

Background: Sphingosine kinase 1 is regulated by oncogenesis.
Results: K-Ras increases SK1 activity leading to increased S1P and decreased ceramide.
Conclusion: Oncogenic K-Ras regulates sphingolipid metabolism through its effect on sphingosine kinase 1.
Significance: This is the first report showing that oncogenic K-Ras regulates sphingolipid metabolism through SK1 and that this regulation is independent of phosphorylation at serine 225.
Keywords: Ceramide, ERK, Oncogene, Sphingosine 1-Phosphate, Transformation, Colony Formation, K-Ras, Senescence, Sphingosine Kinase
Abstract
Sphingosine kinase 1 (SK1) is an important enzyme involved in the production of the bioactive lipid sphingosine 1-phosphate (S1P). SK1 is overexpressed in many forms of cancer, however, the contribution of SK1 to cancer progression is still unclear. One of the best characterized mutations found in several forms of human cancer is an activating point mutation in the Ras oncogene, which disrupts its GTPase activity and leads to stimulation of the MEK/ERK pathway. Because SK1 activity and subcellular localization have been shown to be regulated by ERK, we wished to investigate the effect of oncogenic Ras, a potent activator of the Raf/MEK/ERK pathway, on the activity of SK1 and sphingolipid metabolism. Using HEK293T cells transiently transfected with the K-RasG12V oncogene and both wild type and Sphk1−/− mouse embryonic fibroblasts stably infected with retroviral K-RasG12V, we found that K-RasG12V increases the production of S1P and decreases the production of ceramide in a SK1-dependent manner. In addition, we found that expression of the K-RasG12V oncogene leads to plasma membrane localization of SK1 and a reduction in cytosolic levels of SK1. This effect is likely mediated by the Raf/MEK/ERK pathway as constitutively active B-Raf or MEK1 are able to activate SK1, but constitutively active Akt1 is not. We believe this research has important implications for how sphingolipids may be contributing to oncogenic transformation and provide some of the first evidence for oncogenes inducing specific changes in sphingolipid metabolism through SK1 regulation.
Introduction
Sphingolipids are an important class of membrane constituents that have structural and signaling roles in eukaryotic cells. For approximately two decades, ceramide, sphingosine, and sphingosine 1-phosphate (S1P)2 have been recognized as bioactive lipids that regulate proliferation, survival, and cellular differentiation. An increased cellular level of ceramide and sphingosine often correlate with, and can be the cause of, the induction of apoptosis, differentiation, or senescence (1, 2). S1P, on the other hand, promotes proliferation and survival, regulates migration, and promotes angiogenesis (3, 4). S1P can regulate cell fate through intracellular mechanisms that are beginning to be elucidated (5–9). In addition, S1P can be released from cells to exert autocrine and paracrine effects on nearby cells expressing any of the five S1P-specific G-protein coupled receptors (8). Based on many studies, a model has emerged that proposes that the relative level of ceramide, sphingosine, and S1P can determine cell fate (10).
S1P is generated within cells by the sphingosine kinase (SK) family members that utilize ATP to phosphorylate the long chain base sphingosine. The genomes of all eukaryotic species examined to date contain two SK family members encoded by two distinct genes. In humans these enzymes are referred to as SK1 and SK2. SK1 is the better characterized SK and is thought to be primarily responsible for the extracellular release of S1P; however, SK2 may also contribute to the extracellular level of S1P in vivo (11). SK1 is regulated by many extracellular agonists including growth factors and cytokines (3, 7, 12). Activation of SK1 in response to TNF-α and PMA is thought to require phosphorylation of SK1 at serine 225 by the MAP kinases ERK1/2 (13). In addition, recent studies have suggested that the myristoylated calcium-binding protein, CIB1, plays a role in regulating the membrane translocation of SK1 (14).
Several studies have identified a connection between SK1 expression in tumors and patient survival. Although not definitively established, changes in the level of the bioactive sphingolipids ceramide and S1P may contribute to the progression of cancers (15–20). For example, overexpression of SK1 is sufficient to lead to many properties of transformation such as anchorage independent growth and survival during serum starvation (21, 22). It has also been suggested that SK1 is a downstream target of the H-Ras oncogene, however, what effect this has on sphingolipid metabolism and its implications have not yet been explored (23).
Mutations in the K-Ras oncogene are among the most frequent mutations that occur in cancer. Although closely related to the H-Ras family member, K-Ras differs in its subcellular localization, post-translational modifications, and is the more commonly mutated isoform in human cancers (24). During growth factor stimulation, Ras family members exchange GTP for a bound GDP molecule and are consequently able to bind to various downstream effectors such as Raf, PI3 kinase, and other binding partners that have been less studied. Intrinsic GTPase activity in the Ras proteins leads to a self-limiting activation state. Single nucleotide changes in codons 12, 13, or 61 can result in Ras proteins that are defective in their ability to hydrolyze GTP and therefore remain in their active state in the absence of growth factor stimulation. Importantly, the expression of a mutant K-Ras construct such as K-RasG12V is sufficient to induce many of the phenotypic alterations that occur in human cancer.
In this study we set out to determine the effects of oncogenic K-Ras on sphingolipid regulation. We demonstrate that K-Ras regulates levels of bioactive sphingolipids through activation of SK1. Moreover we show that K-Ras activation of SK1 is independent of its phosphorylation at serine 225, but occurs in an ERK-dependent manner.
EXPERIMENTAL PROCEDURES
Generation of Immortalized Mouse Embryonic Fibroblasts
Mouse embryonic fibroblasts (MEFs) were generated from Sphk1+/− mice littermates in a C57BL/6J/129Sv background (11). Primary MEFs were harvested from embryonic day 12–13.5 embryos. Briefly, pregnant female mice were anesthetized using inhaled halothane prior to cervical spine dislocation. An abdominal incision was made to expose the pregnant uterus. The uterus was opened up and embryos were separated into 10-cm Petri dishes. Fetal membranes were removed from the mice embryos using forceps and scissors. Separated embryos had their heads and livers removed to avoid neural stem cell and hematological stem cell contamination and to obtain DNA for genotyping. Embryos were subsequently incubated at 37 °C in the presence of 0.25% trypsin/EDTA (Invitrogen) for 5 min in a humidified incubator. Trypsinized embryos were homogenized by pipetting up and down until a viscous fluid was obtained with few tissue clumps remaining. Homogenized embryos were allowed to sediment for 5 min, and cells sedimenting in the lower phase were collected and plated in fresh DMEM (Invitrogen) containing 10% FBS (HyClone, Logan, UT). Passage 2–3 primary Sphk1+/+ and Sphk1−/− MEFs were immortalized by retroviral infection with pBabe-hygro DN p53. Stable MEFs expressing DN p53 were selected with 150 μg/ml of hygromycin for 14 days.
Sphingosine Kinase Assay
Cells were harvested in SK1 buffer containing 20 mm Tris-HCl, pH 7.4, 1 mm EDTA, 0.5 mm deoxypyridoxine, 15 mm NaF, 1 mm β-mercaptoethanol, 1 mm sodium orthovanadate, 40 mm β-glycerophosphate, 0.4 mm phenylmethylsulfonyl fluoride, 10% glycerol, and complete protease inhibitors. 0.1% Triton X-100 was added for SK1 activity, and 1 m KCl was added for SK2 activity. MEFs were sonicated twice for 30 s and protein concentrations were determined by BCA assay (Pierce location), 30 μg of sample protein was incubated in 90 μl of reaction mixture with 100 mm sphingosine delivered in 4 mg/ml of fatty acid-free bovine serum albumin, [32P]ATP (5 μCi, 1 mm dissolved in 10 mm MgCl2), and SK1 buffer for 30 min at of 37 °C. The reaction was terminated by the addition of 10 μl of 1 n HCl and 400 μl of chloroform/methanol/HCl (100:200:1, v/v/v). Subsequently, 120 μl of chloroform and 120 μl of 2 m KCl were added, and samples were centrifuged at 3000 × g for 5 min. Then, 200 μl of the organic phase was transferred to new glass tubes and dried. Samples were resuspended in 40 ml of chloroform/methanol/HCl (100:100:1, v/v/v). 20 μl of resuspended samples were then resolved on silica thin layer chromatography plates using 1-butanol/methanol/acetic acid/water (8:2:1:2, v/v/v/v) as a solvent system and visualized by autoradiography. The radioactive spots corresponding to S1P were scraped from the plates and counted for radioactivity in a scintillation counter. Background values were determined in negative controls in which sphingosine was not added to the reaction mixture.
Sphingolipidomic Analysis and C17-Sphingosine Labeling
Adherent cells were washed twice with cold PBS and then collected in cold PBS. A small aliquot was taken for protein concentration determination. Samples were then immediately snap-frozen in a dry ice/methanol bath and frozen at −80 °C prior to submission for sphingolipidomic analysis. Lipids were extracted twice using a 2 ml of ethyl acetate/propan-2-ol/water (60:30:10, by volume) solvent system, dried under a stream of nitrogen, and re-suspended into 150 μl of 1 mm ammonium formate in 0.2% formic acid in methanol. Sphingolipid mass levels were determined by electrospray ionization-mass spectrometry (ESI/MS/MS). Analysis of ceramides, sphingoid bases, and sphingomyelins was performed on a Thermo Finnigan TSQ 7000 triple quadrupole mass spectrometer, operating in a multiple reaction-monitoring positive ionization mode, as described previously (25, 26). For C17-sphingosine labeling, 1 μm C17 sphingosine (1 mm stock in ethanol) was added to the medium for 30 min. Cells were washed two times with PBS and collected in PBS. An aliquot was taken for protein concentration determination and snap-frozen in dry ice/methanol and stored at −80 °C until analysis. Analysis was performed as described previously (27).
Real Time Quantitative PCR
RNA was homogenized and extracted from cultured cells or mouse tissue using QIAShredder and RNeasy kits (Qiagen, Valencia, CA), respectively. cDNA was synthesized from 1.0 mg of RNA using Oligo(dT) primers (Invitrogen) using SuperScript II Reverse Transcriptase (Invitrogen) according to the manufacturer's instructions. The standard RT-PCR included 12.5 ml of SYBR Green (Bio-Rad), 0.5 ml of 10 mm forward primer, 0.5 ml of 10 mm reverse primer, and 1.0 ml of cDNA adjusted to a final volume of 25.0 ml per well. The RT-PCR was performed using an iCycler (Bio-Rad) as follows: 3 min at 95 °C, followed by cycles (n = 40) consisting of a 10-s melt at 95 °C, a 45-s annealing step at 60 °C, and an extension step of 45 s at 68 °C. All reactions were performed in triplicate. Primers were designed using Beacon Primer Design Software and purchased from Integrated DNA Technologies (Coralville, IA). Primers user for real-time PCR are as follows: forward human SK1, 5′-CTGGCAGCTTCCTTGAACCAT-3′; reverse human SK1, 5′-TGTGCAGAGACAGCAGGTTCA-3′; forward human SK2, 5′-CCAGTGTTGGAGAGCTGAAGGT-3′; reverse human SK2, 5′-GTCCATTCATCTGCTGGTCCTC-3′; forward human β-actin, 5′-ATTGGCAATGAGCGGTTCC-3′; reverse human β-actin, 5′-GGTAGTTTCGTGGATGCCACA-3′. Threshold cycle (Ct) values for target genes were normalized to the reference gene using QGene software (Bio-Rad) to determine mean normalized expression.
Immunoblotting
Protein samples were separated on 4–20% gradient gels (Criterion; Bio-Rad) at 60 to 100 V before transferring to nitrocellulose membrane in Tris glycine buffer (100 V, 30 min, 4 °C). Membranes were blocked in 5% milk dissolved in PBS containing 0.1% Tween (PBST) for 30 min. Blots were probed with primary antibody in 5% dry milk dissolved in PBST overnight at 4 °C. Membranes were washed four times with 0.1% PBST. Membranes were subsequently probed with horseradish peroxidase-conjugated secondary antibody 1:5000 mouse, goat, or rabbit in 5% nonfat dry milk PBST for 1 h at room temperature. Blots were washed four times with PBST. Proteins were visualized by enhanced chemiluminescence (Pierce).
Subcellular Fractionation
Whole cell lysates were fractionated into cytosolic and membrane components as previously described (28). Briefly, cells were washed in PBS and collected in a SK activity fractionation buffer containing 20 mm Tris, pH 7.5, 10 mm EDTA, 2 mm EGTA, 250 mm sucrose, 1 mm phenylmethylsulfonyl fluoride, 1 mm dithiothreitol, 5 mm NaF, 1 mm Na3VO4, and 0.5 mm 4-deoxypyridoxine. Samples were sonicated 3 times for 30 s. Unbroken cells were separated by centrifuging at 1,000 × g and collecting the supernatant. The supernatant of disrupted cells was subject to centrifugation at 100,000 × g for 1 h. The supernatant was separated, and the pellet was resuspended in SK activity fractionation buffer as above, but also containing 0.8% Triton X-100.
Immunofluorescence and Confocal Microscopy
MEFs were seeded in 35-mm glass bottom MatTek confocal dishes. Media was aspirated and washed twice with 2 ml of PBS. MEFs were fixed with 3.7% formaldehyde in PBS for 20 min at room temperature followed by two washes with 2 ml of PBS. Fixed cells were permeabilized with 0.1% Triton X-100 for 10 min at room temperature. Samples were blocked with 2% human serum for 30 min. Samples were then stained with monoclonal M2 anti-FLAG antibody (1:1000) (Sigma) in 2% human serum for 2 h. Dishes were washed 4 times with 2 ml of PBS. Alexa Fluor 488-conjugated anti-mouse antibody (1:1000) (Abnova, Littleton, CA) was added in 2% human serum and incubated at room temperature for 1 h. Dishes were washed 4 times with PBS. Following washes with PBS, MatTek dishes were stored at 4 °C prior to image collection. Fixed and stained samples were viewed on an LSM 510 Meta Confocal Microscope (Carl Zeiss, Thornwood, NY). Each microscopic image is representative of 10–20 fields over three experiments, and all images were taken at the equatorial plane of the cell. Raw data images were cropped in Adobe Photoshop CS2® for publication.
Statistical Analysis
GraphPad Prism software was used for all statistical analysis. Comparisons between two groups were analyzed by Student's t test. A Bonferroni correction was performed to correct for multiple comparisons.
RESULTS
Oncogenic K-RasG12V Decreases C16-Ceramide and Increases S1P Levels in Cells
First, we set out to determine whether the expression of oncogenic K-RasG12V could regulate bioactive sphingolipid levels or alter sphingolipid metabolism. To test this, we transfected HEK293T cells with K-RasG12V or empty vector and quantified the amount of S1P, ceramide, and dihydroceramide using a modified Bligh and Dyer extraction followed by HPLC-tandem mass spectrometry analysis. The results showed that HEK293T cells expressing oncogenic K-RasG12V had a 2-fold increase in endogenous S1P (Fig. 1A) and a 30% decrease in the level of endogenous C16-ceramide (Fig. 1B) when compared with vector-transfected cells. Interestingly, the very long chain C24 and C24:1 ceramide were not altered by the expression of K-RasG12V in a statistically significant manner (Fig. 1B). Sphingosine levels were not statistically different between the vector-transfected cells and K-RasG12V-transfected cells (Fig. 1C). Altogether, these data suggest that oncogenic K-Ras modulates bioactive sphingolipid levels toward a pro-proliferative profile through an increase in S1P and a decrease in ceramide levels.
FIGURE 1.
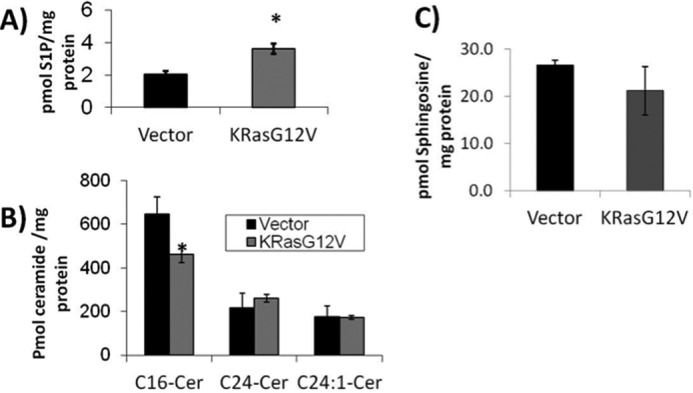
Oncogenic K-RasG12V increases intracellular S1P and decreases C16-ceramide. HEK293T cells were transfected with empty vector or K-RasG12V. 48 h post-transfection cells were collected and analyzed for (A) S1P, (B) ceramide, and (C) sphingosine content using HPLC coupled tandem mass spectrometry (n = 6). Error bars represent S.D. *, denotes p < 0.05.
K-RasG12V Enhances the Production of Both Intracellular and Extracellular Sphingosine 1-Phosphate and Decreases Ceramide Production
Next we wanted to probe how the oncogene K-RasG12V regulates sphingolipid metabolism to alter the levels of these two bioactive sphingolipids. We and others have established a method for evaluating sphingolipid metabolism using a natural but very low abundance sphingosine containing only 17 carbons as a label (C17-sphingosine) (27). Vector or K-RasG12V-transfected HEK293T were labeled with 1 mm C17-sphingosine for 30 min. Cells and media were collected and ceramide, sphingosine, and sphingosine 1-phosphate containing the C17-sphingosine backbone were quantified in each sample using tandem mass spectrometry coupled to HPLC. We found that K-RasG12V-transfected cells had a ∼1.8-fold increase in both intracellular and extracellular C17-S1P (supplemental Figs. S1A and S2B, respectively), suggesting that K-RasG12V expression leads to an increase in S1P production with subsequent release. In addition, K-RasG12V-transfected cells had a decreased amount of the C17-sphingosine label incorporated into all ceramide species (supplemental Fig. S1B), suggesting that the presence of K-RasG12V leads to preferential incorporation of sphingosine into S1P rather than ceramide. Interestingly, the level of intracellular C17-sphingosine was increased when oncogenic K-RasG12V was expressed (supplemental Fig. S1C). The increase in intracellular C17-sphingosine is unlikely to be due to an increase in uptake of exogenous C17-sphingosine label because the extracellular levels of C17-sphingosine (supplemental Fig. S2A) were similar in the vector-transfected cells. Rather, it is possible that the increase in C17-sphingosine may be due to its increased phosphorylation/dephosphorylation by activation of SK, which would be consistent with reports in the literature on activation of SK by H-Ras (23).
Oncogenic K-RasG12V Regulates Sphingosine Kinase 1 Activity
As mentioned above, previous reports have suggested a link between SK activity and oncogenic H-Ras (23). Therefore, we next elected to test if oncogenic K-RasG12V affected SK1 and/or SK2 activity. HEK293T cells were transfected with either empty vector (pcDNA3) or oncogenic K-RasG12V, and SK activity was measured. As early as 48 h post-transfection, it was observed that expression of oncogenic K-RasG12V increased SK1 activity by ∼2.6-fold over vector-transfected cells, but had no effect on SK2 activity (Fig. 2A).
FIGURE 2.
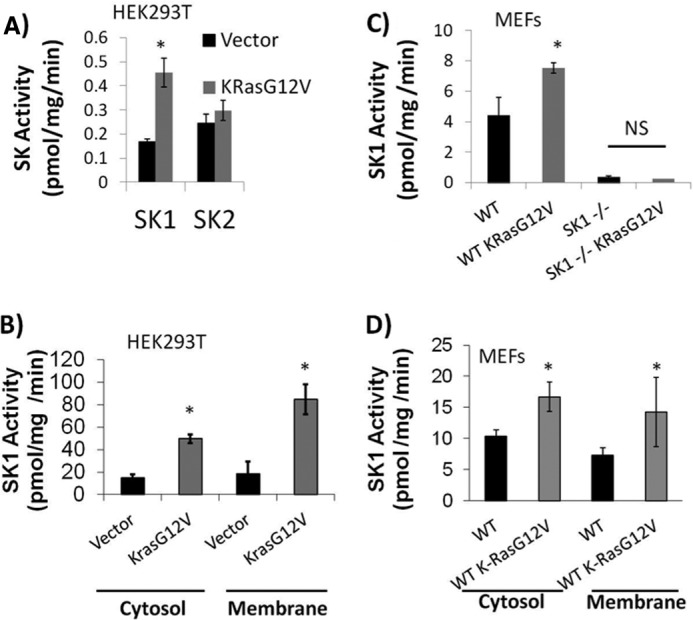
Oncogenic K-RasG12V increases SK1 activity, but not SK2 activity. A, HEK293T cells were transfected with either empty pcDNA3 (vector) or pcDNA3 expressing K-RasG12V. 48 h post-transfection cells were collected and assayed for sphingosine kinase activity in the presence of either 0.5% Triton X-100 (SK1 activity on the left) or 1 m KCl (SK2 activity on the right). B, HEK293T cells were transfected with empty vector or oncogenic K-RasG12V. 48 h post-transfection cells were sonicated followed by centrifugation at 1,000 × g. Supernatants were collected and centrifuged at 100,000 × g for 60 min. Both the pellet (membrane) and supernatant (cytosol) were collected and an SK1 assay was performed (n = 6). Error bars represent S.D. *, denotes p < 0.05. C, immortalized WT or Sphk1−/− mouse embryonic fibroblasts were infected with either empty pBABE retroviral vector or retrovirus expressing K-RasG12V. Infected cells were selected with puromycin. After 1 week of selection, MEFs were passaged and lysates were collected. An SK1 assay was performed as described under “Experimental Procedures.” Error bars represent S.D. *, denotes p < 0.01 (n = 6). D, WT MEFs were infected with retrovirus expressing either empty vector or K-RasG12V. Stable retrovirus-infected cells were selected for with puromycin. MEFs were grown in 10% FBS and collected in PBS. Cells were sonicated followed by centrifugation at 1,000 × g. Supernatants were collected and centrifuged at 100,000 × g for 60 min. Both the pellet (membrane) and supernatant (cytosol) were collected and an SK1 assay was performed (n = 6). Error bars represent S.D. *, denotes p < 0.05. NS, not statistically significant.
To confirm that SK1, and not SK2, was necessary for the change in SK activity observed with oncogenic K-RasG12V, we infected WT or Sphk1−/− MEFs with either retroviruses expressing empty vector or oncogenic K-RasG12V. K-RasG12V-infected WT MEFs had a ∼1.7-fold increase in SK1 activity over vector-infected cells. This effect was not observed in Sphk1−/− MEFs suggesting that the observed effect was not due to background SK2 activity (Fig. 2C).
Activation of SK1 is often accompanied by a translocation of SK1 from the cytosolic compartment to the membrane compartment (21, 28). Therefore, we wanted to test if the presence of oncogenic K-RasG12V alters the relative amount of SK1 activity present in the membrane and cytosolic subcellular fractions. Vector and K-RasG12V-transfected HEK293T cells were harvested, and cytosolic and membrane fractions were obtained as described under “Experimental Procedures.” The fractions were normalized to equal protein and were assayed for SK1 activity in the presence of 0.5% Triton X-100. The results showed that in HEK293T cells expression of K-RasG12V led to an ∼3.5-fold increase in cytosolic SK1 activity and ∼4.3-fold increase in membrane SK1 activity when compared with empty vector expression alone (Fig. 2B). The results also showed that in MEFs, SK1 activity increased in the cytosolic and membrane fractions by ∼1.6- and ∼1.9-fold, respectively, when K-RasG12V was expressed (Fig. 2D). This profile of SK1 activation by K-RasG12V appears to be similar to what occurs by other known SK activators (13, 28, 29).
SK1 Message Is Not Up-regulated by K-RasG12V
Increases in SK1 mRNA have been reported in many human cancers, however, no relationship between oncogenic Ras and SK1 message levels in cancer has been shown (30). In addition, SK1 has been demonstrated to be transcriptionally regulated in response to PMA through the AP-2 and Sp1 transcription factors (31). In addition, v-Src has been shown to regulate AU-rich proteins to alter SK1 mRNA stability (32). Therefore, it became important to evaluate if oncogenic Ras could also regulate SK1 message levels. RNA was collected from HEK293T cells 48 h after being transfected with either empty vector or K-RasG12V. Expression of SK1 and SK2 were quantified using quantitative real time PCR. Actin-specific primers were used as standards to normalize the samples. The results showed that neither SK1 nor SK2 mRNA levels in the K-RasG12V-transfected cells were significantly different from those in vector-transfected cells (Fig. 3). This suggests that the increase in SK1 activity in response to oncogenic K-RasG12V is likely to be independent of transcription or message stabilization.
FIGURE 3.
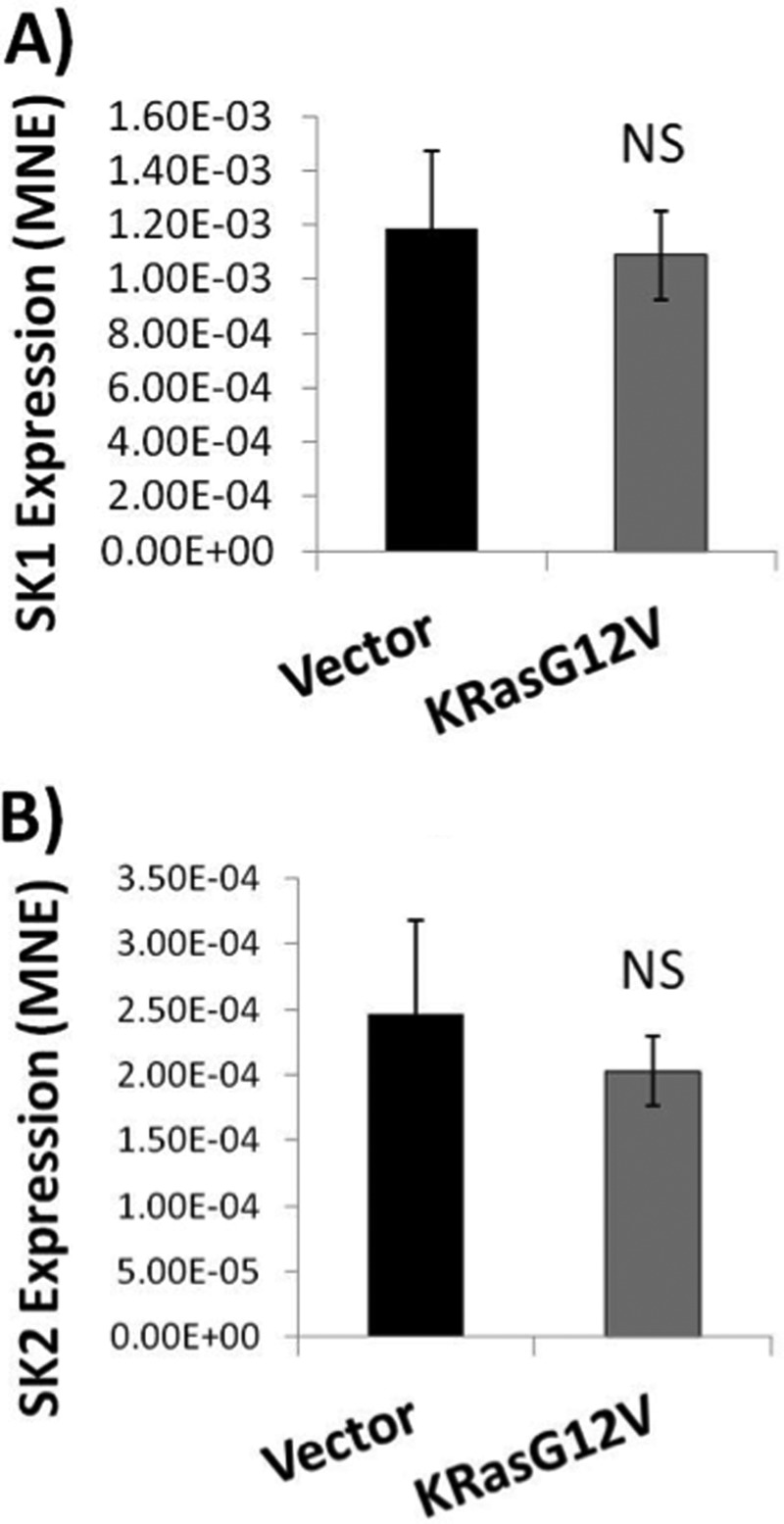
Oncogenic K-RasG12V does not increase SK1 or SK2 mRNA expression. HEK293T cells were plated and transfected with either pcDNA3 (Vector) or pcDNA3 expressing K-RasG12V. 48 h post-transfection cells were collected for RNA isolation. cDNA was created and real time PCR was performed using SK1, SK2, and β-actin specific primers. Both (A) SK1 and (B) SK2 mRNA levels were normalized to β-actin expression (n = 6). Error bars represent S.D. NS denotes p > 0.05.
K-RasG12V Alters Sphingosine Incorporation from Ceramide to S1P in a SK1-dependent Manner
To assess whether or not SK1 was necessary for changes in sphingolipid metabolism observed with oncogenic K-RasG12V, we infected WT, Sphk1−/−, and Sphk2−/− MEFs with either retroviruses expressing empty vector or oncogenic K-RasG12V. MEFs were labeled with C17-sphingosine for 30 min and their C17-labeled sphingolipid content was analyzed. Oncogenic K-RasG12V increased the amount of label incorporated into C17-S1P by ∼1.5-fold in the WT and Sphk2−/− MEFs, but not the Sphk1−/− MEFs suggesting that oncogenic K-RasG12V requires SK1 for the increase in S1P produced (Fig. 4A). In addition, the Sphk1−/− and Sphk2−/− MEFs had a higher level of C17 label incorporated into ceramide when compared with WT MEFs (Fig. 4B). Oncogenic K-RasG12V decreased the level of C17-ceramide in both the WT and Sphk2−/− MEFs, but not the Sphk1−/− MEFs. Interestingly, this effect was more dramatic in the Sphk2−/− MEFs compared with WT MEFs suggesting that K-RasG12V can reverse the increased incorporation of sphingosine into ceramide observed at baseline in the Sphk2−/− MEFs. Therefore, the down-regulation of ceramide and induction of S1P induced by K-RasG12V appears to be dependent primarily on SK1.
FIGURE 4.
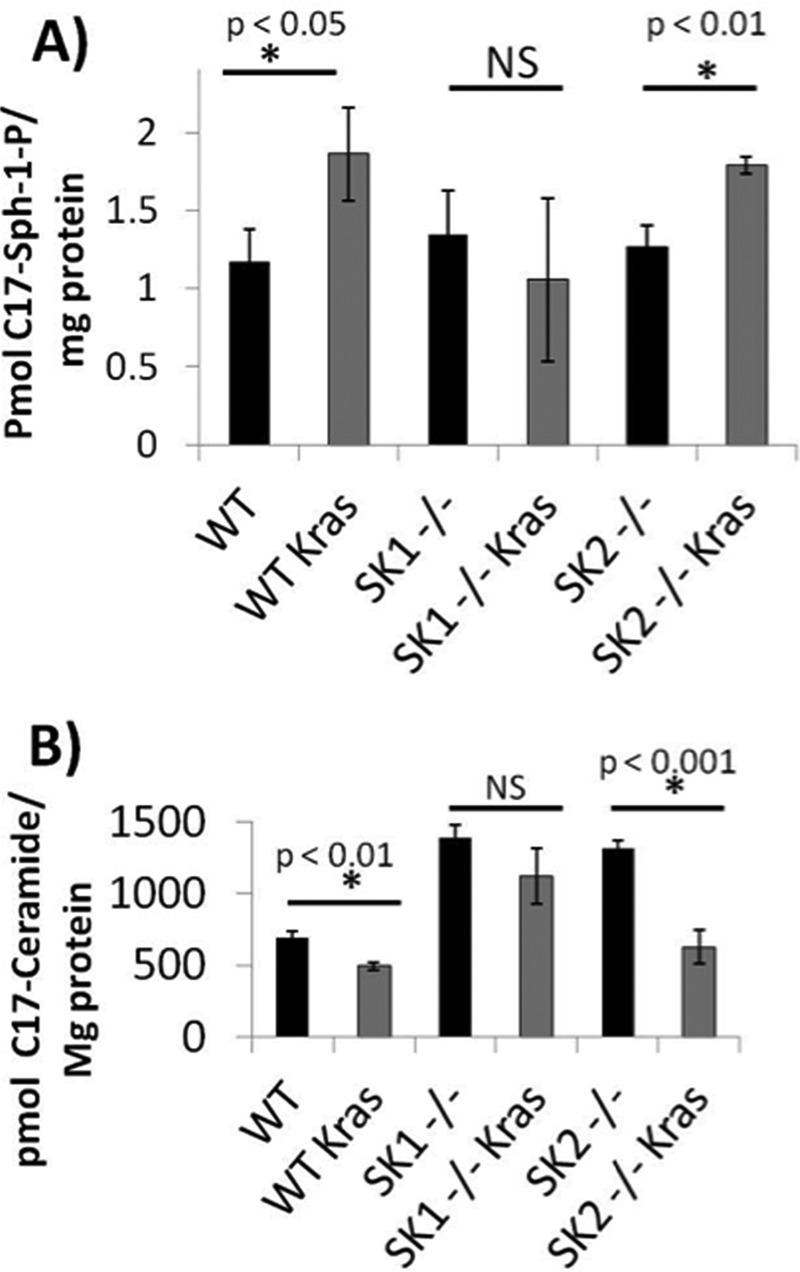
Oncogenic K-RasG12V increases S1P production and decreases ceramide production in a SK1-dependent manner. Wild type, Sphk1−/−, or Sphk2−/− MEFs were isolated and immortalized with dominant-negative p53. MEFs were then infected with retroviruses containing either pBABE empty vector or pBABE expressing K-RasG12V. Vector or K-RasG12V expressing MEFs were plated in 10-cm dishes and labeled with 1 μm C17-sphingosine for 30 min. The media was aspirated and cells were washed with cold PBS. Lipids were extracted and C17-labeled sphingolipids were analyzed using LC-MS-mass spectrometry. A, C17-S1P; B, C17-ceramide. *, denotes statistical significance compared with vector using Student's t test; NS denotes not statistically significant p > 0.05.
Requirement for SK1 Activity but Not Phosphorylation for Mediating the Metabolic Effects of K-RasG12V on Sphingolipid Metabolism
Because H-Ras has been shown to induce phosphorylation of SK1, it became important to determine whether a similar phosphorylation plays a role in activation of SK1 in response to K-Ras. For these experiments, Sphk1−/− MEFs were reconstituted with wild type, phosphorylation-defective (S225A), or a kinase-dead SK1, and their ability to rescue the metabolic properties was evaluated. To test this, we developed retroviral constructs expressing FLAG-SK1, FLAG-SK1S225A, or FLAG-SK1G82D using a pBABE-Puro plasmid as the vector. After stable selection, MEFs were labeled with C17-sphingosine for 30 min, and C17-containing sphingolipids were analyzed in both the cells and media. Overexpression of wild type SK1 or phosphorylation-defective SK1, but not kinase-dead SK1, resulted in an increase in extracellular C17-labeled S1P when K-RasG12V was co-expressed (Fig. 5A). C17-labeled intracellular S1P was increased in wild type MEFs, but not Sphk1−/− MEFs when K-RasG12V was expressed. This defect was reversed with the expression of wild type SK1 or phosphorylation-defective SK1, but not catalytically inactive SK1 (Fig. 5B). In addition, overexpression of wild type or S225A SK1, but not catalytically inactive SK1 lead to a restoration of K-RasG12V-dependent decreases in ceramide (Fig. 5C). Altogether, these data suggest that SK1 activity is required for K-Ras-induced regulation of metabolism of bioactive sphingolipid. Interestingly, however, and in contrast to results with H-Ras, the current results show that this requirement appears independent of serine 225 phosphorylation. To test this, we expressed either retroviral wild type FLAG-SK1 or FLAG-SK1S225A in Sphk1−/− MEFs. Stable FLAG-SK1 or FLAG-SK1S225A expressing MEFs were then infected with either empty vector or K-RasG12V expressing retroviruses. The results showed that the FLAG-SK1 and FLAG-SK1S225A expressing cell lines both had similar enzyme activities and that K-RasG12V expressing cell lines both had an ∼2–3-fold increase in SK1 activity compared with vector alone (Fig. 6A). In addition, the expression of FLAG-SK1 was similar to that of FLAG-SK1S225A in both vector and K-RasG12V expressing cell lines (Fig. 6B, top left panel).
FIGURE 5.

Oncogenic K-RasG12V increases intracellular and extracellular S1P and decreases ceramide in a SK1-dependent manner independent of serine 225. Sphk1−/− MEFs were infected with retrovirus containing pBABE expressing FLAG-SK1, FLAG-SK1 S225A, or FLAG-SK1G82D. After selection with puromycin, MEFs were infected with retrovirus containing either pBABE or pBABE expressing K-RasG12V. Vector or K-RasG12V expressing MEFs were plated in 10-cm dishes and labeled with 1 mm C17-sphingosine for 30 min. The media were collected and cells were washed with cold PBS. Lipids were extracted from both cells and media and C17-labeled sphingolipids were analyzed using LC-MS-mass spectrometry. A, extracellular C17-sphingosine 1-phosphate; B, intracellular C17-S1P; and C, C17-ceramide. *, denotes statistical significance compared with vector using Student's t test; NS denotes not statistically significant p > 0.05.
FIGURE 6.
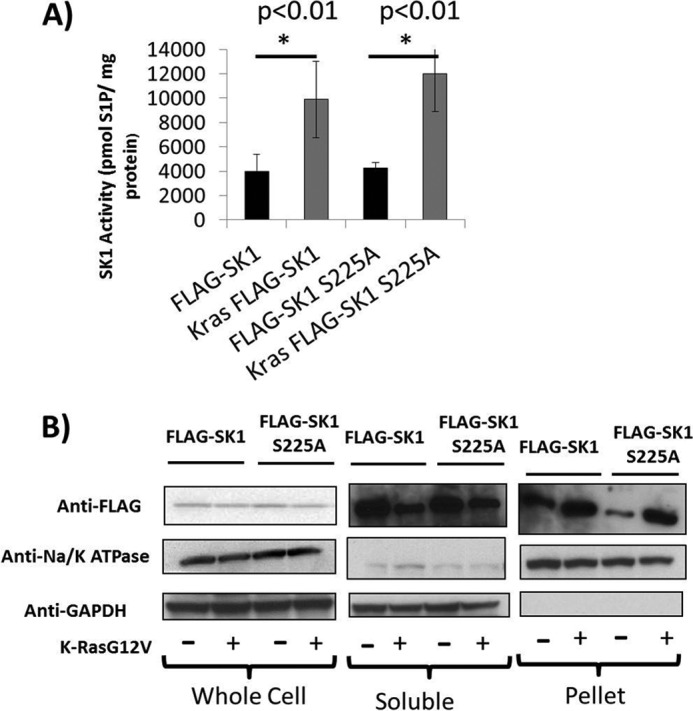
K-RasG12V activates and induces the membrane localization of both SK1 and SK1S225A. Sphk1−/− MEFs were infected with retrovirus containing either pBABE expressing FLAG-SK1 or FLAG-SK1 S225A. After selection with puromycin, MEFs were infected with retrovirus containing either pBABE or pBABE expressing K-RasG12V. MEFs were plated and collected for (A) SK1 activity or (B) subcellular fractionation. Whole cell lysates were separated into whole cell, soluble, and pellet fractions as described under “Experimental Procedures.” All samples were normalized for total protein. 30 mg of total protein from each sample was analyzed using SDS-PAGE. Membranes were probed with anti-FLAG to assess exogenously expressed SK1 localization. GAPDH and Na+/K+-ATPase probes were used to assess equal loading and to determine the quality of the fractionation (n = 3). Error bars represent S.D. in panel A.
K-RasG12V Induces SK1 to Localize to the Plasma Membrane
Previous studies have shown that SK1 localizes to the plasma membrane in a serine 225-dependent manner. Therefore, it became important to test if SK1 was being translocated to the plasma membrane from the cytosolic fraction in response to oncogenic K-RasG12V and if this also required phosphorylation of SK1 on Ser-225. The results showed that in both the FLAG-SK1 and FLAG-SK1S225A expressing MEFs that K-RasG12V induced an increase in FLAG-SK1 in the membrane fraction (Fig. 6B, upper right panel) and a decrease in the cytosolic fraction (Fig. 6B, upper middle panel) despite the total level of SK1 remaining the same (Fig. 6B, top left panel). GAPDH and Na/K-ATPase were used as normalization markers for cytosolic and membrane fractions, respectively. We next wished to visualize which membrane compartment SK1 was translocating to in response to K-RasG12V. Sphk1−/− MEFs expressing vector, FLAG-SK1, or FLAG-SK1 S225A with or without K-RasG12V were fixed and stained for anti-FLAG followed by an Alexa 488-conjugated anti-mouse antibody. The results showed that oncogenic K-RasG12V induced a significant increase in the expression of SK1 specifically on the plasma membrane (Fig. 7). This effect was observed with wild type SK1 (Fig. 7, middle panels), phosphorylation-defective SK1 (Fig. 7, right panels), or kinase-dead SK1 (not shown). Therefore, K-Ras appears to activate SK1 by a mechanism involving translocation to the plasma membrane even in the absence of SK1 kinase activity or phosphorylation of serine 225.
FIGURE 7.

Oncogenic K-RasG12V induces SK1 to localize to the plasma membrane. Sphk1−/− MEFs were infected with retrovirus containing either pBABE or pBABE expressing FLAG-SK1 or FLAG-SK1 S225A. After selection with puromycin, vector, FLAG-SK1, or FLAG-SK1S225A expressing MEFs were infected with retrovirus containing either pBABE or pBABE expressing K-RasG12V. MEFs were fixed and stained with anti-FLAG antibody followed by Alexa 488-conjugated secondary antibody for visualization using confocal microscopy. Representative cells of >90% of the cell populations on each dish are shown in compilation images (n = 3).
Requirement for the ERK Pathway, but Not Akt in Activation of SK1
Two of the major downstream targets of activated K-RasG12 are Raf and PI3K. Therefore, it became important to determine the signaling pathway by which oncogenic K-Ras activates SK1. For these experiments, the effects of constitutively active components of the MAP kinase pathway or activated Akt can activate SK1. HEK293T cells were transfected with empty vector, oncogenic K-RasG12V, active B-RafV600E, constitutively active MEK1-DD, or myristoylated Akt1. The results showed that the expression of K-RasG12V strongly activated SK1 (Fig. 8, second lane). In addition transfection of HEK293 cells with either constitutively active B-RafV600E (Fig. 8, third lane) or MEK1-DD also activated SK1 (Fig. 8, fourth lane), but to a lesser degree than oncogenic K-RasG12V (Fig. 8, second lane). Myristoylated Akt1 had no effect on SK1 activity, suggesting that K-RasG12V does not activate SK1 through Akt1 (Fig. 8, fifth lane).
FIGURE 8.
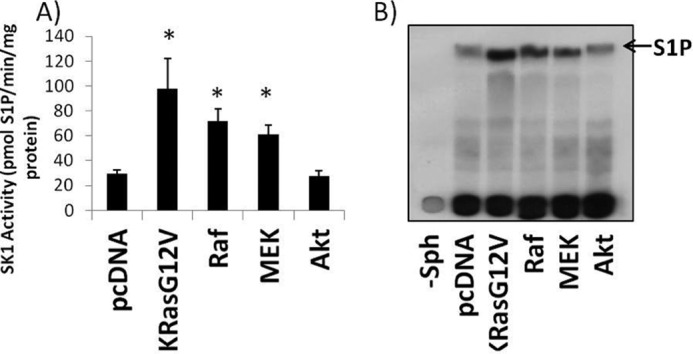
Oncogenic K-RasG12V, B-RafV600E, or MEK1-DD increase SK1 activity, but myr-Akt1 does not affect SK1 activity. HEK293T cells were plated and transfected with vector (pcDNA), K-RasG12V, B-RafV600E, MEK1-DD, or myristoylated Akt1. 48 h post-transfection cells were collected and assayed for sphingosine kinase activity in the presence of 0.5% Triton X-100. Briefly, 30 mg of protein was assayed per sample in the presence of 32P-labeled 20 mm ATP and 50 mm sphingosine in 0.4% BSA for 30 min at 37 °C. A, scintillation counter quantification of the S1P band from 6 independent experiments. Error bars on represent S.D. *, denotes p < 0.01 when compared with pcDNA. B, representative radiograph. Far left lane (−Sph) is pcDNA without the addition of sphingosine to the reaction.
Activation of SK1 by K-RasG12V Requires ERK
Because phosphorylation of SK1 at serine 225 was not required for SK1 activation by oncogenic K-Ras, we wanted to test if ERK itself was required for activation. To address this, HEK293T cells were transfected with oncogenic K-RasG12V or constitutively active MEK in conjunction with empty vector, dominant-negative MEK, or dominant-negative ERK. The results showed that the co-expression of dominant-negative MEK or ERK inhibited the activation of SK1 by K-RasG12V (Fig. 9). In addition, dominant-negative ERK inhibited the activation of SK1 by a constitutively active MEK1 (Fig. 9). These data suggest that ERK is indeed required for the activation of SK1 in response to both oncogenic K-RasG12V and by constitutively active MEK1. K-RasG12V activated SK1 to a greater extent than MEK1-DD, suggesting that an additional signaling pathway may contribute to SK1 activation, although to a minor extent as compared with the MAP kinase pathway.
FIGURE 9.
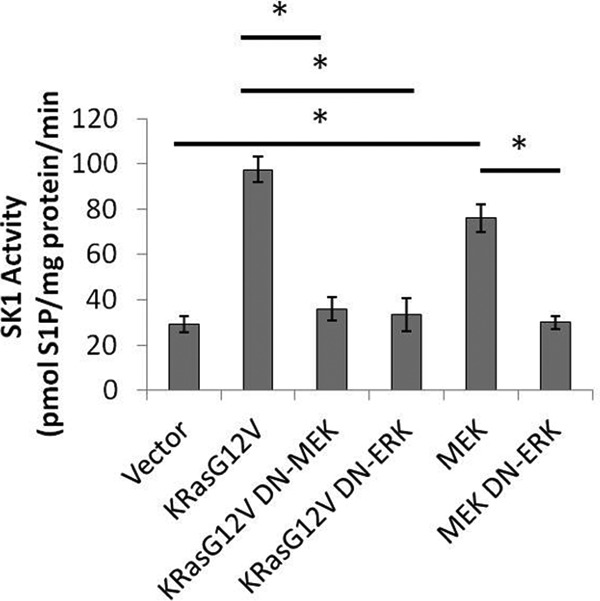
Activation of SK1 by K-RasG12V is dependent on ERK. HEK293T cells were plated in 60-mm dishes. Cells were transfected with empty vector, K-RasG12V, or constitutively active MEK1-DD along with either empty vector, DN-MEK, or DN-ERK. 48 h post-transfection cells were collected and assayed for sphingosine kinase activity in the presence of 0.5% Triton X-100. Briefly, 30 mg of protein was assayed per sample in the presence of 32P-labeled 20 mm ATP and 50 mm sphingosine in 0.4% BSA for 30 min at 37 °C (n = 3). Error bars represent S.D. *, denotes p < 0.01 when compared with vector.
DISCUSSION
The results from this study show that SK1 is a downstream effector of oncogenic K-RasG12V and that expression of oncogenic K-RasG12V influences sphingolipid metabolism primarily through the regulation of SK1. Activation of SK1 by K-RasG12V leads to an increase in both the intracellular and extracellular levels of S1P and a decrease in the intracellular level of ceramide. Using wild type, Sphk1−/−, and Sphk2−/− MEFs we were able to show that this occurs in a SK1-dependent manner.
The results from this study clearly show that activation and translocation of SK1 by oncogenic K-RasG12V is independent of phosphorylation of SK1 at serine 225 because a SK1 construct containing a serine to alanine mutant at position 225 becomes equally localized to the plasma membrane and is similarly activated by oncogenic K-RasG12V. Although the results show that phosphorylation at serine 225 is not necessary for activation by K-RasG12V, they do show that K-RasG12V-dependent activation of SK1 requires the MAP kinase, ERK1/2, as both dominant-negative ERK or dominant-negative MEK inhibited the activation of SK1 by K-RasG12V. In addition, activation of SK1 by constitutively active MEK was also inhibited by a dominant-negative ERK construct. This is in agreement with previous reports suggesting that ERK2 is required for SK1 activation in response to TNF-α or PMA treatment (13, 21). Although myristoylated Akt1 failed to activate SK1 in our system, we cannot rule out PI3K as an important regulator of SK1 activity. Early studies involved with biochemical characterization of SK1 showed a positive effect of phosphatidylinositol on purified rat SK1 activity (43). It is unclear if phosphatidylinositol or phosphatidylinositol polyphosphate have an effect on SK activity in vivo.
Several studies have explored the mechanism of activation of SK1 in response to different extracellular agonists. In HEK293 cells, TNF-α or PMA activate SK1 in an ERK-dependent manner due to its ability to phosphorylate SK1 at serine 225. Activation of SK1 in this manner induces the plasma membrane localization of SK1 leading to an increase in intracellular and extracellular S1P production (28). In a more recent study, localization of SK1 to the plasma membrane was shown to require the calcium-binding protein CIB1, suggesting that an acute calcium signal and phosphorylation at serine 225 are both required for the translocation of SK1 to the plasma membrane (14). The connection between serine 225 and CIB1-dependent plasma membrane localization remains unclear because wild type SK1 and SK1 S225A are able to bind to CIB1 equally well (14). Therefore, the mechanistic basis by which phosphorylation of SK1 at serine 225 regulates the activation and plasma membrane localization of SK1 remains unclear. One hypothesis is that phosphorylation of SK1 at serine 225 enhances its affinity for anionic phospholipids such as phosphatidylserine, which are abundant at the plasma membrane (35). In this study, we find that SK1 can localize to the plasma membrane in the absence of this phosphorylation site suggesting that there are alternative mechanisms by which SK1 can localize to the plasma membrane. It is possible that an alternative phosphorylation site exists on SK1, however, there are no alternative predicted ERK sites on SK1 and therefore ERK may activate SK1 through an indirect mechanism.
In 2000, Xia et al. (23) showed that oncogenic H-RasG12V activates SK1 and that a dominant-negative SK1, SK1G82D, could partially inhibit anchorage independent growth induced by H-RasG12V. Because K-Ras is the most commonly mutated isoform of Ras in human cancers, we wanted to test if the oncogene K-RasG12V could activate SK1 and to determine the consequences of this activation on regulation of bioactive sphingolipids, the latter being a crucial and unexplored area as a potential downstream effector of oncogenic Ras. Because SK1 regulates the production of S1P and the catabolism of ceramide, it has been hypothesized that SK1 may serve as a critical enzyme for regulating the levels of ceramide and sphingosine 1-phosphate; however, this hypothesis has largely been untested in the context of a direct effect of oncogenes. In this study we found that the expression of K-RasG12V leads to an increase in S1P production both intracellularly and in the extracellular environment. In addition, we found that intracellular ceramide is decreased and that the production of ceramide from exogenous sphingosine is reduced. Interestingly, we found that the level of sphingomyelin in K-RasG12V-transformed cells is also decreased, suggesting that K-RasG12V may accelerate sphingomyelin catabolism or inhibit the production of sphingomyelin (data not shown). Because the level of C16-dihydroceramide is actually increased in transformed cells, it is likely that de novo synthesis of sphingolipids is not inhibited by K-RasG12V, but rather catabolism is increased. We cannot, however, rule out that K-RasG12V also has an inhibitory effect on one or more ceramide synthase or sphingomyelin synthase. Further exploration into the specific effects the K-RasG12V oncogene has on a broader selection of sphingolipid metabolizing enzymes will be required to understand how global sphingolipid metabolism is affected in cancer cells. Importantly, this is one of the first studies to identify a mechanism by which the activation of an oncogene can have a direct influence on sphingolipid metabolism.
SK1 has been considered a potential therapeutic target for cancer therapy because it controls the production of the growth-promoting lipid S1P and the catabolism of the pro-apoptotic lipid ceramide. Therefore, SK1 lies at a critical juncture in the regulation of sphingolipid metabolism. A clear connection between oncogenes that become activated through genomic mutations and an alteration in sphingolipid metabolism has not been previously established. In this study we have provided a mechanism by which an oncogene may activate SK1 and alter sphingolipid metabolism. This study may have a significant impact specifically in types of cancer that commonly harbor K-Ras mutations such as colon cancer, non-small cell lung cancer, and pancreatic cancer. Importantly, mice lacking any functional SK1 alleles are resistant to the formation of colonic adenocarcinomas and have a decreased formation of aberrant crypt foci in an azoxymethane chemical carcinogenesis model of colon cancer (15). This is important because colonic adenocarcinomas frequently acquire mutations in the K-Ras allele. In addition, dietary sphingolipids have been shown to play an important role in regulating the development of colon cancer. The presence of an oncogenic K-Ras allele may affect how colonic epithelial cells respond to exogenous sphingolipids. We expect that activation of SK1 by an activated K-Ras oncogene may have a significant impact on how cells metabolize extracellular sphingolipids as revealed by our C17-sphingosine labeling studies. In addition, other oncogenes that activate the MAP kinase pathway such as B-RafV600E, which commonly occurs in melanoma, may have a similar effect on sphingolipid metabolism in the skin. Therefore oncogenes that activate the ERK pathway may also affect sphingolipid metabolism and alter which bioactive sphingolipids are produced.
Ceramide and S1P have long been regarded as having opposing effects on cellular proliferation and survival. In addition, the activity of SK1 has been shown to correlate with the ability of cancer cells and xenografts to resist chemotherapy (36). Activation of SK1 by oncogenic K-RasG12V may have important effects on carcinogenesis through its ability to regulate the levels of the intracellular bioactive lipids, ceramide and S1P. The activation of SK1 has been shown to protect cells from chemotherapeutic agents while inhibition of SK1 induces chemosensitivity to various chemotherapies (37, 38). In addition, S1P has an important paracrine signaling role because it potently stimulates angiogenesis and potentially lymphangiogenesis (39–42). Although, VEGF has long been recognized as an important regulator of tumor angiogenesis, recent studies have shown that S1P is also an important regulator of angiogenesis in the tumor microenvironments. Activation of SK1 and an increased production of extracellular S1P may be an important mechanism by which tumors with oncogenic K-RasG12V recruit blood vessels for their growth. Therefore, activation of SK1 by oncogenic K-RasG12V could have important effects through the alteration of intracellular sphingolipids and through the regulation of extracellular S1P.
Also of interest is our observation that SK1 becomes concentrated at the plasma membrane when oncogenic K-RasG12V is expressed. Previous studies have shown that plasma membrane localization correlates with SK1 activation, although the precise mechanism by which localization and alteration in activity are coordinated is unclear (13, 28). Some have proposed that localization of SK1 to the plasma membrane increases the probability that it interacts with anionic phospholipids, which are known to activate SK1 in vitro (33, 34). Anionic phospholipids might regulate SK1 activity by inducing a conformational change in SK1 that increases its catalytic activity, although until we have a crystal structure the precise mechanism by which this occurs remains unknown.
In summary, we present a mechanism by which an oncogene can alter sphingolipid metabolism through the regulation of SK1. Activation of SK1 by K-RasG12V results in an increase in both intracellular and extracellular S1P, which may have important effects on cancer cell proliferation or have a significant paracrine role in the tumor microenvironment. These effects may include the promotion of angiogenesis and lymphangiogenesis or through the promotion of inflammation, among other possible effects (16, 40, 42). This is the first study to provide a clear mechanism by which an oncogene can have a dramatic effect on sphingolipid metabolism through the regulation of SK1.
Acknowledgments
We thank Marianne Dubard-Gault for help in generating the SK1 mutants and Kathy Wiita-Fisk for administrative assistance in preparing this manuscript. Measurement of sphingolipids was conducted by the Lipidomics Core of Medical University of South Carolina (MUSC) in a facility constructed with support from National Institutes of Health Grant C06 RR018823 from the Extramural Research Facilities Program of the National Center for Research Resources. LC-MS/MS analysis of sphingolipids was performed by Lipidomics Shared Resource, MUSC (25) supported by NCI Grants IPO1CA097132 and P30 CA 138313 and National Institutes of Health NCRR SC COBRE Grant P20 RR017677. Laboratory space in the CRI building of MUSC was supported by National Institutes of Health Grant C06 RR018823 from the Extramural Research Facilities Program of the National Center for Research Resources.
This work was supported, in whole or in part, by National Institutes of Health Grants NHLBI NRSA 5F30 HL093991 (to C. R. G.), NIGMS R01 GM062887 and NCI P01CA097132 (to Y. A. H.), R01CA131-200 from the NCI (to S. E.), K22 ES012985-01 from the NIEHS (to C. N.), P01 CA0971321 from the NCI (to L. M. O.), and by MERIT Award 1I0 BX000156 01A1) (to L. M. O.) from the Office of Research and Development, Department of Veterans Affairs, Ralph H. Johnson Veterans Medical Center, Charleston, SC.

This article contains supplemental Figs. S1 and S2.
- S1P
- sphingosine 1-phosphate
- SK
- sphingosine kinase
- MEF
- mouse embryonic fibroblast
- DN
- dominant-negative.
REFERENCES
- 1. Hannun Y. A., Obeid L. M. (2008) Principles of bioactive lipid signaling. Lessons from sphingolipids. Nat. Rev. Mol. Cell. Biol. 9, 139–150 [DOI] [PubMed] [Google Scholar]
- 2. Kim M. Y., Linardic C., Obeid L., Hannun Y. (1991) Identification of sphingomyelin turnover as an effector mechanism for the action of tumor necrosis factor α- and γ-interferon. Specific role in cell differentiation. J. Biol. Chem. 266, 484–489 [PubMed] [Google Scholar]
- 3. Spiegel S., Olivera A., Zhang H., Thompson E. W., Su Y., Berger A. (1994) Sphingosine 1-phosphate, a novel second messenger involved in cell growth regulation and signal transduction, affects growth and invasiveness of human breast cancer cells. Breast Cancer Res. Treat 31, 337–348 [DOI] [PubMed] [Google Scholar]
- 4. Cuvillier O., Pirianov G., Kleuser B., Vanek P. G., Coso O. A., Gutkind S., Spiegel S. (1996) Suppression of ceramide-mediated programmed cell death by sphingosine 1-phosphate. Nature 381, 800–803 [DOI] [PubMed] [Google Scholar]
- 5. Hait N. C., Allegood J., Maceyka M., Strub G. M., Harikumar K. B., Singh S. K., Luo C., Marmorstein R., Kordula T., Milstien S., Spiegel S. (2009) Regulation of histone acetylation in the nucleus by sphingosine 1-phosphate. Science 325, 1254–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alvarez S. E., Harikumar K. B., Hait N. C., Allegood J., Strub G. M., Kim E. Y., Maceyka M., Jiang H., Luo C., Kordula T., Milstien S., Spiegel S. (2010) Sphingosine 1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465, 1084–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Spiegel S., Milstien S. (2003) Sphingosine 1-phosphate. An enigmatic signaling lipid. Nat. Rev. Mol. Cell Biol. 4, 397–407 [DOI] [PubMed] [Google Scholar]
- 8. Strub G. M., Maceyka M., Hait N. C., Milstien S., Spiegel S. (2010) Extracellular and intracellular actions of sphingosine 1-phosphate. Adv. Exp. Med. Biol. 688, 141–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Strub G. M., Paillard M., Liang J., Gomez L., Allegood J. C., Hait N. C., Maceyka M., Price M. M., Chen Q., Simpson D. C., Kordula T., Milstien S., Lesnefsky E. J., Spiegel S. (2011) Sphingosine 1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 25, 600–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hait N. C., Oskeritzian C. A., Paugh S. W., Milstien S., Spiegel S. (2006) Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim. Biophys. Acta 1758, 2016–2026 [DOI] [PubMed] [Google Scholar]
- 11. Allende M. L., Sasaki T., Kawai H., Olivera A., Mi Y., van Echten-Deckert G., Hajdu R., Rosenbach M., Keohane C. A., Mandala S., Spiegel S., Proia R. L. (2004) Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J. Biol. Chem. 279, 52487–52492 [DOI] [PubMed] [Google Scholar]
- 12. Taha T. A., Hannun Y. A., Obeid L. M. (2006) Sphingosine kinase. Biochemical and cellular regulation and role in disease. J. Biochem. Mol. Biol. 39, 113–131 [DOI] [PubMed] [Google Scholar]
- 13. Pitson S. M., Moretti P. A., Zebol J. R., Lynn H. E., Xia P., Vadas M. A., Wattenberg B. W. (2003) Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 22, 5491–5500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jarman K. E., Moretti P. A., Zebol J. R., Pitson S. M. (2010) Translocation of sphingosine kinase 1 to the plasma membrane is mediated by calcium- and integrin-binding protein 1. J. Biol. Chem. 285, 483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kawamori T., Kaneshiro T., Okumura M., Maalouf S., Uflacker A., Bielawski J., Hannun Y. A., Obeid L. M. (2009) Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J. 23, 405–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kawamori T., Osta W., Johnson K. R., Pettus B. J., Bielawski J., Tanaka T., Wargovich M. J., Reddy B. S., Hannun Y. A., Obeid L. M., Zhou D. (2006) Sphingosine kinase 1 is up-regulated in colon carcinogenesis. FASEB J. 20, 386–388 [DOI] [PubMed] [Google Scholar]
- 17. Van Brocklyn J. R., Jackson C. A., Pearl D. K., Kotur M. S., Snyder P. J., Prior T. W. (2005) Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme. Roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J. Neuropathol. Exp. Neurol. 64, 695–705 [DOI] [PubMed] [Google Scholar]
- 18. Ruckhäberle E., Rody A., Engels K., Gaetje R., von Minckwitz G., Schiffmann S., Grösch S., Geisslinger G., Holtrich U., Karn T., Kaufmann M. (2008) Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res. Treat. 112, 41–52 [DOI] [PubMed] [Google Scholar]
- 19. Li W., Yu C. P., Xia J. T., Zhang L., Weng G. X., Zheng H. Q., Kong Q. L., Hu L. J., Zeng M. S., Zeng Y. X., Li M., Li J., Song L. B. (2009) Sphingosine kinase 1 is associated with gastric cancer progression and poor survival of patients. Clin. Cancer Res. 15, 1393–1399 [DOI] [PubMed] [Google Scholar]
- 20. Knapp P., Baranowski M., Knapp M., Zabielski P., Bl̸achnio-Zabielska A. U., Górski J. (2010) Altered sphingolipid metabolism in human endometrial cancer. Prostaglandins Other Lipid Mediat. 92, 62–66 [DOI] [PubMed] [Google Scholar]
- 21. Pitson S. M., Xia P., Leclercq T. M., Moretti P. A., Zebol J. R., Lynn H. E., Wattenberg B. W., Vadas M. A. (2005) Phosphorylation-dependent translocation of sphingosine kinase to the plasma membrane drives its oncogenic signaling. J. Exp. Med. 201, 49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Olivera A., Kohama T., Edsall L., Nava V., Cuvillier O., Poulton S., Spiegel S. (1999) Sphingosine kinase expression increases intracellular sphingosine 1-phosphate and promotes cell growth and survival. J. Cell Biol. 147, 545–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xia P., Gamble J. R., Wang L., Pitson S. M., Moretti P. A., Wattenberg B. W., D'Andrea R. J., Vadas M. A. (2000) An oncogenic role of sphingosine kinase. Curr. Biol. 10, 1527–1530 [DOI] [PubMed] [Google Scholar]
- 24. Henis Y. I., Hancock J. F., Prior I. A. (2009) Ras acylation, compartmentalization, and signaling nanoclusters (Review). Mol. Membr. Biol. 26, 80–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bielawski J., Szulc Z. M., Hannun Y. A., Bielawska A. (2006) Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods 39, 82–91 [DOI] [PubMed] [Google Scholar]
- 26. Bielawski J., Pierce J. S., Snider J., Rembiesa B., Szulc Z. M., Bielawska A. (2009) Comprehensive quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods Mol. Biol. 579, 443–467 [DOI] [PubMed] [Google Scholar]
- 27. Spassieva S., Bielawski J., Anelli V., Obeid L. M. (2007) Combination of C17 sphingoid base homologues and mass spectrometry analysis as a new approach to study sphingolipid metabolism. Methods Enzymol. 434, 233–241 [DOI] [PubMed] [Google Scholar]
- 28. Johnson K. R., Becker K. P., Facchinetti M. M., Hannun Y. A., Obeid L. M. (2002) PKC-dependent activation of sphingosine kinase 1 and translocation to the plasma membrane. Extracellular release of sphingosine 1-phosphate induced by phorbol 12-myristate 13-acetate (PMA). J. Biol. Chem. 277, 35257–35262 [DOI] [PubMed] [Google Scholar]
- 29. Sukocheva O. A., Wang L., Albanese N., Pitson S. M., Vadas M. A., Xia P. (2003) Sphingosine kinase transmits estrogen signaling in human breast cancer cells. Mol. Endocrinol. 17, 2002–2012 [DOI] [PubMed] [Google Scholar]
- 30. Johnson K. R., Johnson K. Y., Crellin H. G., Ogretmen B., Boylan A. M., Harley R. A., Obeid L. M. (2005) Immunohistochemical distribution of sphingosine kinase 1 in normal and tumor lung tissue. J. Histochem. Cytochem. 53, 1159–1166 [DOI] [PubMed] [Google Scholar]
- 31. Nakade Y., Banno Y., T-Koizumi K., Hagiwara K., Sobue S., Koda M., Suzuki M., Kojima T., Takagi A., Asano H., Nozawa Y., Murate T. (2003) Regulation of sphingosine kinase 1 gene expression by protein kinase C in a human leukemia cell line, MEG-O1. Biochim. Biophys. Acta 1635, 104–116 [DOI] [PubMed] [Google Scholar]
- 32. Sobue S., Murakami M., Banno Y., Ito H., Kimura A., Gao S., Furuhata A., Takagi A., Kojima T., Suzuki M., Nozawa Y., Murate T. (2008) v-Src oncogene product increases sphingosine kinase 1 expression through mRNA stabilization. Alteration of AU-rich element-binding proteins. Oncogene 27, 6023–6033 [DOI] [PubMed] [Google Scholar]
- 33. Olivera A., Kohama T., Tu Z., Milstien S., Spiegel S. (1998) Purification and characterization of rat kidney sphingosine kinase. J. Biol. Chem. 273, 12576–12583 [DOI] [PubMed] [Google Scholar]
- 34. Wattenberg B. W., Pitson S. M., Raben D. M. (2006) The sphingosine and diacylglycerol kinase superfamily of signaling kinases. Localization as a key to signaling function. J. Lipid Res. 47, 1128–1139 [DOI] [PubMed] [Google Scholar]
- 35. Stahelin R. V., Hwang J. H., Kim J. H., Park Z. Y., Johnson K. R., Obeid L. M., Cho W. (2005) The mechanism of membrane targeting of human sphingosine kinase 1. J. Biol. Chem. 280, 43030–43038 [DOI] [PubMed] [Google Scholar]
- 36. Bonhoure E., Pchejetski D., Aouali N., Morjani H., Levade T., Kohama T., Cuvillier O. (2006) Overcoming MDR-associated chemoresistance in HL-60 acute myeloid leukemia cells by targeting sphingosine kinase-1. Leukemia 20, 95–102 [DOI] [PubMed] [Google Scholar]
- 37. Pchejetski D., Doumerc N., Golzio M., Naymark M., Teissié J., Kohama T., Waxman J., Malavaud B., Cuvillier O. (2008) Chemosensitizing effects of sphingosine kinase-1 inhibition in prostate cancer cell and animal models. Mol. Cancer Ther. 7, 1836–1845 [DOI] [PubMed] [Google Scholar]
- 38. Pchejetski D., Golzio M., Bonhoure E., Calvet C., Doumerc N., Garcia V., Mazerolles C., Rischmann P., Teissié J., Malavaud B., Cuvillier O. (2005) Sphingosine kinase-1 as a chemotherapy sensor in prostate adenocarcinoma cell and mouse models. Cancer Res. 65, 11667–11675 [DOI] [PubMed] [Google Scholar]
- 39. Nava V. E., Hobson J. P., Murthy S., Milstien S., Spiegel S. (2002) Sphingosine kinase type 1 promotes estrogen-dependent tumorigenesis of breast cancer MCF-7 cells. Exp. Cell Res. 281, 115–127 [DOI] [PubMed] [Google Scholar]
- 40. Anelli V., Gault C. R., Snider A. J., Obeid L. M. (2010) Role of sphingosine kinase-1 in paracrine/transcellular angiogenesis and lymphangiogenesis in vitro. FASEB J. 24, 2727–2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tengood J. E., Kovach K. M., Vescovi P. E., Russell A. J., Little S. R. (2010) Sequential delivery of vascular endothelial growth factor and sphingosine-1-phosphate for angiogenesis. Biomaterials 30, 7805–7812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Anelli V., Gault C. R., Cheng A. B., Obeid L. M. (2008) Sphingosine kinase 1 is up-regulated during hypoxia in U87MG glioma cells. Role of hypoxia-inducible factors 1 and 2. J. Biol. Chem. 283, 3365–3375 [DOI] [PubMed] [Google Scholar]
- 43. Pitson S. M., D'Andrea R. J., Vandeleur L., Moretti P. A., Xia P., Gamble J. R., Vadas M. A., Wattenberg B. W. (2000) Human sphingosine kinase. Purification, molecular cloning, and characterization of the native and recombinant enzymes. Biochem. J. 350, 429–441 [PMC free article] [PubMed] [Google Scholar]