

Background: The Ω-loop (Arg-164 to Asp-179) is a conserved region among class A β-lactamases.
Results: In KPC-2, a carbapenemase of significant clinical importance, Arg-164 substitutions in the Ω-loop selectively enhanced ceftazidime hydrolysis.
Conclusion: Ceftazidime resistance may proceed by a novel mechanism that uses covalent trapping and hydrolysis.
Significance: Future antibiotic design must consider the distinctive behavior of the Ω-loop of KPC-2.
Keywords: Antibiotic Resistance, Antibiotics Action, Drug Resistance, Infectious Diseases, Peptidoglycan, Beta-Lactamase, KPC-2, Carbapenemase, Ceftazidime
Abstract
Gram-negative bacteria harboring KPC-2, a class A β-lactamase, are resistant to all β-lactam antibiotics and pose a major public health threat. Arg-164 is a conserved residue in all class A β-lactamases and is located in the solvent-exposed Ω-loop of KPC-2. To probe the role of this amino acid in KPC-2, we performed site-saturation mutagenesis. When compared with wild type, 11 of 19 variants at position Arg-164 in KPC-2 conferred increased resistance to the oxyimino-cephalosporin, ceftazidime (minimum inhibitory concentration; 32→128 mg/liter) when expressed in Escherichia coli. Using the R164S variant of KPC-2 as a representative β-lactamase for more detailed analysis, we observed only a modest 25% increase in kcat/Km for ceftazidime (0.015→0.019 μm−1 s−1). Employing pre-steady-state kinetics and mass spectrometry, we determined that acylation is rate-limiting for ceftazidime hydrolysis by KPC-2, whereas deacylation is rate-limiting in the R164S variant, leading to accumulation of acyl-enzyme at steady-state. CD spectroscopy revealed that a conformational change occurred in the turnover of ceftazidime by KPC-2, but not the R164S variant, providing evidence for a different form of the enzyme at steady state. Molecular models constructed to explain these findings suggest that ceftazidime adopts a unique conformation, despite preservation of Ω-loop structure. We propose that the R164S substitution in KPC-2 enhances ceftazidime resistance by proceeding through “covalent trapping” of the substrate by a deacylation impaired enzyme with a lower Km. Future antibiotic design must consider the distinctive behavior of the Ω-loop of KPC-2.
Introduction
Resistance to β-lactam antibiotics (e.g., penicillins, cephalosporins, and carbapenems; Fig. 1) remains a major clinical challenge. In Gram-negative pathogens, β-lactamases (EC 3.5.2.6) represent the most common enzymatic mechanism of antibiotic resistance (1). As a consequence of facile transfer of β-lactamase (bla) genes among bacteria, the commercialization of novel classes of β-lactams has repeatedly prompted widespread resistance (2–5). Presently, carbapenemase-type β-lactamases are emerging that hydrolyze carbapenem antibiotics, which until recently were used as “last resort” drugs for treatment of life-threatening infections (3, 6).
FIGURE 1.

Chemical structures of β-lactams used in this study. On ceftazidime, R1 and R2 side chains are indicated. Note the bulky, oxyimino R1 side chains characteristic of the extended spectrum cephalosporins cefotaxime, ceftazidime, and cefepime, and the monobactam aztreonam.
Among carbapenemases, the Ambler class A Klebsiella pneumoniae carbapenemase (KPC)5 family is the most widespread (1, 7). The blaKPC-2 gene is encoded on a plasmid and has been found in Enterobacteriaceae (e.g., K. pneumoniae and Escherichia coli) and Pseudomonas aeruginosa (8–10). Moreover, pathogens harboring KPC-2 are resistant to all β-lactams and β-lactamase inhibitors, severely limiting treatment options and leading to high fatality rates (9, 11–14). Therefore, the biochemical characterization of KPC-2 is critical to inform the design of novel β-lactams with efficacy against KPC-2-producing pathogens.
In an effort to understand the structural basis for catalysis by class A carbapenemases, crystal structures of KPC-2, NmcA, and SME-1 have been solved (Fig. 2A) (15–18). Distinct from non-carbapenemase class A enzymes, KPC-2 β-lactamase exhibits a widened active site and a recessed position of the catalytic nucleophile, Ser-70. An additional notable feature is a Cys-69–Cys-238 disulfide bond, covalently linking the active site b3 β-strand and the helix encompassing Ser-70. Complementary mutagenesis studies have determined that Trp-105, Thr-237, and Arg-220 of KPC-2 define novel catalytic properties (19–21).
FIGURE 2.

A, structure of KPC-2 (Protein Data Bank code 2OV1). The nucleophile Ser-70 and residue of focus in this study, Arg-164, are indicated. Active site regions are color-coded as follows: orange, SXXK motif (residues 70–73); green, SDN loop (130–132); blue, Ω-loop (164–179); red, b3 β-strand (234–242). B, sequence alignment of the Ω-loops of KPC-2 with other class A carbapenemases (blue), penicillinases (red), and cephalosporinases (green). Note the strict conservation of Arg-164 and Asp-179. C, overlay of Ω-loops from TEM-1 (red) and KPC-2 (blue), showing the salt bridge between Arg-164 and Asp-179.
The Ω-loop is a “hot spot” for substitutions that extend the substrate spectrum of many class A enzymes (22, 23). This region (residues 164–179 in class A β-lactamases) forms the “floor” of the active site and contains Glu-166 and Asn-170, two residues that participate in priming a water molecule for deacylation of the β-lactam (Fig. 2B) (15, 24–26). In the class A penicillinases TEM-1 and SHV-1, the flexibility of the Ω-loop is restricted by a network of hydrogen and ionic bonds, one of which connects Arg-164 and Asp-179 at opposite poles of the “neck” of the Ω-loop (Fig. 2C) (27, 28). Disruption of the Arg-164/Asp-179 salt bridge by substitution at either residue enhances the “flexibility” of the Ω-loop, thereby allowing for hydrolysis of extended-spectrum cephalosporins (29, 30). In some variants, this occurs in the apo enzyme (e.g., the R164S variant of TEM-1), whereas in others, it requires ligand binding (e.g., SHV R164H or -S) (31, 32).
There exists a high degree of both sequence and structural conservation in the Ω-loop region of KPC-2 and TEM-1, including conservation of the salt bridge between Arg-164 and Asp-179 (Fig. 2, B and C). Therefore, we were compelled to generate a full library of substitutions at Arg-164 in KPC-2 and perform a complete biochemical characterization of the Arg-164 variant enzymes. The data reveal that Ω-loop substitutions at this conserved residue distinctly impact catalysis of ceftazidime an important advanced generation antibiotic and argue for a “covalent trapping” mechanism of resistance in addition to hydrolysis.
MATERIALS AND METHODS
Plasmids, PCR Primers, and Mutagenesis
For antimicrobial susceptibility testing, all KPC-2 variants were generated in the pBR322-catI plasmid harboring the blaKPC-2 gene as described previously (9). For protein purification the R164S substitution was generated in pET24a(+) harboring blaKPC-2 without the sequence encoding the leader peptide, a plasmid generated previously (21). The QuikChange XL site-directed mutagenesis kit (Agilent, Santa Clara, CA) was used to generate all variants, following the manufacturer's protocol. For PCR mutagenesis, seven degenerate oligonucleotide sets covering all 19 amino acid substitutions (Thermo Fisher Scientific, Hampton, NH) at Arg-164 were generated and yielded 15 of the 19 variants. The R164H, R164S, R164I, and R164F variants were constructed by site-directed mutagenesis. All primers were designed using Agilent genomics software. All of the mutations were verified by sequencing.
Antimicrobial Susceptibility Testing
Lysogeny broth (LB) minimal inhibitory concentration (MICs) determinations for β-lactams were performed on single clones of E. coli DH10B (Invitrogen) harboring pBR322-catI-blaKPC-2 or a variant. We performed our experiments according to the criteria of the Clinical and Laboratory Standards Institute (33). The MICs shown in Table 1 are the mode of at least three independent experiments.
TABLE 1.
MIC values for β-lactams (mg/liter)
AMP, ampicillin; THIN, cephalothin; TAX, cefotaxime; TAZ, ceftazidime; FEP, cefepime; AZT, aztreonam; IMI, imipenem.
| AMP | THIN | TAX | TAZ | FEP | AZT | IMI | |
|---|---|---|---|---|---|---|---|
| E. coli DH10B | 1 | 4 | .06 | 0.25 | 0.06 | 0.25 | 0.12 |
| pBR322-catI-blaKPC-2 | 4096 | 1024 | 8 | 32 | 8 | 256 | 4 |
| Hydrophobic | |||||||
| R164G | 2048 | 256 | 4 | 128 | 4 | 128 | 1 |
| R164A | 2048 | 256 | 4 | 128 | 4 | 64 | 0.5 |
| R164L | 1024 | 256 | 0.12 | 64 | 1 | 16 | 0.12 |
| R164V | 256 | 64 | 0.06 | 64 | 0.5 | 2 | 0.25 |
| R164I | 256 | 16 | 0.12 | 128 | 0.25 | 1 | 0.12 |
| R164M | 1024 | 256 | 0.12 | 64 | 1 | 16 | 0.25 |
| Positive | |||||||
| R164K | 2048 | 256 | 4 | 128 | 4 | 128 | 1 |
| R164H | 2048 | 512 | 4 | 64 | 8 | 128 | 2 |
| Negative | |||||||
| R164D | 2048 | 256 | 4 | 128 | 4 | 128 | 1 |
| R164E | 2048 | 256 | 4 | 128 | 4 | 64 | 0.5 |
| Aromatic | |||||||
| R164F | 1024 | 128 | 0.12 | 64 | 1 | 16 | 0.25 |
| R164Y | 1 | 8 | .06 | 4 | 0.12 | 0.5 | 0.12 |
| R164W | 1 | 4 | .06 | 0.25 | 0.06 | 0.25 | 0.12 |
| Polar | |||||||
| R164S | 2048 | 256 | 4 | 128 | 4 | 64 | 1 |
| R164T | 2048 | 256 | 4 | 128 | 4 | 64 | 1 |
| R164N | 2048 | 512 | 4 | 128 | 4 | 256 | 2 |
| R164Q | 2048 | 512 | 4 | 128 | 4 | 128 | 1 |
| Unique | |||||||
| R164P | 1 | 8 | 0.5 | 128 | 0.5 | 2 | 0.12 |
| R164C | 1 | 8 | 0.06 | 0.25 | 0.06 | 0.25 | 0.12 |
Immunoblotting
E. coli DH10B harboring pBR322-catI-blaKPC-2 or a variant were grown to stationary phase in LB containing 20 mg/liter chloramphenicol selection. The cells were pelleted and boiled for 10 min in Laemmli buffer to induce lysis. The details of the immunoblotting procedure were described previously (21). The membranes were probed with a rabbit anti-KPC-2 polyclonal antibody (Sigma-Genosys) and, as a loading control, an anti-DNA-K rabbit polyclonal antibody (Stressgene, San Diego, CA).
β-Lactamase Purification
KPC-2 and R164S proteins were purified from E. coli Origami2 DE3 (Novagen, Darmstadt, Germany) harboring pET24a(+) blaKPC-2 or the variant as described previously (21). Briefly, bacteria grown in super optimal broth were induced to express β-lactamase with 500 μm isopropyl-β-d-thiogalactoside for 2 h at 37 °C, at a starting A600 of 0.6. Bacterial cells were pelleted and frozen at −20 °C for at least 12 h. The pellets were lysed in a buffer containing 40 mg/ml lysozyme, and the cleared supernatant was subjected to preparative isoelectric focusing. Gel segments containing β-lactamase were identified by assaying for activity against nitrocefin. The purity of active fractions was assessed by SDS-PAGE followed by staining with Coomassie Brilliant Blue R250. Fractions that were qualitatively determined to be more than ∼80% pure were pooled and concentrated in preparation for anion exchange chromatography, which was performed using a HiTrap Q HP column (GE Healthcare). Purity was reassessed, and fractions containing more than ∼90% purity were pooled, exchanged into 10 mm PBS (pH 7.4), and concentrated. Protein concentration was calculated by measuring absorbance at λ280 and using the extinction coefficient of the protein. The identity of each protein preparation was confirmed by mass spectrometry (described below). The proteins were stored at either 4 °C for ∼1 week or, for longer periods, at −20 °C in 50% glycerol.
Kinetics
β-Lactamases hydrolyze β-lactam substrates through a two-step mechanism, which is commonly represented as follows.
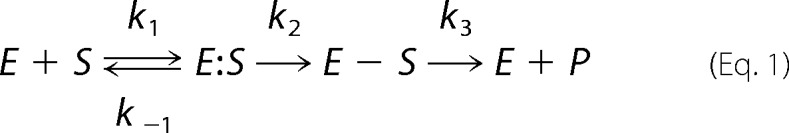 |
In this scheme, k1 is a second order rate constant governing the formation of the precovalent Henri-Michaelis complex, E:S, and k−1 is a first order rate governing its dissociation. k2 represents the first order rate constant for formation of the acyl-enzyme, E − S. k3 represents the first order rate constant for product deacylation and release.
The relationship between reaction velocity (v) and substrate concentration can be represented by the Henri-Michaelis-Menten equation.
Qualitatively, kcat represents the first order rate constant for reaction of the E:S to generate product and regenerate free enzyme, Km is the concentration of substrate required to reach ½ Vmax, and kcat/Km is the second order rate constant for reaction of the free E and S to form free E and P. To measure the steady-state parameters kcat, Km, kcat/Km, reactions were initiated at room temperature in 10 mm PBS (pH 7.4) containing variable amounts of enzyme depending on the enzyme-substrate pair, where conditions always conform to the steady-state assumption (<10% of the substrate is consumed in the reaction). Initial velocities were obtained on an Agilent 8453 diode array spectrophotometer by monitoring changes in absorption upon β-lactam ring opening, using the following extinction coefficients: ampicillin, Δϵ235 = −900 m−1 cm−1; cephalothin, Δϵ262 = −7660 m−1 cm−1; nitrocefin, Δϵ482 = 17,400 m−1 cm−1; cefoxitin, Δϵ260 = −6225 m−1 cm−1; cefotaxime, Δϵ262 = −7250 m−1 cm−1; ceftazidime, Δϵ256 = −7600 m−1 cm−1; cefepime, Δϵ260 = −750 m−1 cm−1; aztreonam, Δϵ318 = −640 m−1 cm−1; and imipenem, Δϵ299 = −9000 m−1 cm−1. For velocity determinations for ampicillin, cephalothin, nitrocefin, and imipenem, a 1-cm path length quartz cuvette was used. For cefoxitin, cefotaxime, ceftazidime, cefepime, and aztreonam, a 0.2-cm path length quartz cuvette was used.
Steady-state kinetic parameters were derived by a nonlinear least squares fit of the data to Equation 2 using the program Origin 8.1 (OriginLab, Northampton, MA). The velocities for cefepime hydrolysis by KPC-2 did not approach saturation at testable concentrations. In this case, the slope of the velocity versus cefepime concentration line is the second order rate constant for hydrolysis at steady state, kcat/Km.
Pre-steady-state Kinetics
Pre-steady-state kinetics were carried out on an Applied Photophysics π*-180 stopped flow spectrophotometer. 100 μm ceftazidime was reacted with either 2 or 4 μm KPC-2 or the R164S variant at 25 °C for times ranging from 2 to 20 s. At least four reactions were performed under each condition. The data were analyzed using Pro K software. To determine the burst, π, the linear portion of the curve corresponding to the steady-state velocity was extrapolated back to the y-intercept at time = 0 s. The following equation describes the burst.
Based on the reaction in Equation 1, a is the net rate constant for formation of E-S from free E and S (a = k2[S]/(K′ + [S]) where K′ = (k−1 + k2)/k1), and b is k3 (34).
Electrospray Ionization (ESI)-MS
In these experiments, 19 μm (10 μg) β-lactamase was incubated with ceftazidime at a molar ratio of 1:100 at room temperature in 10 mm PBS (pH 7.4). For KPC-2 and the R164S variant, the reactions proceeded for 5, 15, and 60 s. The reactions were quenched by equilibration to 0.2% formic acid. The samples were desalted and concentrated using a C18 ZipTip (Millipore, Bedford, MA) according to the manufacturer's protocol. ESI-MS of the eluted sample was performed on a Q-STAR Elite quadrupole time of flight mass spectrometer equipped with a TurboIon spray source (Applied Biosystems, Framingham, MA), and spectra were deconvoluted using the Applied Biosystems Analyst program.
CD and Thermal Denaturation
CD experiments were carried out in a Jasco (Easton, MD) J-815 spectrometer with a Peltier effect temperature controller. Quartz cells with a 0.1-cm path length were used for all experiments. For thermal denaturation, 19 μm KPC-2 or the R164S variant with or without 250 μm ceftazidime were monitored for helical content by CD at λ220 between 25 and 65 °C with a heating rate of 2 °C/min. Two-state, reversible denaturation was verified by the return of the original CD signal of the protein upon cooling and by comparison of the enzyme activity prior to denaturation and then after cooling.
For CD spectra, 19 μm KPC-2 or the R164S variant was incubated with 250 μm ceftazidime in PBS (pH 7.4). CD spectra were obtained at room temperature, and data points were recorded every 0.1 nm between λ200 and λ260 with a scan rate of 20 nm/min. CD spectra for ceftazidime alone were taken and subtracted from the spectra of enzyme and ceftazidime.
Molecular Modeling
The KPC-2 β-lactamase crystal coordinates (Protein Data Bank accession code 2OV5) were used to generate the R164S variant by substituting serine for arginine at position 164. The KPC-2 structure and the generated R164S model were optimized by energy minimization using Discovery Studio 3.1 software (Accelrys, San Diego, CA) (19). The minimization was performed in several steps, using steepest descent and conjugate gradient algorithms to reach the minimum convergence (0.01 kcal/mol*Å). The protein was immersed in a water box, 7 Å from any face of the box, and the solvation model employed was with periodic boundary conditions. The force field parameters of CHARMm were used for minimization, and the particle mesh Ewald method addressed long range electrostatics. The minimized structures were utilized for constructing the Michaelis-Menten and acyl complexes with ceftazidime. The ligand structure was built using D.S. Fragment Builder tools. The CHARMm force field was applied; the molecule was solvated with periodic boundary conditions and minimized using a Standard Dynamics Cascade protocol (one minimization using the steepest descent algorithm, followed by adopted basis Newton-Raphson algorithm and three subsequent dynamics stages at a constant pressure and 300 K). The minimized ligand was docked in the active site of the enzyme/model using flexible docking algorithm (19). The generated conformations were visually inspected, the most favorable were solvated, and energy was minimized using the conjugate gradient algorithm with periodic boundary conditions to 0.001 minimum derivatives. To check the stability and look for possible conformational changes of the complex, molecular dynamics simulation was conducted (19). The molecular dynamics simulation was run for 6 ps.
RESULTS AND DISCUSSION
Consequences of the Arg-164 Substitution on the Phenotype, Protein Expression, and Stability of KPC-2 Variants Expressed in E. coli
Site-saturation and site-directed mutagenesis were performed in the pBR322-catI vector harboring blaKPC-2 yielding all 19 variants at position Arg-164 (Ambler numbering scheme). To evaluate how substitutions at this site impact bacterial susceptibility to β-lactams, we transformed pBR322-catI-blaKPC-2-R164X into the uniform E. coli DH10B background and measured MICs. Penicillins (ampicillin) narrow and extended spectrum cephalosporins (cephalothin, cefoxitin, cefotaxime, ceftazidime, and cefepime), aztreonam, and carbapenems (imipenem, meropenem, ertapenem, and doripenem) were tested. The results are presented in Table 1.
KPC-2 expressed in E. coli conferred high level resistance to ampicillin (MIC = 4096 mg/liter) and cephalothin (1024 mg/liter). In general, all the engineered Arg-164 substitutions resulted in reduced MICs to ampicillin, cephalothin, cefotaxime, cefepime, aztreonam, and imipenem. In contrast, 11 of 19 substitutions conferred elevated MICs for ceftazidime versus KPC-2 (32 to 128 mg/liter). The R164S variant of KPC-2 expressed in E. coli was chosen for further study (see below) because this substitution demonstrates the highest level of resistance to ceftazidime and possesses clinical correlates in other class A enzymes (35, 36).
This pattern of susceptibility findings prompted us to examine expression levels of the Arg-164 variants grown in E. coli DH10B. Here we asked whether loss or gain of resistance resulting from altering the amino acid at Arg-164 was a consequence of changes in protein expression or catalytic properties. With some exceptions (R164L, R164I, R164M, R164E, R164V, and R164F), our immunoblotting experiments showed that steady-state expression levels paralleled the resistance to ampicillin and cephalothin (Fig. 3). These observations indicated that, in general, the phenotype of the KPC variants expressed in bacteria may be partly governed by the expression level of the enzyme.
FIGURE 3.

Immunoblot for KPC-2 and, as a loading control, DNA-K, probing E. coli DH10B cells harboring pBR322-catI-blaKPC-2 R164X grown in LB.
As a biochemical correlate, we next determined whether the change in the nature of the amino acid across the “neck” of the Ω-loop had any “cost” on protein thermal stability. We purified the R164S variant for analysis because this substitution in other class A β-lactamases is predicted to disrupt the key salt bridge with Asp-179. Melting temperature (Tm) was determined by monitoring CD difference at λ220 as the temperature was raised from 25 to 65 °C, and affirmed a two-state, reversible denaturation. The Tm was 56 °C for KPC-2 and 52 °C for the R164S variant (Fig. 4). These observations support the notion that loss of the ionic bond between Ω-loop “neck” residues Arg-164 and Asp-179 destabilizes class A enzymes while enhancing their activity against ceftazidime. This “activity/stability tradeoff” is common among β-lactamases (37, 38).
FIGURE 4.

Thermal denaturation experiment. 19 μm KPC-2 or the R164S variant was incubated with or without 250 μm ceftazidime and tested by circular dichroism at λ220. Mean residue molar ellipticity values (MRE) were converted to a fraction of folded protein at each temperature by the formula: Fraction folded = MREtime point/MREminimum. The calculated Tm values for KPC-2 and the R164S variant were 56 and 52 °C, respectively, whereas ceftazidime (TAZ) stabilized both proteins by 1 °C.
Because Arg-164 substitutions both decreased protein expression and enhanced resistance to ceftazidime, we hypothesized that ceftazidime may increase the stability of the R164S variant, leading to a greater intracellular abundance of the enzyme and, perhaps, higher ceftazidime resistance. Incubation with ceftazidime raised the Tm = ∼1 °C for both KPC-2 and R164S, suggesting that ceftazidime did not substantially stabilize the R164S variant (Fig. 4). We interpret from this finding that the more ceftazidime resistance in the variant stems not from enhanced protein stability but is likely a result of changes in the catalytic properties conferred by the R164S substitution.
β-Lactam Kinetics
To understand how disruption of the salt bridge between Arg-164 and Asp-179 impacts catalysis by KPC-2, we performed steady-state kinetic analysis on KPC-2 and the R164S variant. In concert with the relatively moderate MIC changes, the R164S variant did not exhibit more than a 3-fold change in any steady-state kinetic parameter for all of the substrates tested (Table 2). For ampicillin and cephalothin, the substitution slightly decreased kcat/Km because of an increased Km. For oxyimino cephalosporins cefotaxime, ceftazidime, and cefepime, we observed decreased kcat values and Km values (∼2-fold), indicating that the substitution lowers Km but inhibits catalysis for these substrates (Table 2). The kcat/Km was altered only for ceftazidime, with the variant exhibiting a modest increase of 25% in catalytic efficiency.
TABLE 2.
Kinetics with β-lactams
The units of measure are μm for Km, s−1 for kcat, and μm−1 s−1 for kcat/Km.
| KPC-2 | R164S | Fold change (R164S/KPC-2) | |
|---|---|---|---|
| Ampicillin | |||
| Km | 51 ± 7 | 93 ± 10 | 1.8 |
| kcat | 26 ± 1 | 26 ± 1 | 1 |
| kcat/Km | 0.5 | 0.3 | 0.6 |
| Cephalothin | |||
| Km | 30 ± 6 | 40 ± 8 | 1.3 |
| kcat | 250 ± 20 | 280 ± 20 | 1.1 |
| kcat/Km | 8.3 | 7 | 0.8 |
| Nitrocefin | |||
| Km | 7 ± 0.7 | 8 ± 1 | 1.1 |
| kcat | 95 ± 10 | 66 ± 7 | 0.7 |
| kcat/Km | 13 | 8 | 0.6 |
| Cefoxitin | |||
| Km | 340 ± 100 | 260 ± 50 | 0.8 |
| kcat | 8 ± 1 | 3.2 ± 0.5 | 0.4 |
| kcat/Km | 0.024 | 0.012 | 0.5 |
| Cefotaxime | |||
| Km | 515 ± 160 | 260 ± 70 | 0.5 |
| kcat | 180 ± 20 | 90 ± 10 | 0.5 |
| kcat/Km | 0.35 | 0.35 | 1 |
| Ceftazidime | |||
| Km | 375 ± 60 | 180 ± 30 | 0.5 |
| kcat | 5.8 ± 0.6 | 3.5 ± 0.3 | 0.6 |
| kcat/Km | 0.015 | 0.019 | 1.3 |
| Cefepime | |||
| Km | >1000 | 900 ± 150 | NA |
| kcat | >6 | 6.1 ± 0.6 | NA |
| kcat/Km | 0.0067 | 0.0067 | 1 |
| Aztreonam | |||
| Km | 920 ± 160 | 700 ± 115 | 0.8 |
| kcat | 280 ± 30 | 120 ± 10 | 0.4 |
| kcat/Km | 0.30 | 0.17 | 0.6 |
| Imipenem | |||
| Km | 14 ± 2 | 11 ± 2 | 0.8 |
| kcat | 13 ± 1 | 5.5 ± 0.5 | 0.4 |
| kcat/Km | 1 | 0.5 | 0.5 |
Mechanistic Insights into Ceftazidime Hydrolysis and Resistance
Given that the modest increase of the R164S enzyme in kcat/Km for ceftazidime was not completely sufficient to explain the 4-fold enhanced resistance phenotype when expressed in bacteria, we addressed the reaction mechanism in more detail.
To determine whether conformational changes accompany ceftazidime hydrolysis, we obtained CD spectra during the course of the reaction. KPC-2 underwent a subtle loss of helicity throughout λ200–240 upon ceftazidime hydrolysis (Fig. 5A). In contrast, the R164S variant CD spectrum did not change appreciably upon ceftazidime hydrolysis (Fig. 5B). Previously, Taibi-Tronche et al. (39) observed a loss of helical content of TEM-1 during the course of cefepime hydrolysis. This similarity between KPC-2 and TEM-1 raises the possibility that conformational changes may be an important feature of extended spectrum cephalosporin hydrolysis by class A enzymes. Our findings suggest that the CD change observed may be a “slow” relaxation of KPC-2 required to accommodate the bulky oxyimino R1 substituent of the substrate. In stark contrast, the CD spectrum of the R164S variant did not change in the reaction, suggesting that the variant may be “primed” to accommodate ceftazidime and does not require the conformational change, resulting in an enhanced Km. The differences exhibited in this experiment may be further evidence for a lower Km of the R164S variant for ceftazidime. Importantly, these data raise the possibility that a different form of the enzyme is populated at steady-state for the Arg-164 variant.
FIGURE 5.
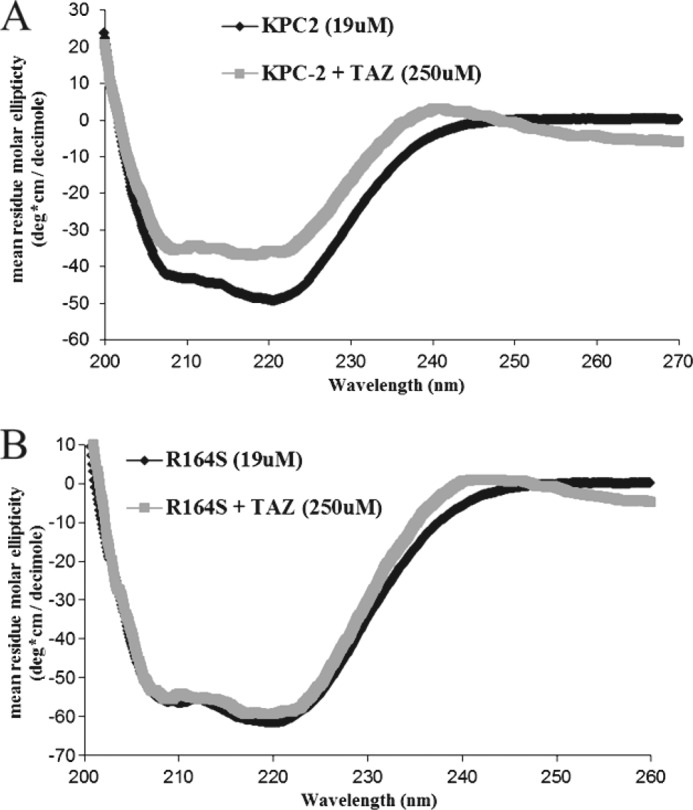
Determination of protein secondary structure by circular dichroism difference spectrum. 19 μm KPC-2 (A) or the R164S variant (B) was incubated either alone or with 250 μm ceftazidime (TAZ), and CD spectra were gathered and plotted as described under “Materials and Methods.”
To understand how the KPC-2 R164S substitution impacts the kinetics of acylation and deacylation, we performed timed ESI-MS and pre-steady-state kinetic experiments. By ESI-MS (t = 5, 15, or 60 s) and a ceftazidime:enzyme molar ratio (100:1), we determined the proportion of acyl-enzyme to Michaelis-Menten complex and used this ratio as an approximation of the relative rate constants for acylation (k2) and deacylation (k3). We observed that acyl-enzyme species were not captured with the KPC-2 enzyme at any time point, suggesting that k3 is faster relative to k2 (Fig. 6A). Surprisingly, for the R164S variant, a species corresponding to an acyl-enzyme adduct appeared at 5 s, diminished at 15 s, and disappeared by 60 s, indicative of a slower k3 (Fig. 6B) and accumulation of the acyl-enzyme at steady-state.
FIGURE 6.

Deconvoluted ESI-MS spectra of ceftazidime reaction coordinate. A and B, 19 μm KPC-2 (A) or the R164S variant (B) was incubated with ceftazidime (TAZ) at a 100:1 substrate to enzyme ratio, and reactions were quenched at 5, 15, or 60 s in 0.2% formic acid, processed, and then analyzed as described under “Materials and Methods.” All of the measurements have an error of ± 5 atomic mass units. C, ceftazidime undergoing R2 elimination to obtain the +Δ468 adduct.
The m/z of the R164S acyl-enzyme adducts at 5 and 15 s were +Δ468 (Fig. 6C). The +Δ468 adduct corresponds to a ceftazidime adduct in which the R2 group is eliminated (R2 elimination has been described previously and occurs rapidly with ceftazidime upon β-lactam ring opening) (31). An unexpected mass of 29,264 Da, which we are unable to identify, was also observed.
Stimulated by the results of the ESI-MS analysis and to verify the results independently, pre-steady-state reactions were performed with ceftazidime. Here 100 μm ceftazidime was reacted with either 2 or 4 μm of KPC-2 or the R164S variant for 2–20 s; a burst of substrate hydrolysis was detected with both enzymes. For KPC-2, the burst, π, was low (0.20 for 2 μm KPC-2) and slightly increased with increasing enzyme concentration (Fig. 7). These values suggest that k3 is fast relative to k2. Therefore, k3 has a negligible impact on kcat, and k2 is the rate-limiting step in ceftazidime hydrolysis. On the other hand, the π value of the R164S variant was much higher (1.43 for 2 μm R164S), and when the concentration of enzyme was raised, the π value increased by 0.3 (Fig. 7). Because of the difficulty achieving saturating substrate concentrations as a result of weak binding, under these conditions the observed burst will not quantitatively reflect the maximal amplitude for either enzyme. However, the mutant clearly shows a burst of product formation occurring pre-steady-state, providing evidence that k3 is slow relative to the net rate constant for formation of E:S and E − S. These results are perfectly consistent with the direct observation of acyl-enzyme by MS and the CD data showing distinct enzyme forms for the mutant and wild type at steady-state.
FIGURE 7.
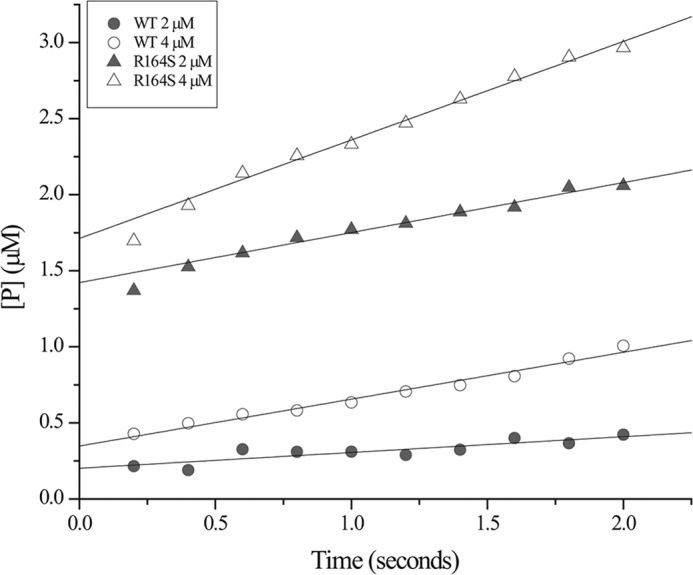
Pre-steady-state kinetic traces of ceftazidime hydrolysis. In a stopped flow spectrophotometer, 100 μm ceftazidime was incubated with 2 and 4 μm KPC-2 or 2 and 4 μm the R164S variant, and the rate of product (P) formation (converted from absorbance readings, Δϵ256 = −7600 m−1 cm−1) was followed for 2 s. Representative traces are shown.
Overall, we propose that the R164S substitution in KPC-2 augments ceftazidime resistance by enhancing kcat/Km (only 25%) and also through a change in the kinetic mechanism of the reaction. The data suggest that a mechanism favoring slow hydrolysis relative to acylation (i.e., enhanced accumulation of acyl-enzyme) as observed in the variant is dominant over hydrolysis, as seen in KPC-2, in determining phenotype. Although the R164S substitution has only a small effect on the measured steady-state kinetic parameters, it likely results in a very large decrease in the hydrolysis rate, such that it becomes at least partially rate-limiting for kcat. These characteristics are reminiscent of those of the TEM-1 M182T/E166R variant, which binds ceftazidime with low Km but is completely deacylation-deficient yet still confers a high level resistance (40). This study in TEM advanced a “covalent trapping” mechanism of resistance, in which the enzyme serves as a “sink” for substrate rather than as a catalyst. We infer from this that regeneration of existing apo-enzyme via deacylation, which is faster for KPC-2, is less important for determining resistance levels compared with the translation of nascent enzyme.
We next considered whether “covalent trapping” of ceftazidime by KPC-2 is substrate-specific in E. coli. In answering this question, we point out that resistance to cefotaxime, a similar substrate, is not enhanced by the R164S substitution. The substitution causes a similar ∼2-fold reduction in kcat and Km for cefotaxime, resulting in an unchanged catalytic efficiency. Unlike ceftazidime, however, cefotaxime is a “fast” substrate (kcat = ∼200 s−1), such that the absolute reduction in kcat observed in the variant for cefotaxime (∼100 s−1) is much greater than for ceftazidime (∼3 s−1). On the basis of this key difference, we posit that under our experimental conditions, deacylation is dominant in clearing “fast” substrates in vivo, whereas commitment to catalysis may be the dominant determinant of resistance for “slow” substrates.
Modeling of Ceftazidime Michaelis-Menten Complexes
To anticipate the structural basis for the enhanced ceftazidime resistance, we constructed molecular models of the Michaelis-Menten complex of ceftazidime with KPC-2 and the R164S variant to help explain our findings. To start, our model predicts that the disposition of the Ω-loop is similar, but not identical (Fig. 8A). Our representations did not suggest that there is disorder in R164S, as observed in other structures (30, 32). The mini helix encompassing Glu-166 and Asn-170 is slightly more mobile in the R164S variant compared with KPC-2, but we do not observe a major reconfiguration of this segment as observed in the R164S-substituted TEM-64 enzyme. We suspect that the Ω-loop of KPC-2 is likely stabilized by as yet unidentified interactions between Ω-loop residues and other active site motifs or a network of water molecules that reside in the Ω-loop cavity (27).
FIGURE 8.

Molecular representations of the Michaelis-Menten complexes of ceftazidime with KPC-2 or the R164S variant. A, superimposition of the KPC-2 (beige) and R164S (green) KPC-2 models, with the Ω-loops and positions of Ser-70 highlighted. The Ω-loop demonstrates slightly increased flexibility in the R164S variant, but the spatial positions of all side chains are within 1 Å in the two models. Conversely, the Oγ of Ser-70 has shifted 2.3 Å in the model for KPC-2. B, superimposition of the models, highlighting the conformations of ceftazidime in the KPC-2 (beige) and R164S (green) models. The arrow indicates the flipping of the ceftazidime aminothiazole and di-methyl carboxyl groups in the two models. C and D, representation of the predicted interactions by DS Studio 3.1 between ceftazidime and indicated active site residues in KPC-2 (C) and the R164S variant (D), indicated by blue dotted lines. In particular, note the hydrogen bond between the main chain oxygen of Asn-170 and the R1 carboxyl group of ceftazidime in the R164S, but not KPC-2 model. Also note the more extensive stabilization of the β-lactam carbonyl in the KPC-2, but C3 carboxylate in the R164S variant.
A second notable feature of the model is the 2.3 Å offset of Oγ of Ser-70 between the two structures (Fig. 8A). By superimposing our models on the KPC-2 crystal structure (Protein Data Bank code 2OV1), we find that a shift occurs in KPC-2, not the variant (data not shown). The Ser-70 shift recalls the crystal structure of Ke et al. (15), who found evidence of enhanced flexibility of Ser-70, likely imparting the range of motion necessary to position the nucleophile for hydrolysis of many β-lactams.
Most importantly, ceftazidime adopts different conformations in the two models (Fig. 8B). In the KPC-2 β-lactamase, the aminothiazole ring in the R1 group of ceftazidime is oriented toward the Ω-loop, whereas the di-methyl carboxyl group is facing away, exposed to solvent. This conformation is reminiscent of previous crystal structures of d,d-peptidase/transpeptidase with cephalothin and cefotaxime (31, 41). In the R164S variant, however, the position of the aminothiazole ring and the dimethyl carboxyl group are reversed, with the di-methyl carboxyl oriented toward the Ω-loop.
Because of these possible conformational differences, the stabilizing interactions between ceftazidime and the two enzymes are quite different. In KPC-2, the β-lactam carbonyl is situated in the oxyanion hole created by the main chain nitrogens of Ser-70 and Thr-237 (Fig. 8C). In contrast, in the R164S variant, the carbonyl oxygen is not in the oxyanion hole, coordinated by Lys-73 Nζ and the nucleophile Ser-70 Oγ (Fig. 8D). These differences in the position of the β-lactam carbonyl oxygen likely have important effects on catalysis, which can only proceed if the incipient negative charge on the oxygen in the tetrahedral transition state is properly stabilized by the oxyanion hole. Furthermore, in the R164S variant, the interaction between the ceftazidime carbonyl oxygen and the nucleophile (Oγ) would likely inhibit its activation for attack on the carbon of the β-lactam carbonyl, slowing catalysis.
Lastly, steric crowding between the side chain of Asn-170 and the aminothiazole group of ceftazidime contributes to the poor affinity of class A non-carbapenemases for extended spectrum cephalosporins (31). Similarly, in the KPC-2 model, the terminal amino group of the aminothiazole ring resides within 3.2 Å of the main chain oxygen of Asn-170 (Fig. 8C). This interaction imposes steric and possibly electrostatic constraints. In contrast, in the R164S variant, the ceftazidime carboxyl group is located 2.8 Å from the main chain oxygen of Asn-170 and is predicted to hydrogen bond with the ceftazidime moiety (Fig. 8D). This “flipping” of the ceftazidime carboxyl and aminothiazole in the active site likely facilitates an electrostatically favorable interaction in the R164S variant, contributing to the enhanced affinity for ceftazidime.
CONCLUSION
In this work we have evaluated how substitutions at residue Arg-164, a conserved active site Ω-loop “neck” residue commonly mutated in TEM-type (ESBLs), impact catalysis and stability of the class A carbapenemase KPC-2. The “ceftazidime-selective” resistance compelled us to investigate the kinetic mechanism for ceftazidime hydrolysis by KPC-2 and the highly resistant R164S variant. We feel that two important points emerge from the findings in this paper.
The first relates to structure/function; Ω-loop flexibility is directly coupled to catalytic properties in class A β-lactamases. The microbiological, kinetic, and modeling data for KPC-2 R164S, considered in concert with similar data for SHV-1 and TEM-1, suggest that greater enhancement in Ω-loop flexibility in the penicillinases is accompanied by greater catalytic and phenotypic changes than for KPC-2, where we see more moderate catalytic effects. In contrast, despite relative preservation of Ω-loop structure, the thermal stability of the R164S variant is decreased. TEM-1 Arg-164 and SHV-1 Asp-179 variants display similarly decreased stability, but concomitantly display increased Ω-loop flexibility and a much more dramatic gain in ceftazidime activity. Why this “activity/stability tradeoff” is more favorable to TEM-1 and SHV-1 penicillinases than to the KPC-2 carbapenemase is unknown.
The second important point is that the kinetic mechanism also argues for a “covalent trapping” ceftazidime resistance mechanism by R164S. The kinetic data suggest that for substrates such as ceftazidime, slow hydrolysis and accumulation of the acyl-enzyme is more critical than regeneration of native enzyme via deacylation in determining resistance phenotypes in bacteria. Design of “next generation” oxyimino cephalosporins and monobactams should pay special attention to β-lactamase affinity and acylation kinetics to preserve their clinical longevity.
This work was supported, in whole or in part, by National Institutes of Health Grant 1R01-A1063517 (to R. A. B.).
- KPC
- K. pneumoniae carbapenemase
- MIC
- minimum inhibitory concentration
- ESI
- electrospray ionization.
REFERENCES
- 1. Bush K., Fisher J. F. (2011) Epidemiological expansion, structural studies, and clinical challenges of new β-lactamases from Gram-negative bacteria. Annu. Rev. Microbiol. 65, 455–478 [DOI] [PubMed] [Google Scholar]
- 2. Drawz S. M., Bonomo R. A. (2010) Three decades of β-lactamase inhibitors. Clin. Microbiol. Rev. 23, 160–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Papp-Wallace K. M., Endimiani A., Taracila M. A., Bonomo R. A. (2011) Carbapenems. Past, present, and future. Antimicrob. Agents Chemother. 55, 4943–4960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Perez F., Endimiani A., Hujer K. M., Bonomo R. A. (2007) The continuing challenge of ESBLs. Curr. Opin. Pharmacol. 7, 459–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barlow M. (2009) What antimicrobial resistance has taught us about horizontal gene transfer. Methods Mol. Biol. 532, 397–411 [DOI] [PubMed] [Google Scholar]
- 6. Queenan A. M., Bush K. (2007) Carbapenemases. The versatile β-lactamases. Clin. Microbiol. Rev. 20, 440–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nordmann P., Cuzon G., Naas T. (2009) The real threat of Klebsiella pneumoniae carbapenemase-producing bacteria. Lancet Infect. Dis. 9, 228–236 [DOI] [PubMed] [Google Scholar]
- 8. Villegas M. V., Lolans K., Correa A., Kattan J. N., Lopez J. A., Quinn J. P. (2007) First identification of Pseudomonas aeruginosa isolates producing a KPC-type carbapenem-hydrolyzing β-lactamase. Antimicrob. Agents Chemother. 51, 1553–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yigit H., Queenan A. M., Anderson G. J., Domenech-Sanchez A., Biddle J. W., Steward C. D., Alberti S., Bush K., Tenover F. C. (2001) Novel carbapenem-hydrolyzing β-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob. Agents Chemother. 45, 1151–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alba J., Ishii Y., Thomson K., Moland E. S., Yamaguchi K. (2005) Kinetics study of KPC-3, a plasmid-encoded class A carbapenem-hydrolyzing β-lactamase. Antimicrob. Agents Chemother. 49, 4760–4762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Papp-Wallace K. M., Bethel C. R., Distler A. M., Kasuboski C., Taracila M., Bonomo R. A. (2010) Inhibitor resistance in the KPC-2 β-lactamase, a preeminent property of this class A β-lactamase. Antimicrob. Agents Chemother. 54, 890–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bergamasco M. D., Barroso Barbosa M., de Oliveira Garcia D., Cipullo R., Moreira J. C., Baia C., Barbosa V., Abboud C. S. (2012) Infection with KPC-producing K. pneumoniae in solid organ transplantation. Transpl. Infect. Dis. 14, 198–205 [DOI] [PubMed] [Google Scholar]
- 13. Kontopoulou K., Protonotariou E., Vasilakos K., Kriti M., Koteli A., Antoniadou E., Sofianou D. (2010) Hospital outbreak caused by Klebsiella pneumoniae producing KPC-2 β-lactamase resistant to colistin. J. Hosp. Infect. 76, 70–73 [DOI] [PubMed] [Google Scholar]
- 14. Mouloudi E., Protonotariou E., Zagorianou A., Iosifidis E., Karapanagiotou A., Giasnetsova T., Tsioka A., Roilides E., Sofianou D., Gritsi-Gerogianni N. (2010) Bloodstream infections caused by MBL/KPC-producing K. pneumoniae among intensive care unit patients in Greece. Risk factors for infection and impact of type of resistance on outcomes. Infect. Control Hosp. Epidemiol. 31, 1250–1256 [DOI] [PubMed] [Google Scholar]
- 15. Ke W., Bethel C. R., Thomson J. M., Bonomo R. A., van den Akker F. (2007) Crystal structure of KPC-2. Insights into carbapenemase activity in class A β-lactamases. Biochemistry 46, 5732–5740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Petrella S., Ziental-Gelus N., Mayer C., Renard M., Jarlier V., Sougakoff W. (2008) Genetic and structural insights into the dissemination potential of the extremely broad-spectrum class A β-lactamase KPC-2 identified in an Escherichia coli strain and an Enterobacter cloacae strain isolated from the same patient in France. Antimicrob. Agents Chemother. 52, 3725–3736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sougakoff W., L'Hermite G., Pernot L., Naas T., Guillet V., Nordmann P., Jarlier V., Delettré J. (2002) Structure of the imipenem-hydrolyzing class A β-lactamase SME-1 from Serratia marcescens. Acta Crystallogr. D Biol. Crystallogr. 58, 267–274 [DOI] [PubMed] [Google Scholar]
- 18. Swarén P., Maveyraud L., Raquet X., Cabantous S., Duez C., Pédelacq J. D., Mariotte-Boyer S., Mourey L., Labia R., Nicolas-Chanoine M. H., Nordmann P., Frère J. M., Samama J. P. (1998) X-ray analysis of the NMC-A β-lactamase at 1.64-A resolution, a class A carbapenemase with broad substrate specificity. J. Biol. Chem. 273, 26714–26721 [DOI] [PubMed] [Google Scholar]
- 19. Papp-Wallace K. M., Taracila M. A., Wallace C. J., Hujer K. M., Bethel C. R., Hornick J. M., Bonomo R. A. (2010) Elucidating the role of Trp105 in the KPC β-lactamase. Protein Sci. 19, 1714–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Papp-Wallace K. M., Taracila M. A., Smith K. M., Xu Y., Bonomo R. A. (2012) Understanding the molecular determinants of substrate and inhibitor specificity in the carbapenamase, KPC-2. Exploring the roles of Arg220 and Glu276. Antimicrob. Agents Chemother. 56, 4428–4438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Papp-Wallace K. M., Taracila M., Hornick J. M., Hujer A. M., Hujer K. M., Distler A. M., Endimiani A., Bonomo R. A. (2010) Substrate selectivity and a novel role in inhibitor discrimination by residue 237 in the KPC-2 β-lactamase. Antimicrob. Agents Chemother. 54, 2867–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Palzkill T., Le Q. Q., Venkatachalam K. V., LaRocco M., Ocera H. (1994) Evolution of antibiotic resistance. Several different amino acid substitutions in an active site loop alter the substrate profile of β-lactamase. Mol. Microbiol. 12, 217–229 [DOI] [PubMed] [Google Scholar]
- 23. Petrosino J. F., Palzkill T. (1996) Systematic mutagenesis of the active site Ω loop of TEM-1 β-lactamase. J. Bacteriol. 178, 1821–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Strynadka N. C., Adachi H., Jensen S. E., Johns K., Sielecki A., Betzel C., Sutoh K., James M. N. (1992) Molecular structure of the acyl-enzyme intermediate in β-lactam hydrolysis at 1.7 A resolution. Nature 359, 700–705 [DOI] [PubMed] [Google Scholar]
- 25. Zawadzke L. E., Chen C. C., Banerjee S., Li Z., Wäsch S., Kapadia G., Moult J., Herzberg O. (1996) Elimination of the hydrolytic water molecule in a class A β-lactamase mutant. Crystal structure and kinetics. Biochemistry 35, 16475–16482 [DOI] [PubMed] [Google Scholar]
- 26. Brown N. G., Shanker S., Prasad B. V., Palzkill T. (2009) Structural and biochemical evidence that a TEM-1 β-lactamase N170G active site mutant acts via substrate-assisted catalysis. J. Biol. Chem. 284, 33703–33712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bös F., Pleiss J. (2008) Conserved water molecules stabilize the Ω-loop in class A β-lactamases. Antimicrob. Agents Chemother. 52, 1072–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuzin A. P., Nukaga M., Nukaga Y., Hujer A. M., Bonomo R. A., Knox J. R. (1999) Structure of the SHV-1 β-lactamase. Biochemistry 38, 5720–5727 [DOI] [PubMed] [Google Scholar]
- 29. Raquet X., Lamotte-Brasseur J., Fonzé E., Goussard S., Courvalin P., Frère J. M. (1994) TEM β-lactamase mutants hydrolysing third-generation cephalosporins. A kinetic and molecular modelling analysis. J. Mol. Biol. 244, 625–639 [DOI] [PubMed] [Google Scholar]
- 30. Herzberg O., Kapadia G., Blanco B., Smith T. S., Coulson A. (1991) Structural basis for the inactivation of the P54 mutant of β-lactamase from Staphylococcus aureus PC1. Biochemistry 30, 9503–9509 [DOI] [PubMed] [Google Scholar]
- 31. Vakulenko S. B., Taibi-Tronche P., Tóth M., Massova I., Lerner S. A., Mobashery S. (1999) Effects on substrate profile by mutational substitutions at positions 164 and 179 of the class A TEM(pUC19) β-lactamase from Escherichia coli. J. Biol. Chem. 274, 23052–23060 [DOI] [PubMed] [Google Scholar]
- 32. Sampson J. M., Ke W., Bethel C. R., Pagadala S. R., Nottingham M. D., Bonomo R. A., Buynak J. D., van den Akker F. (2011) Ligand-dependent disorder of the Ω-loop observed in extended-spectrum SHV-type β-lactamase. Antimicrob. Agents Chemother. 55, 2303–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.(2008) Clinical and Laboratory Standards Institute Performance Standards for Antimicrobial Susceptibility Testing, Eighteenth Informational Supplement, CLSI Document M100-S18, Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 34. Fersht A. (1999) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding, 9th Ed., W.H. Freeman and Company, New York, NY [Google Scholar]
- 35. Sougakoff W., Petit A., Goussard S., Sirot D., Bure A., Courvalin P. (1989) Characterization of the plasmid genes blaT-4 and blaT-5 which encode the broad-spectrum β-lactamases TEM-4 and TEM-5 in Enterobacteriaceae. Gene 78, 339–348 [DOI] [PubMed] [Google Scholar]
- 36. Weber D. A., Sanders C. C., Bakken J. S., Quinn J. P. (1990) A novel chromosomal TEM derivative and alterations in outer membrane proteins together mediate selective ceftazidime resistance in Escherichia coli. J. Infect. Dis. 162, 460–465 [DOI] [PubMed] [Google Scholar]
- 37. Huang W., Palzkill T. (1997) A natural polymorphism in β-lactamase is a global suppressor. Proc. Natl. Acad. Sci. U.S.A. 94, 8801–8806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang X., Minasov G., Shoichet B. K. (2002) Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J. Mol. Biol. 320, 85–95 [DOI] [PubMed] [Google Scholar]
- 39. Taibi-Tronche P., Massova I., Vakulenko S. B., Lerner S. A., Mobashery S. (1996) Evidence for structural elasticity of class A β-lactamases in the course of catalytic turnover of the novel cephalosporin cefepime. J. Am. Chem. Soc. 118, 7441–7448 [Google Scholar]
- 40. Antunes N. T., Frase H., Toth M., Mobashery S., Vakulenko S. B. (2011) Resistance to the third-generation cephalosporin ceftazidime by a deacylation-deficient mutant of the TEM β-lactamase by the uncommon covalent-trapping mechanism. Biochemistry 50, 6387–6395 [DOI] [PubMed] [Google Scholar]
- 41. Kuzin A. P., Liu H., Kelly J. A., Knox J. R. (1995) Binding of cephalothin and cefotaxime to d-Ala-d-Ala-peptidase reveals a functional basis of a natural mutation in a low-affinity penicillin-binding protein and in extended-spectrum β-lactamases. Biochemistry 34, 9532–9540 [DOI] [PubMed] [Google Scholar]