives were compared and found to be highly similar (supplemental Fig. S2).
The CD spectra were fitted using the DichroWeb (23) suite of fitting programs. All of these programs indicated that HDx1 fibrils contain ∼35–45% β-sheet structure and ∼5–10% α-helical structure. These data are consistent with previous findings which suggest the presence of β-sheet structure in the polyQ region and α-helical structure in the N terminus (21). 30–50% of the structure was fit to be disordered. It should be noted, however, that these programs do not distinguish between polyproline II helices and disordered structure, which result in similar CD spectra (see below).
The PolyQ Domain Becomes Highly Ordered in Fibrils, but It Does Not Have a Parallel, in-Register Structure
Upon fibril formation, the most significant changes in the EPR spectra are within the polyQ domain (Fig. 3, red spectra). All sites in this domain display significant line broadening and increased separation of the outer peaks, indicating that the polyQ domain becomes strongly immobilized upon aggregation. Such immobilization is typical for core regions of all amyloid fibrils that have been investigated by EPR (38). What is different about the spectra for HDx1 fibrils, however, is that no site in any region of the protein displays the highly characteristic, single-line EPR spectra that have been seen in α-synuclein, tau, β2m, human prion protein, and IAPP (38–41). Single-line EPR spectra are the consequence of the spin exchange that occurs in a parallel, in-register structure in which four or more spin labels stack on top of another and come into physical contact (38, 39). Thus, unlike other amyloid fibrils, the HDx1 fibrils formed in this study do not have a parallel, in-register structure in which each protein takes up a new layer along the fibril axis, and the same sites come into close contact with one another.
FIGURE 3.

EPR spectra of spin-labeled HDx1 fibrils. 100% (red) and 10% (blue) labeled fibrils for each site are overlaid. All spectra of 10% labeled fibrils are shown at the same amplitude with respect to each other. To illustrate the effects of spin-spin interaction, the corresponding pairs of spectra at 10 and 100% labeling are shown normalized to the same number of spins. The arrows denote a mobile component in the last residue of the polyQ stretch as well as the first residue in the polyproline region. X-band EPR spectra were recorded at 150-gauss scan width at room temperature. The numbers indicate the locations of the spin label in the amino acid sequences of HDx1 containing 46 Gln.
To test whether any spin-spin interaction could be detected between the same labeled residues, we performed a dilution experiment in which fibrils were grown from a mixture of 10% labeled and 90% unlabeled protein. By reducing the density of spin-labeled proteins, any effects of spin-spin interaction become strongly diminished. The spectra of diluted HDx1 fibrils are qualitatively similar to those of the fully labeled forms, but spin dilution causes an increase in the spectral amplitude at most sites, including the polyQ domain (Fig. 3, blue spectra). Such spectral changes only occur when labels come within 20 Å of each other (42). Thus, although sites in the polyQ are not parallel and in-register, they are still within 20 Å of each other in the fibril. This might be expected if the polyQ domains of different molecules interact with each other. These data also indicate the spin-labeled and native, unlabeled HDx1 can co-mix in fibrils and take up equivalent structures.
HDx1 C Terminus Lies Outside the Fibril Core
All of our C-terminal sites have three, easily discernible, relatively sharp resonance lines upon fibril formation (Fig. 3). Hence, these sites have elevated mobility and do not engage in significant tertiary or quaternary contacts. Despite the overall high mobility, there is a notable increase in mobility within this domain toward the extreme C terminus. These data indicate that the C terminus is anchored at the polyQ domain, from where it becomes increasingly dynamic. In agreement with this notion, the spin-spin interactions become weaker toward the C terminus and become barely detectable at residues 76 and 111 (Fig. 3). Thus, especially in the most C terminus, same residues are mostly separated by more than 20 Å.
The transition from a highly immobilized polyQ domain to the more dynamic C-terminal domain can also be seen in the spectra of 63R1 and 64R1, which are at the border between the polyQ and C-terminal domains. Both spectra exhibit a mobile (see arrows in Fig. 3) as well as a strongly immobilized component, suggesting that they have structural features of both domains. Moreover, the C terminus seems to be capable of locally loosening up the packing in the adjacent polyQ domain.
Polyproline II Structure in HDx1 Fibrils
Despite the high overall mobility, the EPR spectra of C-terminal sites reveal a gradual increase in mobility spanning a stretch of about 40 amino acids (Fig. 3, also see below). This behavior could be due to residual polyproline II structure. The highly proline-rich C terminus has been predicted (21, 43, 44), but not shown directly, to take up such a structure in HDx1 fibrils.
To test whether polyproline II structure might be present in fibrils, we recorded CD spectra at different temperatures. It is well established that polyproline II structures become more ordered upon cooling. This ordering is typically very gradual and occurs over a broad temperature range between 100 and 0 ºC (45, 46). Although heating HDx1 fibrils to higher temperatures caused aggregation, we found that it was possible to record CD spectra of HDx1 fibrils between 0 and 50 ºC without causing irreversible changes to the sample. The spectra obtained at 0 and 50 ºC are given in Fig. 4A. The corresponding difference spectrum (Fig. 4B) has the typical features of a polyproline II structure including a minimum at 206 nm and a maximum near 224 nm (47). As commonly observed for polyproline II (45, 47), both spectral features increased with decreasing temperature. It is important to note that the opposite trend would have been observed for the unfolding of α-helical or β-sheet structures into a random coil. To test whether the formation of polyproline II exhibits the typical gradual temperature transition (45, 46), we systematically varied the temperature and monitored the mean residue ellipticity at 206 nm. As expected, the amount of polyproline II structure changed almost linearly without exhibiting a defined folding/unfolding transition (Fig. 4C).
FIGURE 4.
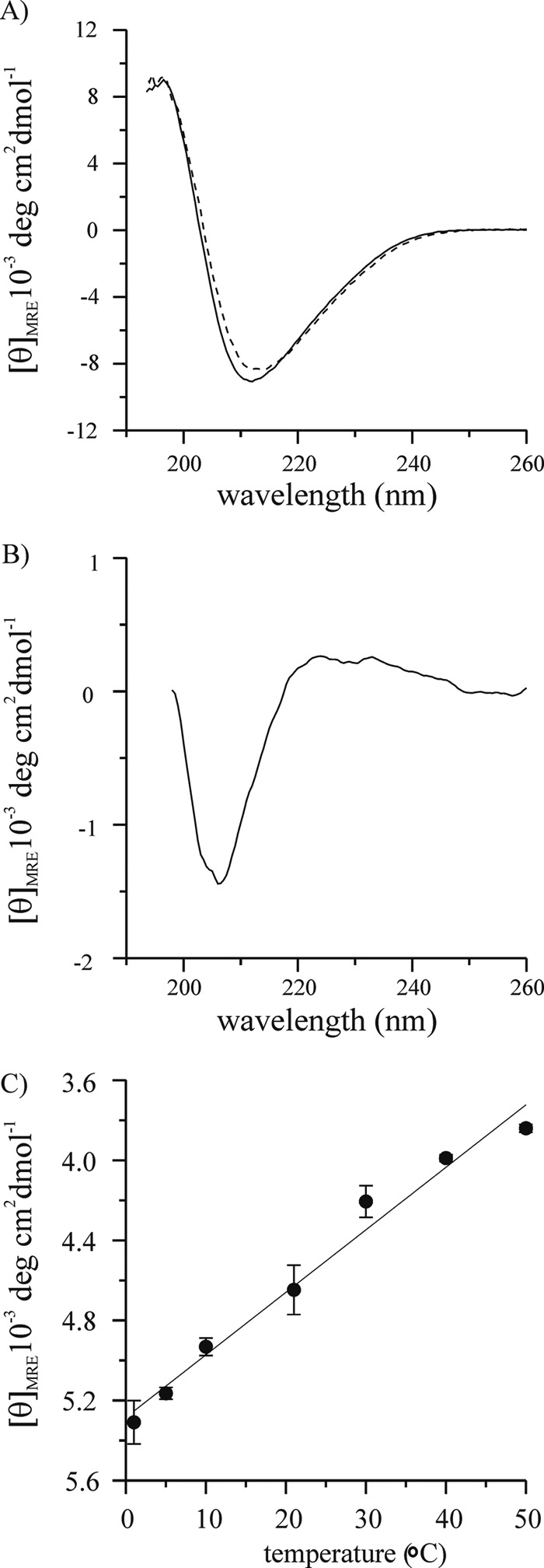
Temperature dependence of HDx1 fibrils indicates polyproline II structure. A, CD spectra of HDx1 fibrils (30 μm) at 0 °C (solid line) and 50 °C (dashed line) shown using mean residue ellipticity. B, difference spectrum for the spectra of A. C, mean residue ellipticity values at 206 nm for CD spectra of HDx1 obtained at different temperatures.
It is generally difficult to quantify the amount of polyproline II structure, as the reference spectra for the fully folded and unfolded states cannot readily be obtained (45). This is particularly difficult in the present study, as we presumably cannot fully denature the polyproline II structure at 50 ºC. Thus, only rather approximate estimates of polyproline II structure can be given in the present case. Prior studies on polyproline model peptides found that the mean residue ellipticity for the 224 nm peak changed by about 1000 ºC cm2 dmol−1 as the temperature was varied between 0 and 50 ºC (45, 46). Here, we observe about a quarter of that value suggesting that about a quarter (∼20–30 residues) in HDx1 fibrils might be able to take up polyproline II structure.
The N Terminus Becomes Immobilized in the Fibrils
A recent solid-state NMR study found that the HDx1 N terminus in fibrils takes up an α-helical structure (21). In agreement with this predicted structural ordering, the EPR spectra of N-terminal sites exhibit significant immobilization (Fig. 3). However, the immobilization exceeds that typically observed on the surface of an α-helix, indicating the presence of tertiary and quaternary contacts on both hydrophobic (residues 3 and 11) and hydrophilic (residues 5 and 9) faces. There are also significant spin-spin interactions for each of these sites, suggesting that the N termini from different molecules come into proximity.
DISCUSSION
In the present study we define the domain organization of mutant HDx1 in soluble and fibrillar form in terms of mobility and intermolecular proximity. To summarize the mobility information contained in the data, we used the semiquantitative mobility parameter, the inverse of the width of the central resonance line, ΔH0−1 (48). As in previous studies (40, 41, 49), this analysis was performed on the spectra from the spin-diluted fibrils to minimize any effects on line broadening due to spin-spin interaction. Although the soluble protein is rather dynamic, significant immobilization can be seen upon fibril formation. This is especially the case for the N terminus and the polyQ domain (Fig. 5A).
FIGURE 5.

A, inverse central line width from EPR spectra of HDx1 fusion protein (circles) and sparsely labeled fibrils (triangles). The inverse of the width of the central resonance line is plotted as function of residue number to give a measure of mobility. Individual domains are highlighted. The orange, blue, and green shaded regions represent the N-terminal, polyQ, and C-terminal domains, respectively. B, sketch of the domain organization within HDx1 fibrils. The N termini (orange) and polyQ (blue) form the fibril core region (gray cylinder), from which the solvated, highly mobile C termini (green) protrude. The polyQ region is represented as generic stacked rectangles and is not meant to imply any statement about the actual number or arrangement of individual β-strands. Whether the N-terminal region is located at the center or the periphery of the core region is unknown.
The polyQ domain has long been thought to form the amyloid core. As expected, we found that this domain is tightly packed, but there are significant differences in the motilities at its N and C termini. These differences in mobility depend on the nature of the flanking domain. The N terminus, which is almost as immobilized as the polyQ domain, has previously been shown to take up an α-helical structure (21). Our data are consistent with a structuring of this domain. In addition, we find that this domain makes tertiary or quaternary contacts. According to the detectable spin-spin interaction, these contacts are very likely with other N termini. This notion is consistent with cross-linking data (50) as well as a recent analytical ultracentrifugation study demonstrating that N termini form α-helix-rich cores of oligomers (19). Thus, the N terminus is not only important for oligomer formation but it also stabilizes the fibril via its packing interactions, and it must be buried within the fibril (Fig. 5B). The present study did not determine whether the N terminus is located at the periphery or the center of the core region.
Compared with the N terminus, the C terminus is much more mobile. This mobility gradually increases with increasing distance from the polyQ core domain. Although the C terminus is very dynamic, with no tertiary or quaternary interactions, the increase in mobility occurs gradually such that the mobility does not reach that of the soluble fusion protein until about residue 111 (Fig. 5A). This increase in mobility, which occurs over more than 40 amino acids, again suggests the presence of a residual secondary structure, rather than merely a tethering effect. Our CD data indicate a polyproline II helical structure in HDx1 fibrils. Given that the N terminus and the polyQ region are devoid of proline residues whereas the C terminus is rich in proline (∼60%), we propose that the C terminus adopts a polyproline II structure in HDx1 fibrils. This polyproline II structure lies outside the fibril core; it does not make any significant intermolecular contacts and must be largely solvated (Fig. 5B). In contrast to the N terminus, the C terminus does not engage in contacts that could promote fibril stability. Rather, it appears to destabilize the adjacent glutamine residues. Gln-63 is the final glutamine of the polyQ domain, and Pro-64 is the first proline residue of the proline-rich C terminus. Both residues reside in a region of intermediate mobility, and their spectra reflect the properties both of the polyQ as and the C-terminal domains. Consistent with these structural data, biochemical experiments show that the C-terminal proline stretches inhibit aggregation of polyQ (51) and HDx1 (16, 22, 44). A potential role of the polyproline domain in influencing the local structure of the polyQ domain is further supported by a recent crystallography study of soluble wild-type HDx1, which found that the four glutamines adjacent to the proline stretch are in an extended, rather than a random coil conformation (52). Our data may offer insight into the observation that MW7, an antibody that binds the polyproline region immediately adjacent to the polyQ domain (53), is capable of disaggregating preformed fibrils (54). Although further studies will be required to map and characterize the precise interactions of antibodies with the fibrils, epitopes in this region may be especially promising targets for therapies designed to reduce aggregation. It is accessible to the antibody and binding may enhance the naturally destabilizing effect of the C-terminal tail.
Whereas the mobility profile described here has many similarities to those of amyloid fibrils, the spin-spin interactions contained in the EPR spectra of HDx1 fibrils are different from those of previously described, disease-related amyloid fibrils. No regions of the HDx1 fibrils display detectable evidence of spin exchange, indicating that there is no stacking of multiple residues in a parallel, in-register arrangement of β-strands (Fig. 6A). Although unusual, because nearly all reported fibrils with amyloid cores over 20 amino acids in length have a parallel, in-register β-strand arrangement, this is not necessarily surprising. Many proteins may prefer the latter arrangement because it maximizes hydrophobic contacts by allowing like hydrophobic residues to stack on top of one another (38). The homopolymer nature of polyQ could make it more difficult for identical sites in the primary sequence to find each other and stack in this manner. Thus, although the commonly seen parallel, in-register structure is inconsistent with the present data, we cannot exclude a more loosely arranged parallel structure in which each molecule takes up more than one layer (Fig. 6B), residues are not perfectly in-register (Fig. 6C), or there is some combination of the two. Our data are also consistent with an antiparallel β-sheet structure (55–57) (Fig. 6D).
FIGURE 6.

Sketches of different types of cross-β structures. A, HDx1 does not adopt a parallel, in-register structure common to other disease-causing amyloid proteins, in which multiple spin labels (yellow dots) come into close contact. B, one potential structure in which spin labels approach within 20 Å is a parallel arrangement of β-strands (arrows) in which each molecule takes up more than one layer and spin labels are separated by one or more intervening layers of β-strands. C, another possibility would be a parallel structure in which each molecule takes up a separate layer, but, due to the promiscuous nature of polyQ, labels are not in register. D, alternatively, several antiparallel models are possible, with or without intervening β-strands. The fibril axis is indicated, which is also the direction of main chain hydrogen bonds. Different shades of arrows represent β-strands from different HDx1 molecules.
We have begun to elucidate the structural features of the various domains in HDx1 fibrils in the biologically relevant full-length version of the protein. Together with a recent solid-state NMR study, our findings represent a first step toward a high resolution structure of HDx1 fibrils. Future EPR experiments using a more exhaustive set of distance constraints from single- and double-labeled mutants should allow us to obtain a more precise idea of the arrangement of glutamine strands, as well as the orientations of the N and C termini. Recent spin-labeling work, which combined continuous wave EPR, pulsed EPR, and computational refinement, demonstrated that detailed, three-dimensional structural information can be obtained using such an approach (58, 59).
Acknowledgments
We thank Konstantin Piatkov for advice regarding protein purification and Ansgar Siemer for helpful discussion.
This work was supported, in whole or in part, by a grant from the National Institutes of Health through the NINDS (to P. H. P.). This work was also supported by grants from the Hereditary Disease Foundation (to R. L. and P. H. P.).

This article contains supplemental Figs. S1 and S2.
- HD
- Huntington disease
- HDx1
- huntingtin exon 1
- Htt
- huntingtin
- MTSL
- S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate
- polyQ
- polyglutamine.
REFERENCES
- 1. The Huntington Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington disease chromosomes. Cell 72, 971–983 [DOI] [PubMed] [Google Scholar]
- 2. Zoghbi H. Y., Orr H. T. (2000) Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 23, 217–247 [DOI] [PubMed] [Google Scholar]
- 3. Brinkman R. R., Mezei M. M., Theilmann J., Almqvist E., Hayden M. R. (1997) The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am. J. Hum. Genet. 60, 1202–1210 [PMC free article] [PubMed] [Google Scholar]
- 4. Vonsattel J. P., Myers R. H., Stevens T. J., Ferrante R. J., Bird E. D., Richardson E. P., Jr. (1985) Neuropathological classification of Huntington disease. J. Neuropathol. Exp. Neurol. 44, 559–577 [DOI] [PubMed] [Google Scholar]
- 5. Rosas H. D., Goodman J., Chen Y. I., Jenkins B. G., Kennedy D. N., Makris N., Patti M., Seidman L. J., Beal M. F., Koroshetz W. J. (2001) Striatal volume loss in HD as measured by MRI and the influence of CAG repeat. Neurology 57, 1025–1028 [DOI] [PubMed] [Google Scholar]
- 6. Mangiarini L., Sathasivam K., Seller M., Cozens B., Harper A., Hetherington C., Lawton M., Trottier Y., Lehrach H., Davies S. W., Bates G. P. (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506 [DOI] [PubMed] [Google Scholar]
- 7. DiFiglia M., Sapp E., Chase K. O., Davies S. W., Bates G. P., Vonsattel J. P., Aronin N. (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993 [DOI] [PubMed] [Google Scholar]
- 8. Sieradzan K. A., Mechan A. O., Jones L., Wanker E. E., Nukina N., Mann D. M. (1999) Huntington disease intranuclear inclusions contain truncated, ubiquitinated huntingtin protein. Exp. Neurol. 156, 92–99 [DOI] [PubMed] [Google Scholar]
- 9. Huang C. C., Faber P. W., Persichetti F., Mittal V., Vonsattel J. P., MacDonald M. E., Gusella J. F. (1998) Amyloid formation by mutant huntingtin: threshold, progressivity and recruitment of normal polyglutamine proteins. Somat. Cell. Mol. Genet. 24, 217–233 [DOI] [PubMed] [Google Scholar]
- 10. Poirier M. A., Li H., Macosko J., Cai S., Amzel M., Ross C. A. (2002) Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J. Biol. Chem. 277, 41032–41037 [DOI] [PubMed] [Google Scholar]
- 11. Perutz M. F., Pope B. J., Owen D., Wanker E. E., Scherzinger E. (2002) Aggregation of proteins with expanded glutamine and alanine repeats of the glutamine-rich and asparagine-rich domains of Sup35 and of the amyloid β-peptide of amyloid plaques. Proc. Natl. Acad. Sci. U.S.A. 99, 5596–5600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jahn T. R., Makin O. S., Morris K. L., Marshall K. E., Tian P., Sikorski P., Serpell L. C. (2010) The common architecture of cross-β amyloid. J. Mol. Biol. 395, 717–727 [DOI] [PubMed] [Google Scholar]
- 13. Chen S., Berthelier V., Yang W., Wetzel R. (2001) Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J. Mol. Biol. 311, 173–182 [DOI] [PubMed] [Google Scholar]
- 14. Chen S., Berthelier V., Hamilton J. B., O'Nuallain B., Wetzel R. (2002) Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry 41, 7391–7399 [DOI] [PubMed] [Google Scholar]
- 15. Gu X., Greiner E. R., Mishra R., Kodali R., Osmand A., Finkbeiner S., Steffan J. S., Thompson L. M., Wetzel R., Yang X. W. (2009) Serines 13 and 16 are critical determinants of full-length human mutant huntingtin-induced disease pathogenesis in HD mice. Neuron 64, 828–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thakur A. K., Jayaraman M., Mishra R., Thakur M., Chellgren V. M., Byeon I. J., Anjum D. H., Kodali R., Creamer T. P., Conway J. F., Gronenborn A. M., Wetzel R. (2009) Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 16, 380–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Williamson T. E., Vitalis A., Crick S. L., Pappu R. V. (2010) Modulation of polyglutamine conformations and dimer formation by the N terminus of huntingtin. J. Mol. Biol. 396, 1295–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rockabrand E., Slepko N., Pantalone A., Nukala V. N., Kazantsev A., Marsh J. L., Sullivan P. G., Steffan J. S., Sensi S. L., Thompson L. M. (2007) The first 17 amino acids of Huntingtin modulate its subcellular localization, aggregation and effects on calcium homeostasis. Hum. Mol. Genet. 16, 61–77 [DOI] [PubMed] [Google Scholar]
- 19. Jayaraman M., Kodali R., Sahoo B., Thakur A. K., Mayasundari A., Mishra R., Peterson C. B., Wetzel R. (2012) Slow amyloid nucleation via α-helix-rich oligomeric intermediates in short polyglutamine-containing huntingtin fragments. J. Mol. Biol. 415, 881–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mishra R., Jayaraman M., Roland B. P., Landrum E., Fullam T., Kodali R., Thakur A. K., Arduini I., Wetzel R. (2012) Inhibiting the nucleation of amyloid structure in a huntingtin fragment by targeting α-helix-rich oligomeric intermediates. J. Mol. Biol. 415, 900–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sivanandam V. N., Jayaraman M., Hoop C. L., Kodali R., Wetzel R., van der Wel P. C. (2011) The aggregation-enhancing huntingtin N terminus is helical in amyloid fibrils. J. Am. Chem. Soc. 133, 4558–4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hollenbach B., Scherzinger E., Schweiger K., Lurz R., Lehrach H., Wanker E. E. (1999) Aggregation of truncated GST-HD exon 1 fusion proteins containing normal range and expanded glutamine repeats. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 991–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Whitmore L., Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89, 392–400 [DOI] [PubMed] [Google Scholar]
- 24. Compton L. A., Johnson W. C., Jr. (1986) Analysis of protein circular dichroism spectra for secondary structure using a simple matrix multiplication. Anal. Biochem. 155, 155–167 [DOI] [PubMed] [Google Scholar]
- 25. Manavalan P., Johnson W. C., Jr. (1987) Variable selection method improves the prediction of protein secondary structure from circular dichroism spectra. Anal. Biochem. 167, 76–85 [DOI] [PubMed] [Google Scholar]
- 26. Sreerama N., Woody R. W. (2000) Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 287, 252–260 [DOI] [PubMed] [Google Scholar]
- 27. Sreerama N., Woody R. W. (1993) A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal. Biochem. 209, 32–44 [DOI] [PubMed] [Google Scholar]
- 28. Andrade M. A., Chacón P., Merelo J. J., Morán F. (1993) Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 6, 383–390 [DOI] [PubMed] [Google Scholar]
- 29. Sreerama N., Venyaminov S. Y., Woody R. W. (1999) Estimation of the number of α-helical and β-strand segments in proteins using circular dichroism spectroscopy. Protein Sci. 8, 370–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Provencher S. W., Glöckner J. (1981) Estimation of globular protein secondary structure from circular dichroism. Biochemistry 20, 33–37 [DOI] [PubMed] [Google Scholar]
- 31. van Stokkum I. H., Spoelder H. J., Bloemendal M., van Grondelle R., Groen F. C. (1990) Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal. Biochem. 191, 110–118 [DOI] [PubMed] [Google Scholar]
- 32. Scherzinger E., Lurz R., Turmaine M., Mangiarini L., Hollenbach B., Hasenbank R., Bates G. P., Davies S. W., Lehrach H., Wanker E. E. (1997) Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90, 549–558 [DOI] [PubMed] [Google Scholar]
- 33. Scherzinger E., Sittler A., Schweiger K., Heiser V., Lurz R., Hasenbank R., Bates G. P., Lehrach H., Wanker E. E. (1999) Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington disease pathology. Proc. Natl. Acad. Sci. U.S.A. 96, 4604–4609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bennett M. J., Huey-Tubman K. E., Herr A. B., West A. P., Jr., Ross S. A., Bjorkman P. J. (2002) Inaugural Article: a linear lattice model for polyglutamine in CAG-expansion diseases. Proc. Natl. Acad. Sci. U.S.A. 99, 11634–11639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Langen R., Cai K., Altenbach C., Khorana H. G., Hubbell W. L. (1999) Structural features of the C-terminal domain of bovine rhodopsin: a site-directed spin-labeling study. Biochemistry 38, 7918–7924 [DOI] [PubMed] [Google Scholar]
- 36. Margittai M., Fasshauer D., Jahn R., Langen R. (2003) The Habc domain and the SNARE core complex are connected by a highly flexible linker. Biochemistry 42, 4009–4014 [DOI] [PubMed] [Google Scholar]
- 37. Nekooki-Machida Y., Kurosawa M., Nukina N., Ito K., Oda T., Tanaka M. (2009) Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 106, 9679–9684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Margittai M., Langen R. (2008) Fibrils with parallel in-register structure constitute a major class of amyloid fibrils: molecular insights from electron paramagnetic resonance spectroscopy. Q. Rev. Biophys. 41, 265–297 [DOI] [PubMed] [Google Scholar]
- 39. Ladner C. L., Chen M., Smith D. P., Platt G. W., Radford S. E., Langen R. (2010) Stacked sets of parallel, in-register β-strands of β2-microglobulin in amyloid fibrils revealed by site-directed spin labeling and chemical labeling. J. Biol. Chem. 285, 17137–17147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen M., Margittai M., Chen J., Langen R. (2007) Investigation of α-synuclein fibril structure by site-directed spin labeling. J. Biol. Chem. 282, 24970–24979 [DOI] [PubMed] [Google Scholar]
- 41. Margittai M., Langen R. (2004) Template-assisted filament growth by parallel stacking of tau. Proc. Natl. Acad. Sci. U.S.A. 101, 10278–10283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hubbell W. L., Cafiso D. S., Altenbach C. (2000) Identifying conformational changes with site-directed spin labeling. Nat. Struct. Biol. 7, 735–739 [DOI] [PubMed] [Google Scholar]
- 43. Lakhani V. V., Ding F., Dokholyan N. V. (2010). Polyglutamine-induced misfolding of huntingtin exon1 is modulated by the flanking sequences. PLoS Comput. Biol. 6, e1000772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Darnell G., Orgel J. P., Pahl R., Meredith S. C. (2007) Flanking polyproline sequences inhibit β-sheet structure in polyglutamine segments by inducing PPII-like helix structure. J. Mol. Biol. 374, 688–704 [DOI] [PubMed] [Google Scholar]
- 45. Kelly M. A., Chellgren B. W., Rucker A. L., Troutman J. M., Fried M. G., Miller A. F., Creamer T. P. (2001) Host-guest study of left-handed polyproline II helix formation. Biochemistry 40, 14376–14383 [DOI] [PubMed] [Google Scholar]
- 46. Horng J. C., Raines R. T. (2006) Stereoelectronic effects on polyproline conformation. Protein Sci. 15, 74–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Woody R. W. (1992) Circular dichroism of unordered peptides. Adv. Biophys. Chem. 2, 37–79 [Google Scholar]
- 48. Mchaourab H. S., Lietzow M. A., Hideg K., Hubbell W. L. (1996) Motion of spin-labeled side chains in T4 lysozyme: correlation with protein structure and dynamics. Biochemistry 35, 7692–7704 [DOI] [PubMed] [Google Scholar]
- 49. Török M., Milton S., Kayed R., Wu P., McIntire T., Glabe C. G., Langen R. (2002) Structural and dynamic features of Alzheimer's Aβ peptide in amyloid fibrils studied by site-directed spin labeling. J. Biol. Chem. 277, 40810–40815 [DOI] [PubMed] [Google Scholar]
- 50. Tam S., Spiess C., Auyeung W., Joachimiak L., Chen B., Poirier M. A., Frydman J. (2009) The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol. 16, 1279–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bhattacharyya A., Thakur A. K., Chellgren V. M., Thiagarajan G., Williams A. D., Chellgren B. W., Creamer T. P., Wetzel R. (2006) Oligoproline effects on polyglutamine conformation and aggregation. J. Mol. Biol. 355, 524–535 [DOI] [PubMed] [Google Scholar]
- 52. Kim M. W., Chelliah Y., Kim S. W., Otwinowski Z., Bezprozvanny I. (2009) Secondary structure of Huntingtin amino-terminal region. Structure 17, 1205–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ko J., Ou S., Patterson P. H. (2001) New anti-huntingtin monoclonal antibodies: implications for huntingtin conformation and its binding proteins. Brain Res. Bull. 56, 319–329 [DOI] [PubMed] [Google Scholar]
- 54. Legleiter J., Lotz G. P., Miller J., Ko J., Ng C., Williams G. L., Finkbeiner S., Patterson P. H., Muchowski P. J. (2009) Monoclonal antibodies recognize distinct conformational epitopes formed by polyglutamine in a mutant huntingtin fragment. J. Biol. Chem. 284, 21647–21658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Thakur A. K., Wetzel R. (2002) Mutational analysis of the structural organization of polyglutamine aggregates. Proc. Natl. Acad. Sci. U.S.A. 99, 17014–17019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sharma D., Shinchuk L. M., Inouye H., Wetzel R., Kirschner D. A. (2005) Polyglutamine homopolymers having 8–45 residues form slablike β-crystallite assemblies. Proteins 61, 398–411 [DOI] [PubMed] [Google Scholar]
- 57. Poirier M. A., Jiang H., Ross C. A. (2005) A structure-based analysis of huntingtin mutant polyglutamine aggregation and toxicity: evidence for a compact β-sheet structure. Hum. Mol. Genet. 14, 765–774 [DOI] [PubMed] [Google Scholar]
- 58. Jao C. C., Hegde B. G., Chen J., Haworth I. S., Langen R. (2008) Structure of membrane-bound α-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. U.S.A. 105, 19666–19671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bedrood S., Li Y., Isas J. M., Hegde B. G., Baxa U., Haworth I. S., Langen R. (2012) Fibril structure of human islet amyloid polypeptide. J. Biol. Chem. 287, 5235–5241 [DOI] [PMC free article] [PubMed] [Google Scholar]