

Background: Cardiac troponin C mutations are rare causes of HCM. A novel mutation in TNNC1 gene was identified in a pediatric HCM patient.
Results: Functional characterization demonstrated increased myofilament Ca2+ affinity.
Conclusion: The proband presented with ventricular fibrillation, aborted sudden cardiac death associated with myofilament dysregulation.
Significance: The newly identified cardiac troponin C mutation predisposes to pathogenesis of a fatal arrhythmogenic subtype of HCM.
Keywords: Cardiac Hypertrophy, Cardiac Muscle, Cardiomyopathy, Genetic Diseases, Protein Structure, Troponin, Actomyosin ATPase, Circular Dichroism, Skinned Fibers
Abstract
Defined as clinically unexplained hypertrophy of the left ventricle, hypertrophic cardiomyopathy (HCM) is traditionally understood as a disease of the cardiac sarcomere. Mutations in TNNC1-encoded cardiac troponin C (cTnC) are a relatively rare cause of HCM. Here, we report clinical and functional characterization of a novel TNNC1 mutation, A31S, identified in a pediatric HCM proband with multiple episodes of ventricular fibrillation and aborted sudden cardiac death. Diagnosed at age 5, the proband is family history-negative for HCM or sudden cardiac death, suggesting a de novo mutation. TnC-extracted cardiac skinned fibers were reconstituted with the cTnC-A31S mutant, which increased Ca2+ sensitivity with no effect on the maximal contractile force generation. Reconstituted actomyosin ATPase assays with 50% cTnC-A31S:50% cTnC-WT demonstrated Ca2+ sensitivity that was intermediate between 100% cTnC-A31S and 100% cTnC-WT, whereas the mutant increased the activation of the actomyosin ATPase without affecting the inhibitory qualities of the ATPase. The secondary structure of the cTnC mutant was evaluated by circular dichroism, which did not indicate global changes in structure. Fluorescence studies demonstrated increased Ca2+ affinity in isolated cTnC, the troponin complex, thin filament, and to a lesser degree, thin filament with myosin subfragment 1. These results suggest that this mutation has a direct effect on the Ca2+ sensitivity of the myofilament, which may alter Ca2+ handling and contribute to the arrhythmogenesis observed in the proband. In summary, we report a novel mutation in the TNNC1 gene that is associated with HCM pathogenesis and may predispose to the pathogenesis of a fatal arrhythmogenic subtype of HCM.
Introduction
Hypertrophic cardiomyopathy (HCM),7 defined as left ventricular hypertrophy without a clinically identifiable origin, affects ∼1 of 500 individuals and is the most common cause of sudden death in the young athlete (1–3). Thought to be of genetic origin, HCM is inherited typically in an autosomal dominant fashion. Investigations over the past 20 years have led to the identification of hundreds of mutations associated with dozens of genes that have been linked to the pathogenesis of HCM (4, 5). Principal among these are genes encoding components of the sarcomeric myofilament, which host the majority of HCM-associated mutations. Recently, mutations identified in TNNC1-encoded cardiac troponin C (cTnC), part of the sarcomeric thin filament, have been implicated as a rare cause of HCM (6–8). Despite this progress, little is known about the arrhythmogenic role of TNNC1 mutations in the setting of cardiomyopathic disease. Furthermore, there is little mechanistic explanation for the increased risk of sudden cardiac death in some patients with this clinically heterogeneous disease (9).
Troponin C is part of the heterotrimeric regulatory troponin complex of the sarcomeric thin filament and serves as the Ca2+ sensor of muscle contraction. The Ca2+-binding protein TnC works in concert with inhibitor troponin I (TnI) and troponin T (TnT), which provides a direct link to tropomyosin and assists in transducing the contractile signal to the rest of the thin filament. This process is initiated by the binding of cytosolic Ca2+ during Ca2+-induced Ca2+ release, the beginning of the Ca2+ transient, which increases the binding affinity of TnC for TnI, thus pulling the TnI inhibitory domain away from its binding site on actin (10). The release of TnC allows the troponin-tropomyosin complex to move farther into the actin groove fully exposing the myosin binding sites on actin. The formation of active cross-bridges may then occur, allowing muscle tension to develop (10).
Cardiac TnC is a Ca2+-binding protein that belongs to the EF-hand superfamily which consists of two globular functional domains attached by a flexible linker (11). The N-domain is considered the regulatory domain and has one active Ca2+ binding site (site II) that binds Ca2+ with low affinity (∼105 m−1). The C-domain is known as the structural domain and contains two Ca2+/Mg2+ binding sites (sites III and IV) that bind Ca2+ at low concentrations (∼107 m−1) while also competitively binding Mg2+ (∼103 m−1) (12). Therefore, only site II of the regulatory N-domain reversibly binds cytosolic Ca2+ during cardiac contraction, and mutations that affect the function of cTnC may alter its global structure thus modifying its Ca2+ affinity and/or interfering with protein-protein interactions necessary to appropriately transmit the Ca2+ binding signal. Recently, we identified a novel TNNC1 mutation alanine substituted by serine at position 31 (A31S) in the non-functional Ca2+ binding site I. This mutation may alter Ca2+ binding to site II, which regulates the sensitivity of muscle contraction. Here, we investigated the source of the primary defect (increased Ca2+ sensitivity in skinned muscles and ventricular tachycardia in the patient) that occurs in the presence of this mutation.
In addition to initiating cardiac contraction, cTnC is an important Ca2+ buffer that assists in maintaining Ca2+ homeostasis in the myocyte, and increased Ca2+ binding affinity may result in arrhythmogenic Ca2+ mishandling (13). Traditionally associated with mutations in cardiac ion channels, ventricular tachycardia has been identified in mouse models hosting Ca2+-sensitizing cTnT mutations (14). Furthermore, mouse models of a cTnI-R145G mutation have been shown to increase Ca2+ sensitivity and prolong Ca2+ transients (15). Despite these studies, the link between myofilament mutations and arrhythmogenesis, particularly in the context of HCM, remains relatively uncharacterized. To this end, we investigated the effects of a novel TNNC1 mutation identified in a host demonstrating youthful HCM presentation and repeated episodes of medically refractory ventricular fibrillation.
EXPERIMENTAL PROCEDURES
Genetic Analysis
The proband underwent HCM genetic testing utilizing comprehensive direct sequencing analysis of the nine canonical genes associated with sarcomeric HCM including MYH7-encoded β myosin heavy chain, MYBPC3-cardiac myosin binding protein C, MYL2-encoded regulatory myosin light chain, MYL3-encoded essential myosin light chain, TNNT2-, TNNI3-, TNNC1-, and TPM1-encoded α-tropomyosin, and ACTC-encoded α-cardiac actin (Transgenomic Inc, New Haven, CT). Genetic causes of cardiac disease, which can mimic HCM including PRKAG2-encoded γ-regulatory subunit of AMP-activated protein kinase, GLA-encoded α-galactosidase A, and LAMP2-encoded lysosome-associated membrane protein, were also comprehensively genotyped.
Study Control Cohorts
The absence of the mutation was confirmed in >26,600 reference alleles. Among these, were >800 unrelated, ethnically diverse, ostensibly healthy individuals recruited by Transgenomic Inc., 100 ostensibly health African American and 200 Caucasian American individuals from the Coriell Institute for Medical Research (Camden, NJ), and 200 additional Caucasian subjects with normal screening electro- and echocardiograms recruited from Olmsted County, Minnesota. Publically available databases from the Exome Chip Project (12,031 exomes), including the 1000 Genome Project (1,128 exomes) and NHLBI, National Institutes of Health Exome Sequencing Project (4,260 exomes), were also searched for the presence of the identified mutation (16).
Clinical Evaluation of TNNC1-A31S Proband
Clinical data were collected including pertinent personal and family history, physical examination, 12-lead electrocardiogram analysis, QT interval corrected for heart rate (QTc) measurement, and echocardiographic testing to determine mean left ventricular wall thickness, maximum left ventricular outflow tract gradient, and other parameters.
Functional and Structural Studies
Cloning, Expression, and Purification of Human Cardiac Troponin T, Troponin I, and Troponin C Mutants
The cTnI and cTnT cDNAs were cloned as previously described (17). The cTnC cDNA was cloned previously from total RNA obtained from human heart tissue. The sequential overlapping PCR method was used to introduce the A31S mutation into the cDNA (18). Standard methods previously used in this laboratory were utilized for the expression and purification of wild-type and mutant cTnC (19).
Fiber Preparation and Ca2+ Dependence of Force Development Measurements
Fresh cardiac tissue was obtained from slaughterhouse pigs. Strips of papillary muscle 3–5 mm in diameter and ∼5 mm in length were isolated from the left ventricle and skinned overnight in a 50% glycerol relaxing solution (10−8 m [Ca2+]free) (6). Fibers were then transferred to a similar solution without Triton X-100 and stored at −20 °C. Briefly, a skinned fiber bundle ∼75–100 μm in diameter was mounted using stainless steel clips to a force transducer and then immersed in a pCa 8.0 relaxation solution (conditions described in Ref. 6. The Ca2+ dependence of force development was tested in skinned fibers at low, intermediate, and high concentration Ca2+ solutions (pCa 8.0–4.0) and calculated using the pCa calculator program developed in our laboratory (20). The native cTnC was depleted upon incubation of the fiber in a 1,2-cyclohexylenenitrilotetraacetic acid (CDTA) extracting solution (5 mm CDTA and 25 mm Tris (pH 8.4)) for ∼ 1.5 h. Fibers were considered extracted of cTnC when residual tension remaining in the fiber in pCa 4.0 was 15% or below. Fibers were then incubated with 28 μm concentrations of mutant or WT cTnC diluted in pCa 8.0 for 1 h. The following equation was used to analyze data: % change in force = 100 × [Ca2+]n/([Ca2+]n + [Ca2+50]n), where [Ca2+50] is the free [Ca2+] that produces 50% force, and nHill is the Hill coefficient. All fiber experiments were performed at room temperature.
Formation of Troponin Complexes
The purified individual troponin subunits including 2-(4′-iodoacetamidoanilino)naphthalene-6-sulfonic acid (IAANS)-labeled cTnC were first dialyzed against 3 m urea, 1 m KCl, 10 mm MOPS, 1 mm DTT, and 0.1 mm phenylmethanesulfonyl fluoride and then twice against the same buffer excluding urea. The protein concentrations of the individual subunits were determined using the Coomassie Plus kit and then mixed in a 1.3:1.3:1 cTnT:cTnI:cTnC molar ratio. After 1 h, the complexes were successively dialyzed against solutions containing decreasing concentrations of KCl (0.7, 0.5, 0.3, 0.1, 0.05, 0.025 m). Precipitated excess proteins during complex formation were removed by centrifugation. Proper stoichiometry was verified by SDS-PAGE before storing the troponin complexes at −80 °C. Ternary troponin complexes were utilized in the actin-tropomyosin-activated myosin-ATPase assays containing the cTnC mutant.
Actin-Tropomyosin-activated Myosin-ATPase Assays; Minimum and Maximum ATPase
Porcine cardiac myosin, rabbit skeletal F-actin, and porcine cardiac tropomyosin were prepared as previously described (18). The protein concentrations used for actomyosin ATPase assays were: 0.6 μm porcine cardiac myosin, 3.5 μm rabbit skeletal F-actin, 1 μm porcine cardiac tropomyosin, and 0–2 μm preformed troponin complexes as prepared above and were performed as single point assays that are linear over time (21). The proteins were in the following buffer conditions: myosin in 10 mm MOPS (pH 7.0), 400 mm KCl, 1 mm DTT; actin in 10 mm MOPS (pH 7.0), 40 mm KCl; tropomyosin in 10 mm MOPS (pH 7.0), 300 mm KCl, 1 mm DTT. The final ionic strength of the reactions was ∼75 mm when considering the combined ionic contributions from all buffers. ATPase inhibition measurements were performed in a 0.1 ml reaction mixture: 3.4 mm MgCl2, 0.13 mm CaCl2, 1.5 mm EGTA, 3.5 mm ATP, 1 mm DTT, 11.5 mm MOPS (pH 7.0) at 25 °C. The ATPase activation measurements were conducted using the same 0.1 ml buffer mixture with: 3.3 mm MgCl2 and 1.7 mm CaCl2. ATP was added to initiate the reaction, which was quenched after 20 min using trichloroacetic acid at a final concentration of 35%. The precipitated assay proteins were removed by centrifugation. The amount of ATPase hydrolysis was determined by measuring the release of inorganic phosphate in the supernatant using methods established by Fiske and Subbarow (22).
Ca2+ Sensitivity of the ATPase
The pCa curves were performed using 1 μm preformed troponin complex containing 100% WT or cTnC-A31S as well as 50:50 WT:cTnC-A31S. Also, the same concentrations of porcine β-myosin, rabbit skeletal F-actin, and porcine cardiac tropomyosin were used as described above. The conditions of the assay varied slightly: 11.55 mm MOPS, 2 mm EGTA, 1 mm Nitrilotriacetic Acid, 3.7 mm MgCl2 (pH 7.0 at 25 °C) and the following pCa values: pCa 7.0 (0.448 mm CaCl2), 6.5 (0.955 mm CaCl2), 6.2 (1.293 mm CaCl2), 6.0 (1.489 mm CaCl2), 5.9 (1.574 mm CaCl2), 5.7 (1.714 mm CaCl2), 5.6 (1.770 mm CaCl2), 5.4 (1.860 mm CaCl2), 5.2 (1.929 mm CaCl2), and 4.5 (2.140 mm CaCl2).
Fluorescence Labeling of TNNC1
The TNNC1 was double-labeled with IAANS at Cys-35 and Cys-84 and mono-labeled at Cys-84. IAANS was obtained from Molecular Probes, Plano, TX. Fluorescent labeling and purification of IAANS-labeled cTnC was performed according to established methods (23).
Determination of Apparent Ca2+ Affinities by Fluorescence
IAANS-labeled cTnCs (WT and cTnC-A31S) were dialyzed into fluorescence buffer containing 2 mm EGTA, 5 mm Nitrilotriacetic Acid, 120 mm MOPS, 90 mm KCl. Before titration of isolated cTnC, 1.25 mm MgCl2 and 1 mm freshly prepared DTT were added. For troponin complex formation and fluorescence experiments, fresh DTT was added before the titration because the dialysis buffer already contained 1.25 mm MgCl2 that is needed for complex formation. Fluorescence measurements with isolated cTnC and the troponin complex were performed using double-labeled cTnC, with the IAANS label at Cys-35 and Cys-84. Steady state fluorescence measurements were performed using a Jasco 6500 spectrofluorimeter where IAANS fluorescence was excited at 330 nm, and emission was detected at 450 nm. The thin filaments were made according to Pinto et al. (24). The IAANS mono-labeling configuration was utilized for thin filament and thin filament + subfragment 1 (S1) measurements, where Cys-35 in cTnC was mutated to Ser; therefore, only Cys-84 would be labeled. Putkey et al. (23) first described this method for Ca2+ affinity measurements. The need for two labeling configurations, i.e. TnC labeled at both Cys-35 and -84 (double label) or with cTnC labeled only at Cys-84 (single label) is that in the presence of the other thin filament proteins, only one configuration responds to Ca2+. Therefore, we used the labeling configuration that provides the largest change in fluorescent signal at each given level of thin filament complexity (e.g. cTnC combined with the other troponin subunits (ternary complex) in the presence of tropomyosin and actin). The concentration of proteins used for fluorescence measurements was 10 μm for isolated cTnC, 0.5 μm for troponin, 0.05 mg/ml for thin filament, and 0.02 mg/ml for S1. This gives an overall stoichiometry between the thin filament and S1 as 1:1.58, respectively. We have previously determined that this is the ideal concentration/ratio of S1 that caused the half-maximal change in fluorescence from thin filaments (24). The change in the fluorescence spectra was recorded during the titration of microliter amounts of CaCl2. The concentration of free Ca2+ and amounts of titrated Ca2+ were obtained using the pCa calculator program (20). The program made corrections for dilution effects that occur during titration of Ca2+. The data were fit to a version of the Hill equation that accounted for the spectral changes that occur at a low Ca2+ concentration and plotted using SigmaPlot 11.0.
Circular Dichroism Measurements
Far UV circular dichroism spectra (CD) were collected using a 1-mm-path quartz cell in a Jasco J-720 spectropolarimeter. Spectra were recorded at 195–250 nm with a bandwidth of 1 nm at a speed of 50 nm/min, whereas the resolution was 0.5 nm at room temperature. Ten scans were averaged, and numerical smoothing was not applied. The optical activity of the buffer was subtracted from relevant protein spectra. Mean residue ellipticity for the spectra was calculated utilizing the same Jasco system software using the following equation: [θ]MRE = [θ]/(10 × Cr × l), where [θ] is the measured ellipticity in millidegrees, Cr is the mean residue molar concentration, and l is the path length in cm (25). Protein concentrations were determined by the biuret reaction using bovine serum albumin as a standard. The CD experiments were performed using three different conditions: 1) apo state (divalent cation free) (1 mm EGTA, 20 mm MOPS, 100 mm KCl (pH 7.0)); 2) Mg2+-bound state (1 mm EGTA, 20 mm MOPS, 100 mm KCl, 2.075 mm MgCl2 (pH 7.0)); 3) Ca2+-bound state (1 mm EGTA, 20 mm MOPS, 100 mm KCl, 2.075 mm MgCl2, 1.096 mm CaCl2 (pH 7.0)). The experimental protein concentration for the WT and the A31S mutant cTnC was 0.2 mg/ml.
Three-dimensional Visualization
The cTnC-A31S mutation was visualized in the 1AJ4 Protein Data Bank file using PyMol software. PyMol is an open source molecular visualization program that allows manipulation of PDB files that contain molecular coordinates from x-ray crystallography- or nuclear magnetic resonance-based structures. The program allows mutagenesis of selected residues, portrays potential side chain interactions and potential for hydrogen bonding due to changes in the nature of and proximity of side chains.
Statistical Analysis
The experimental results were reported as x ± S.E. and analyzed for significance using Student's t test at p < 0.05.
RESULTS
Genetic Analysis
Comprehensive HCM genetic analysis identified a heterozygous TNNC1 G → T mutation at nucleotide 91 resulting in a GCT → TCT alanine to serine alteration at residue 31 (TNNC1-A31S). This mutation was not identified in >26,600 reference alleles derived from ostensibly healthy individuals from a variety of racial and ethnic backgrounds. The proband did not host a compound mutation in eight other sarcomeric genes (MYH7, MYBPC3, MYL2, MYL3, TNNT2, TNNI3, TPM1, and ACTC) as well as three HCM phenocopy-associated genes (PRKAG2, GLA, and LAMP2).
Clinical Evaluation
The TNNC1-A31S mutation was identified in a Caucasian male who was symptom-free until his sentinel event of ventricular fibrillation at 3.75 years of age while sleeping at night. He arose, expressed concern to his parents, became syncopal, and was defibrillated successfully by paramedics with an automatic external defibrillator from ventricular fibrillation. He underwent intracardioverter defibrillator implantation and was maintained on β blockade. Despite this therapy, he had five episodes of breakthrough ventricular fibrillation generally when emotionally excited and physically active, with single intracardioverter defibrillator shock restoring normal sinus rhythm in each case. He presented at age 5 to our institution for further evaluation.
He had a negative family history for HCM and sudden cardiac death. Both parents, at 47 and 48 years of age, were negative for HCM by echocardiography (Fig. 1A). On echocardiographic examination, the proband demonstrated asymmetric septal wall hypertrophy with a mean left ventricular wall thickness of 20 mm (6–8 mm normal range) with reverse curve morphology, an ejection fraction of 65%, diastolic dysfunction, and no left ventricular outflow tract obstruction (Fig. 1B). He had moderate left atrial enlargement. Electrocardiographic analysis demonstrated significant voltage criteria for biventricular hypertrophy, ST segment depression in anterior leads, and borderline QT prolongation with a QTc of 460 ms (Fig. 1C).
FIGURE 1.

TNNC1-A31S proband clinical characteristics. A, shown is a pedigree depicting the family of the proband (arrow) who hosts the TNNC1-A31S heterozygous allele as well as his two parents, who are negative for a family history of HCM and sudden cardiac death and are unremarkable upon echocardiographic evaluation. LA, left atrium; LV, left ventricle; VF, ventricular fibrillation; bar, 10 mm. B, shown are representative echocardiographic images demonstrating asymmetric left ventricular hypertrophy and left atrial dilatation during end diastole (left image) and systole (right). C, shown is representative 12-lead electrocardiographic tracing demonstrating voltage criteria for HCM and QT prolongation. Bar, 0.4 s.
Cardiac Skinned Fiber Experiments
To assess whether the A31S mutation perturbs myofilament function, the Ca2+ sensitivity and force recovery were evaluated using cTnC-depleted cardiac porcine fibers reconstituted with WT cTnC and cTnC-A31S. Incorporation of cTnC-A31S caused a leftward shift corresponding to an increase in Ca2+ sensitivity of 0.17 pCa units with a pCa50 of 5.63 in the WT to 5.80 in the mutant (Fig. 2A and Table 1). The cooperativity of thin filament activation (nHill) (Fig. 2A and Table 1) and force recovery % (P/P0) (Fig. 2B) was unchanged.
FIGURE 2.
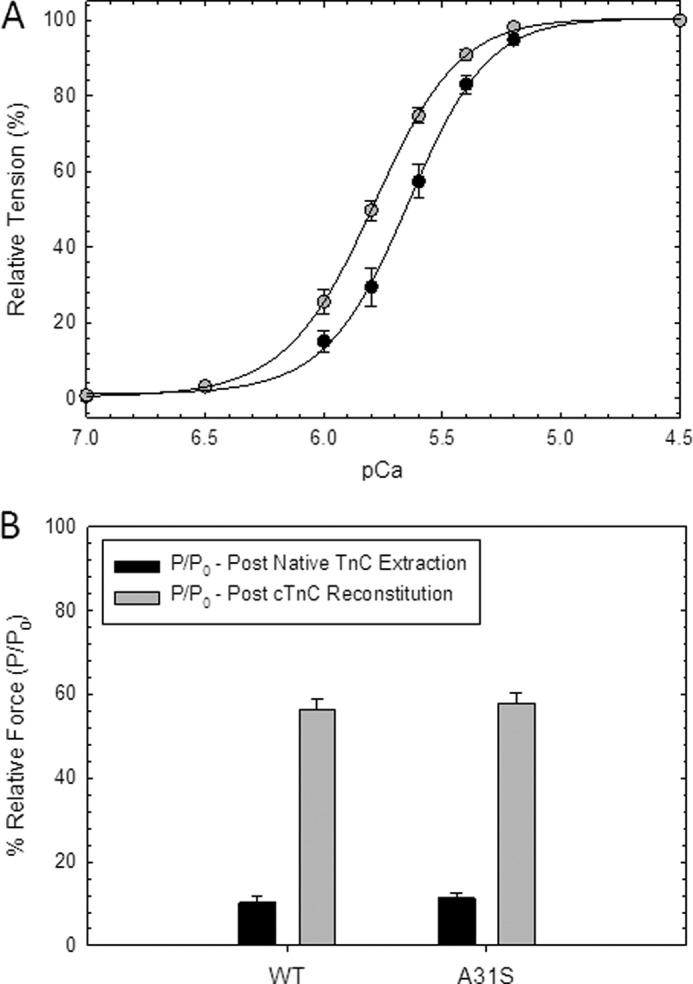
Skinned cardiac fiber experiments reconstituted with the HCM cTnC mutant. A, porcine cardiac fibers were reconstituted with mutant (gray circles) and WT cTnC (black circles), and Ca2+ sensitivity of force development was reported as pCa50 values in Table 1; B). Force recovery values (P) obtained after reconstitution with mutant and WT cTnC were compared with the level of force present before extraction of native cTnC (P0) and were reported as % (P/P0). Data are reported as the mean ± S.E. (n = 8–10).
TABLE 1.
Summary of the Ca2+ sensitivity data obtained in situ and reconstituted assays
WT:A31S in ATPase measurements was 50% WT cTnC and 50% A31S cTnC mutant Tn complexes. ND, not determined.
| TnC mutant | Skinned fiber |
Reconstituted ATPase |
||||||
|---|---|---|---|---|---|---|---|---|
| pCa50 | nHill | pCa50 | nHill | |||||
| WT | 5.63 | 0.01 | 2.71 | 0.14 | 5.79 | 0.02 | 2.35 | 0.16 |
| A31S | 5.80 | 0.02a | 2.45 | 0.11 | 6.17 | 0.02a | 1.95 | 0.18a |
| WT:A31S | ND | ND | 6.02 | 0.03a | 1.95 | 0.08a | ||
a p < 0.05 compared with their respective WT.
Actomyosin ATPase Assays Using Reconstituted Troponin Complexes Containing cTnC-A31S
We next measured the ability of the troponin complex to activate or inhibit the actomyosin ATPase in the presence or absence of Ca2+, respectively. The activation of the ATPase (pCa 4) was measured with increasing amounts of preformed troponin complex (0–2.0 μm) (each point represents, n = 6, performed in triplicate). The troponin complexes containing cTnC-A31S demonstrated increased thin filament activation compared with WT upon increasing concentrations of troponin. Specifically, there was an increase in the level of activation in the mutant cTnC-A31S-reconstituted thin filament (∼180%) compared with WT (∼150%) at 1.0, 1.5, and 2.0 μm troponin (Fig. 3A). The inhibitory properties of the mutant cTnC-A31S complex was assessed at low Ca2+ concentrations (pCa 8) by monitoring the ability to inhibit ATPase activity versus WT. The A31S mutant inhibited the actomyosin ATPase in a manner similar to WT although at higher concentrations of troponin (1.0–2.0 μm) (each point represents n = 7, performed in triplicate). However, there was a statistically significant decrease in inhibition of the ATPase by the mutant troponin at the lower concentration range (∼52% at 0.3–0.8 μm) compared with WT (∼40%) as shown in Fig. 3B. The Ca2+ dependence of actomyosin ATPase activation was also evaluated, and the pCa50 values were determined for the reconstituted thin filaments containing troponin complexes with either 100% WT, 100% cTnC-A31S, and 50:50 WT:cTnC-A31S (experiments performed n = 8). When 100% mutant A31S troponin was incorporated into the reconstituted thin filaments, and Ca2+ sensitivity was increased by +0.38 pCa units, whereas when 50% of the mutant A31S troponin complex was utilized, Ca2+ sensitivity increased by +0.23 pCa units (Fig. 3C and Table 1).
FIGURE 3.

Effect of the HCM cTnC mutant on actin-tropomyosin-troponin-activated ATPase activity measurements. A, activation of actomyosin ATPase by preformed troponin WT and HCM-cTnC mutant complex at increasing ratios is indicated on the abscissa in the presence of Ca2+. B, shown is inhibition of the actomyosin ATPase activity by increasing ratios of preformed troponin WT and HCM-cTnC complex in the absence of Ca2+. For A and B, each point represents an average of six to seven experiments performed in triplicate and is expressed as the mean ± S.E. C, shown is actin-tropomyosin-activated myosin ATPase activity of HCM-cTnC mutant as a function of pCa (each point represents an average of eight experiments, performed in triplicate and expressed as mean ± S.E.). The dark gray circles indicate the presence of 100% cTnC mutant complexes, and light gray circles with the dotted line represent experiments performed with a 50:50 ratio of HCM-cTnC mutant to WT complexes. Black circles represent data obtained with WT alone. The myosin ATPase activity that occurs in the absence of troponin complex is considered 100% ATPase activity. The specific ATPase activity in the absence of troponin complexes was measured as 0.35 mol of Pi × mol of myosin−1 × s−1.
IAANS Fluorescence Measurements of cTnC Troponin, Thin Filament, and Thin Filament S1-containing cTnC-A31S
The Ca2+ affinity of isolated cTnC as measured by fluorescence obtained from IAANS, an extrinsic probe, indicated that the mutant cTnC-A31S (double label) had increased Ca2+ affinity of +0.17 pCa units compared with WT (Fig. 4A and Table 2). When additional constituents (tropomyosin and actin) of the thin filament were included, there was a substantial increase in thin filament Ca2+ affinity containing cTnC-A31S that increased +0.56 pCa units compared with WT as shown in Fig. 4C and Table 2. In addition, spectral fluorescence changes from isolated cTnC were detected from the low affinity C-terminal Ca2+ binding sites indicating that the probe picked up both sets of binding events (Fig. 4A). Next, the Ca2+ affinity of WT and cTnC-A31S was measured with the thin filament proteins (tropomyosin and actin) and myosin S1. Myosin S1 was added to each thin filament containing either WT or cTnC-A31S to determine whether the mutation further enhanced myofilament Ca2+ sensitivity when strong cross-bridges formed between S1 and actin. Subsequently, we found that the cTnC-A31S mutant, in the absence of MgATP, increased the Ca2+ affinity of the thin filament by +0.25 compared with WT (Fig. 4D and Table 2). In this way, the thin filament as well as cross-bridge binding may play an important role in modulating the Ca2+ sensitivity when the A31S mutation is present.
FIGURE 4.

Steady state fluorescence to determine the apparent Ca2+ affinities of IAANS-labeled cTnC A31S mutant. A, isolated cTnC double-labeled with IAANS at Cys-35 and Cys-84 compare Ca2+ titration of A31S (dark gray circles) versus WT (black circles). B, troponin complex with cTnC-IAANS double-labeled at Cys-35 and Cys-84 compare A31S versus WT. C, thin filament with cTnC-IAANS single-labeled at Cys-84 of A31S versus WT is shown. D, thin filament with cTnC-IAANS single-labeled at Cys-84 in the presence of myosin S1 with conditions that favor strong cross-bridge formation (−ATP) is shown. Relative fluorescence values (%) are plotted as a function of Ca2+ concentrations in moles. Data are reported as mean ± S.E. (n = 4–7).
TABLE 2.
Summary of pCa50 of Ca2+ affinity measured by IAANS fluorescence at different levels of myofilament complexity
| Mutant | cTnC |
cTn |
Thin filament |
Thin filament + S1 |
||||
|---|---|---|---|---|---|---|---|---|
| pCa50 | nHill | pCa50 | nHill | pCa50 | nHill | pCa50 | nHill | |
| WT | 4.95 ± 0.01 | 0.83 ± 0.01 | 6.71 ± 0.01 | 1.08 ± 0.01 | 6.16 ± 0.02 | 1.42 ± 0.03 | 6.62 ± 0.03 | 1.09 ± 0.09 |
| A31S | 5.12 ± 0.04a | 0.81 ± 0.01 | 6.98 ± 0.01a | 0.91 ± 0.01a | 6.70 ± 0.03a | 1.36 ± 0.08 | 6.83 ± 0.04a | 1.08 ± 0.11 |
a p < 0.05 compared with their respective WT.
Circular Dichroism of the HCM cTnC Mutant
To determine whether cTnC-A31S had an effect on the structure of the protein in the isolated state, the amount of secondary structure was determined in three conditions: apo (unbound), Mg2+ -loaded, and Ca2+/Mg2+-loaded states (Fig. 5 and Table 3). Typically, the greatest change in α-helical content occurs when divalent cations, such as Mg2+, bind to the C terminus. Thus, the Mg2+-bound state is expected to accurately indicate structural changes occurring within the C terminus. The cardiac TnC-A31S mutation did not significantly change the α-helical content of the states examined, although the IAANS probe present in the N terminus was able to detect Ca2+ binding events in the C terminus (Fig. 4A).
FIGURE 5.

Determination of secondary structural characteristics of HCM mutant and WT-cTnC by circular dichroism. These spectra compare the changes in α-helical content of WT versus cTnC-A31S under different conditions. The graph compares cTnC in the apo, Mg2+-bound, and Mg2+/Ca2+- bound states.
TABLE 3.
Summary of circular dichroism results for HCM cTnC mutants
The CD spectrum is reported for apo (no divalent cations bound) and Mg2+-bound and Ca2+/Mg2+-bound states, and error is reported as S.E. deg, degrees.
| TnC | [θ]222 nm |
n | ||
|---|---|---|---|---|
| Apo | Mg2+ | Ca2+/Mg2+ | ||
| deg.cm2.dmol−1 | ||||
| WT | −15,392.7 ± 106.8 | −18,877.3 ± 86.5 | −20,802.7 ± 167.0 | 4 |
| A31S | −15,154.1 ± 290.0 | −18,474.7 ± 101.7 | −21,062.6 ± 274.9 | 4 |
| [θ] 208 nm |
||||
|---|---|---|---|---|
| TnC | Apo | Mg2+ | Ca2+/Mg2+ | n |
| deg.cm2.dmol−1 | ||||
| WT | −19524.2 ± 194.8 | −22420.2 ± 89.3 | −23868.1 ± 193.9 | 4 |
| A31S | −19237.7 ± 344.5 | −22048.9 ± 96.1 | −24082.8 ± 326.6 | 4 |
DISCUSSION
Mutations in Cardiomyopathic and Arrhythmic Disease
Although some HCM-associated mutations have been associated with sudden death susceptibility based on survival studies of the kindred hosting the mutations, there is a paucity of mechanistic insight into the arrhythmogenic state caused by these mutations (26). Early studies identifying “malignant mutations” in families, such as (β-myosin heavy chain) MYH7-R403Q and (cardiac troponin T) TNNT2-R92W, were associated with decreased Kaplan-Meyer survival when compared with families hosting so-called “benign” HCM mutations; however, detailed mechanistic studies elucidating the sudden death susceptibility potentially caused by these mutations are needed (27, 28).
Significant progress has been made in elucidating the genetic underpinnings of HCM; however, only a handful of mutations in TNNC1 have been identified. L29Q was one of the first mutations in TNNC1 to be associated with HCM (7). Although the effects of this mutation remain controversial, functional analysis has suggested that it may be a non-pathogenic variant based on inconsistent findings of altered Ca2+ sensitivity (29–32). We previously characterized four additional missense mutations, TNNC1-A8V, C84Y, E134D, and D145E, identified in a large cohort of unrelated patients with HCM (6, 33). Cardiac fibers reconstituted with these HCM-associated cTnC mutants results in increased Ca2+ sensitivity through mutation-specific alterations of dynamic interactions between cTnC and other components of the cardiac myofilament (33).
None of the earlier reported HCM TNNC1 mutations have been associated with arrhythmia, except that a recent study provides a link between the clinical outcome of sudden cardiac death and a TNNC1 mutation (8). A mutation in TNNC1, c.363dupG or p.Gln122AlafsX30 (truncation at position 122; cTnC-Δ122), has been identified in the proband age 19 in a small multi-generational family with HCM demonstrating reduced penetrance and variable expressivity (8). As with prior HCM-associated mutations, mechanistic studies identifying the pro-arrhythmic perturbation potentially imparted by this mutation have not been done to date (8). Therefore, we were unable to analyze shared functional properties between cTnC-A31S and the truncation mutant cTnC-Δ122 that may induce arrhythmia. The functional properties of this cTnC-Δ122 mutant are likely altered as it lacks Ca2+ binding site IV, eliminating the coupling that exists between sites of the same domain and affecting the Ca2+ buffering capacity of cTnC. Subsequently, the cTnC-A31S mutation located within the inactive Ca2+ binding site I is unlikely to have restored Ca2+ binding to any degree. However, in this case the A31S mutation may affect coupling between sites I and II of the same domain. Additional HCM-linked cTnC mutants may need to be identified and characterized to address how a cTnC mutant might cause arrhythmia.
In general, cardiomyopathic mutations that manifest significant physiological effects by an early age are considered most severe. In this study the patient presented significant cardiac symptoms involving ventricular fibrillation before the age of 4. In our previous study the TNNC1 mutation C84Y caused mild symptoms (syncope on exertion) at 8.4 years and was successfully managed by β-blockade (6). Comparison of clinical presentation of disease in patients bearing TNNC1 mutations with data measuring their functional consequences may provide additional insight.
Functional Properties of the HCM-associated cTnC-A31S Mutant
It has been hypothesized that a defining characteristic of TNNC1 HCM-associated mutations is the increased, or possibly equivalent, Ca2+ sensitivity that may also alter the maximal myocyte force generation (34). To determine whether the mutant increased Ca2+ sensitivity in a dominant-negative manner, we performed reconstituted actomyosin assays using 100% cTnC-A31S mutant and also in the “heterozygous” state using 50:50 cTnC-A31S:WT proteins to best represent the background of the patient. The intermediate pCa50 obtained with 50% mutant cTnC (compared with 100% mutant or 100% WT) better-approximated the pCa50 seen in the more intact skinned fiber system. This suggests that the mutation exerts its influence in a dominant negative manner, although the pCa50 value may be altered by different amounts of cTnC mutant incorporation.
The reconstituted thin filaments containing cTnC-A31S displayed a significant increase in activation levels compared with those containing WT (Fig. 3A). Enhanced thin filament activation coupled with increased Ca2+ sensitization of the myofilament may contribute toward the diastolic function seen in HCM patients. A likely scenario would be that the increase in Ca2+ sensitivity heightens the contractile response and subsequently impairs the degree of relaxation achieved during diastole. Despite the increase in the actomyosin ATPase activation, the maximal force recovery was unchanged in skinned fibers. However, actomyosin ATPase inhibition measurements performed under relaxing conditions (pCa 8.5) showed that cTnC-A31S inhibited ATPase activity to the same degree as WT when (1.0–2.0 μm cTn) was added to the thin filament. This is consistent with bona fide HCM cTn mutations, which usually do not alter ATPase inhibition (33). The basal force in skinned fibers was unaffected (data not shown) (35). Taken together, the functional data support the HCM phenotype found in the patient.
The TNNC1-A31S Proband
In addition to left ventricular hypertrophy, the proband QTc is at the far upper limits of normal at 460 ms for a prepubertal boy. Usually associated with long QT syndrome, it has been previously demonstrated that elevated QTc is directly, albeit weakly, associated with the degree of left ventricular wall thickness (36, 37). Although mutations in KCNQ1-encoded IKs potassium channel, KCNH2-encoded IKr potassium channel, and SCN5A-encoded INa sodium channel (NaV1.5, LQT3, gain-of-function), the three major genes associated with long QT syndrome, cannot be excluded from our genetic analysis, the elevated QTc observed in this proband is likely a reflection of profound hypertrophy rather than the presence of independent and concomitant long QT syndrome (38).
The etiologies of ventricular fibrillation are diverse. In a structurally normal heart, purely electrical diseases can serve as a pathogenic substrate for ventricular fibrillation and sudden cardiac death. This includes channelopathic diseases such as long QT syndrome, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia, and short QT syndrome. The proband hosting the TNNC1 A31S mutation demonstrated concurrent hypertrophic remodeling of the heart; thus, it is more likely that the ventricular fibrillation and QT prolongation is secondary to his significant underlying hypertrophic cardiomyopathy.
Thin Filament Destabilization and HCM-Associated Alterations in Calcium-Affinity
Our identification of a missense mutation associated with both HCM and sudden cardiac death may represent a novel mechanism of thin filament sensitization. To gain mechanistic insight into how the cTnC-A31S mutation induces the myofilament dysfunction that underlies the presentation of HCM in the patient, we examined the manner in which the mutation altered the Ca2+ affinity of the thin filament. These changes in Ca2+ affinity likely involve alterations in the Ca2+ binding capacity of cTnC that are translated into changes in Ca2+ sensitivity within the myofilament. Previously, we found that although mutations occur within cTnC, they can impact the Ca2+ affinity of the thin filament in a variety of ways; 1) directly (locally targeting the cTnC Ca2+ affinity at the level of cTnC or proteins in direct contact) or 2) indirectly but through local interactions (involving proteins that contact cTnC through its interacting partners e.g. cTnI or cTnT) or by occurrences that indirectly influence cTnC Ca2+ affinity such as strong cross-bridge formation (39, 40).
The cTnC-A31S mutant increased the Ca2+ affinity (ΔpCa50 =+0.17) of the regulatory binding site II of isolated cTnC. In comparison, the previously characterized C84Y mutation also found in the N terminus did not measurably alter Ca2+ affinity (ΔpCa50 = −0.01) of site II in isolated cTnC. The effects of the L29Q mutation on Ca2+ affinity of site II indicate that cTnC-L29Q was less sensitive to Ca2+ (ΔpCa50 = −0.12) than the cTnC-A31S mutant reported here (29). In addition, the C-terminal mutant cTnC-D145E increased Ca2+ affinity of site II (ΔpCa50 = +0.11) in a more complex manner, as it indirectly influences Ca2+ binding. The other HCM-linked cTnC mutants N-terminal A8V and C-terminal E134D did not statistically change Ca2+ affinity of site II in isolated cTnC (33).
Furthermore, we found that the cTnC-A31S mutant sensitized the thin filament to Ca2+ (ΔpCa50 = +0.54) to a much larger degree than any cTnC HCM mutants tested to date. In comparison, the previously described HCM cTnC mutants A8V and D145E increased thin filament Ca2+ affinity with (ΔpCa50s of +0.14 and +0.08), respectively (33). It is well known that the number of cross-bridges can modulate both Ca2+ sensitivity and TnC affinity for the thin filament (41–47). Therefore, the addition of myosin S1 was used to assess whether strong cross-bridge formation indirectly (−ATP) modulated the Ca2+ affinity of the mutant containing thin and thick filament differently than the WT (41). S1 decreased the ΔpCa50 of the cTnC-A31S thin filament compared with when it was in a lower level of complexity (thin filament alone), which indicates that the thin filament may not always be the best predictive model. Subsequently, Ca2+ affinity of troponin with the addition of the thin filament and the catalytic myosin S1 more closely recapitulates what is seen in skinned fibers.
When combined, these data indicate that the A31S mutation increased Ca2+ affinity of cTnC of this increasingly complex system through a direct mechanism, such as increased affinity of the cTnC regulatory domain for Ca2+ or another known mechanism such as increased affinity of the mutant for cTnI. Potentially, a dramatically increased Ca2+ affinity of cTnC has a higher likelihood of perturbing its Ca2+ buffering role. The A31S mutant appears to substantially affect interactions between thin filament proteins, and this level may serve as the primary source of increased Ca2+ sensitivity. In summary, the profoundly altered Ca2+ binding properties of the cTnC-A31S mutant in the isolated state, cTn complex and thin filament coupled with early presentation of disease in the proband has never been shown before with a HCM-associated cTnC mutant.
Structural Characterization of the cTnC-A31S Mutant
Visualization of the troponin complex using the PyMol program suggested that A31S mutation may exert its Ca2+-sensitizing effects by locally stabilizing the EF-hand structure of the inactive Ca2+ binding site I located in the N terminus (Fig. 6). The substitution of serine for alanine at position 31 introduces a polar amino acid into the Ca2+ binding loop. Using a mutagenic function of the program without energy minimalization, substitution of Ser-31 in the mutant cTnC introduces a reactive hydroxyl group in close proximity to the backbone NH2 group of Asp-33 (see Fig. 6A). The program predicts formation of an additional H-bond that would provide three H-bonds to stabilize the structure of the inactive Ca2+ binding site (see Fig. 6B). It has been shown in skeletal troponin C that binding of Ca2+ to the EF hands is cooperative, where coordination exists between the Ca2+ binding sites of each domain. This same coordination has not been shown in the cTnC N terminus as Ca2+ cannot bind to Site I. Therefore, we speculate that this mutation helps to order the structure of site I in a manner similar to Ca2+ binding to site I in skeletal troponin C. This will need to be verified by established structural techniques that provide coordinates for the mutant protein compared with WT.
FIGURE 6.

Molecular visualization of the A31S mutation in the crystal structure 1AJ4. A, the H-bonds made by Ala-31 to adjacent residues located in inactive Ca2+ binding site I are shown. B, shown are the putative H-bonds made when the A31S substitution is present.
The CD data in the unbound (apo) state indicate that the mutation does not globally affect cTnC structure. No significant alterations were seen in the Mg2+-bound cTnC structure, indicating that the N-terminal mutation does not perturb the structure of the C terminus, as Mg2+ binds primarily to the C terminus. The mutation also did not detectably alter the secondary structure of Ca2+/Mg2+-bound cTnC; this is not surprising as most of the spectral changes that occur when cTnC binds Ca2+ originate from the C terminus (48, 49). Therefore, we suggest that A31S in isolated cTnC sensitizes Ca2+ binding to site II by locally affecting coordination between the Ca2+ binding sites, which subtly affects overall structure, and results in a dramatic change in function.
In conclusion, the discovery of the TNNC1 mutation A31S represents one of the first cTnC mutants associated with verified episodes of ventricular fibrillation and aborted sudden cardiac death. This mutation may alter Ca2+ handling and result in both hypertrophic and electrophysiologic remodeling of the cardiomyocyte. From our characterization, it appears that the A31S mutation may increase the Ca2+ binding affinity of the regulatory site II of cTnC by stabilizing the N-domain structure. This in turn could bestow a tremendous capacity for Ca2+ sensitization of the mutant containing myofilament. This mechanistic analysis coupled with clinical data provides insight on the source of myofilament dysfunction and the pathogenic nature of the mutation. It is evident that additional TNNC1 mutations will be discovered in the future and that this study along with others is essential to establish defined functional profiles characteristic of disease-causing mutations located within TNNC1.
Acknowledgments
We thank Michelle A. Jones and Jingsheng Liang for their excellent technical assistance during this project.
This work was supported, in whole or in part, by National Institutes of Health Grant HL42325 (to J. D. P.). M. J. A. is a consultant for Biotronik, Boston Scientific, Medtronic, St. Jude Medical, Inc., and Transgenomic. Intellectual property derived from M. J. A.'s research program resulted in license agreements in 2004 between Mayo Clinic Health Solutions (formerly Mayo Medical Ventures) and PGxHealth (formerly Genaissance Pharmaceuticals, now Transgenomic).
- HCM
- hypertrophic cardiomyopathy
- TNNC1
- troponin C gene
- TnC
- troponin C
- cTnC
- cardiac TnC
- QTc
- QT interval corrected for heart rate
- IAANS
- 2-(4′-iodoacetamidoanilino)naphthalene-6-sulfonic acid
- S1
- subfragment 1.
REFERENCES
- 1. Maron B. J., Epstein S. E., Roberts W. C. (1986) Causes of sudden death in competitive athletes. J. Am. Coll. Cardiol. 7, 204–214 [DOI] [PubMed] [Google Scholar]
- 2. Maron B. J., Gardin J. M., Flack J. M., Gidding S. S., Kurosaki T. T., Bild D. E. (1995) Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA study. Coronary artery risk development in (young) adults. Circulation 92, 785–789 [DOI] [PubMed] [Google Scholar]
- 3. Maron B. J., Roberts W. C., McAllister H. A., Rosing D. R., Epstein S. E. (1980) Sudden death in young athletes. Circulation 62, 218–229 [DOI] [PubMed] [Google Scholar]
- 4. Bos J. M., Towbin J. A., Ackerman M. J. (2009) Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 54, 201–211 [DOI] [PubMed] [Google Scholar]
- 5. Landstrom A. P., Ackerman M. J. (2010) Mutation type is not clinically useful in predicting prognosis in hypertrophic cardiomyopathy. Circulation 122, 2441–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Landstrom A. P., Parvatiyar M. S., Pinto J. R., Marquardt M. L., Bos J. M., Tester D. J., Ommen S. R., Potter J. D., Ackerman M. J. (2008) Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J. Mol. Cell. Cardiol. 45, 281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoffmann B., Schmidt-Traub H., Perrot A., Osterziel K. J., Gessner R. (2001) Hum. Mutat. 17, 524. [DOI] [PubMed] [Google Scholar]
- 8. Chung W. K., Kitner C., Maron B. J. (2011) Novel frameshift mutation in troponin C ( TNNC1) associated with hypertrophic cardiomyopathy and sudden death. Cardiol. Young 21, 345–348 [DOI] [PubMed] [Google Scholar]
- 9. Maron B. J. (2002) Hypertrophic cardiomyopathy. A systematic review. JAMA 287, 1308–1320 [DOI] [PubMed] [Google Scholar]
- 10. Gordon A. M., Homsher E., Regnier M. (2000) Regulation of contraction in striated muscle. Physiol. Rev. 80, 853–924 [DOI] [PubMed] [Google Scholar]
- 11. Herzberg O., James M. N. (1985) Structure of the calcium regulatory muscle protein troponin C at 2.8 A resolution. Nature 313, 653–659 [DOI] [PubMed] [Google Scholar]
- 12. Holroyde M. J., Robertson S. P., Johnson J. D., Solaro R. J., Potter J. D. (1980) The calcium and magnesium binding sites on cardiac troponin and their role in the regulation of myofibrillar adenosine triphosphatase. J. Biol. Chem. 255, 11688–11693 [PubMed] [Google Scholar]
- 13. Bers D. M. (2001) Calcium Sources and Sinks, 2nd Ed., Kluwer Academic Publishers, Boston/London [Google Scholar]
- 14. Baudenbacher F., Schober T., Pinto J. R., Sidorov V. Y., Hilliard F., Solaro R. J., Potter J. D., Knollmann B. C. (2008) Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J. Clin. Investig. 118, 3893–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wen Y., Pinto J. R., Gomes A. V., Xu Y., Wang Y., Wang Y., Potter J. D., Kerrick W. G. (2008) Functional consequences of the human cardiac troponin I hypertrophic cardiomyopathy mutation R145G in transgenic mice. J. Biol. Chem. 283, 20484–20494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mills R. E., Walter K., Stewart C., Handsaker R. E., Chen K., Alkan C., Abyzov A., Yoon S. C., Ye K., Cheetham R. K., Chinwalla A., Conrad D. F., Fu Y., Grubert F., Hajirasouliha I., Hormozdiari F., Iakoucheva L. M., Iqbal Z., Kang S., Kidd J. M., Konkel M. K., Korn J., Khurana E., Kural D., Lam H. Y., Leng J., Li R., Li Y., Lin C. Y., Luo R., Mu X. J., Nemesh J., Peckham H. E., Rausch T., Scally A., Shi X., Stromberg M. P., Stütz A. M., Urban A. E., Walker J. A., Wu J., Zhang Y., Zhang Z. D., Batzer M. A., Ding L., Marth G. T., McVean G., Sebat J., Snyder M., Wang J., Eichler E. E., Gerstein M. B., Hurles M. E., Lee C., McCarroll S. A., Korbel J. O., 1000 Genomes Project (2011) Mapping copy number variation by population-scale genome sequencing. Nature 470, 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang R., Zhao J., Potter J. D. (1995) Phosphorylation of both serine residues in cardiac troponin I is required to decrease the Ca2+ affinity of cardiac troponin C. J. Biol. Chem. 270, 30773–30780 [DOI] [PubMed] [Google Scholar]
- 18. Gomes A. V., Guzman G., Zhao J., Potter J. D. (2002) Cardiac troponin T isoforms affect the Ca2+ sensitivity and inhibition of force development. Insights into the role of troponin T isoforms in the heart. J. Biol. Chem. 277, 35341–35349 [DOI] [PubMed] [Google Scholar]
- 19. Pinto J. R., Parvatiyar M. S., Jones M. A., Liang J., Potter J. D. (2008) A troponin T mutation that causes infantile restrictive cardiomyopathy increases Ca2+ sensitivity of force development and impairs the inhibitory properties of troponin. J. Biol. Chem. 283, 2156–2166 [DOI] [PubMed] [Google Scholar]
- 20. Dweck D., Reyes-Alfonso A., Jr., Potter J. D. (2005) Expanding the range of free calcium regulation in biological solutions. Anal. Biochem. 347, 303–315 [DOI] [PubMed] [Google Scholar]
- 21. Hartshorne D. J., Perry S. V., Schaub M. C. (1967) A protein factor inhibiting the magnesium-activated adenosine triphosphatase of desensitized actomyosin. Biochem. J. 104, 907–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fiske C. H., Subbarow Y. (1925) The colorimetric determination of phosphorus J. Biol. Chem. 66, 375–400 [Google Scholar]
- 23. Putkey J. A., Liu W., Lin X., Ahmed S., Zhang M., Potter J. D., Kerrick W. G. (1997) Fluorescent probes attached to Cys-35 or Cys-84 in cardiac troponin C are differentially sensitive to Ca2+-dependent events in vitro and in situ. Biochemistry 36, 970–978 [DOI] [PubMed] [Google Scholar]
- 24. Pinto J. R., Reynaldo D. P., Parvatiyar M. S., Dweck D., Liang J., Jones M. A., Sorenson M. M., Potter J. D. (2011) Strong cross-bridges potentiate the Ca2+ affinity changes produced by hypertrophic cardiomyopathy cardiac troponin C mutants in myofilaments. A fast kinetic approach. J. Biol. Chem. 286, 1005–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Szczesna D., Ghosh D., Li Q., Gomes A. V., Guzman G., Arana C., Zhi G., Stull J. T., Potter J. D. (2001) Familial hypertrophic cardiomyopathy mutations in the regulatory light chains of myosin affect their structure, Ca2+ binding, and phosphorylation. J. Biol. Chem. 276, 7086–7092 [DOI] [PubMed] [Google Scholar]
- 26. Landstrom A. P., Kellen C. A., Dixit S. S., van Oort R. J., Garbino A., Weisleder N., Ma J., Wehrens X. H., Ackerman M. J. (2011) Junctophilin-2 expression silencing causes cardiocyte hypertrophy and abnormal intracellular calcium-handling. Circ. Heart Fail. 4, 214–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Watkins H., McKenna W. J., Thierfelder L., Suk H. J., Anan R., O'Donoghue A., Spirito P., Matsumori A., Moravec C. S., Seidman J. G. (1995) Mutations in the genes for cardiac troponin T and α-tropomyosin in hypertrophic cardiomyopathy. N. Engl. J. Med. 332, 1058–1064 [DOI] [PubMed] [Google Scholar]
- 28. Watkins H., Rosenzweig A., Hwang D. S., Levi T., McKenna W., Seidman C. E., Seidman J. G. (1992) Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N. Engl. J. Med. 326, 1108–1114 [DOI] [PubMed] [Google Scholar]
- 29. Dweck D., Hus N., Potter J. D. (2008) Challenging current paradigms related to cardiomyopathies. Are changes in the Ca2+ sensitivity of myofilaments containing cardiac troponin C mutations (G159D and L29Q) good predictors of the phenotypic outcomes? J. Biol. Chem. 283, 33119–33128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schmidtmann A., Lindow C., Villard S., Heuser A., Mügge A., Gessner R., Granier C., Jaquet K. (2005) Cardiac troponin C-L29Q, related to hypertrophic cardiomyopathy, hinders the transduction of the protein kinase A dependent phosphorylation signal from cardiac troponin I to C. FEBS J. 272, 6087–6097 [DOI] [PubMed] [Google Scholar]
- 31. Neulen A., Stehle R., Pfitzer G. (2009) The cardiac troponin C mutation Leu29Gln found in a patient with hypertrophic cardiomyopathy does not alter contractile parameters in skinned murine myocardium. Basic Res. Cardiol. 104, 751–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liang B., Chung F., Qu Y., Pavlov D., Gillis T. E., Tikunova S. B., Davis J. P., Tibbits G. F. (2008) Familial hypertrophic cardiomyopathy-related cardiac troponin C mutation L29Q affects Ca2+ binding and myofilament contractility. Physiol. Genomics 33, 257–266 [DOI] [PubMed] [Google Scholar]
- 33. Pinto J. R., Parvatiyar M. S., Jones M. A., Liang J., Ackerman M. J., Potter J. D. (2009) A functional and structural study of troponin C mutations related to hypertrophic cardiomyopathy. J. Biol. Chem. 284, 19090–19100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Willott R. H., Gomes A. V., Chang A. N., Parvatiyar M. S., Pinto J. R., Potter J. D. (2010) Mutations in troponin that cause HCM, DCM, and RCM. What can we learn about thin filament function? J. Mol. Cell. Cardiol. 48, 882–892 [DOI] [PubMed] [Google Scholar]
- 35. Parvatiyar M. S., Pinto J. R., Liang J., Potter J. D. (2010) Predicting cardiomyopathic phenotypes by altering Ca2+ affinity of cardiac troponin C. J. Biol. Chem. 285, 27785–27797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martin A. B., Garson A., Jr., Perry J. C. (1994) Prolonged QT interval in hypertrophic and dilated cardiomyopathy in children. Am. Heart J. 127, 64–70 [DOI] [PubMed] [Google Scholar]
- 37. Jouven X., Hagege A., Charron P., Carrier L., Dubourg O., Langlard J. M., Aliaga S., Bouhour J. B., Schwartz K., Desnos M., Komajda M. (2002) Relation between QT duration and maximal wall thickness in familial hypertrophic cardiomyopathy. Heart 88, 153–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tester D. J., Will M. L., Haglund C. M., Ackerman M. J. (2005) Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2, 507–517 [DOI] [PubMed] [Google Scholar]
- 39. Kreutziger K. L., Piroddi N., McMichael J. T., Tesi C., Poggesi C., Regnier M. (2011) Calcium binding kinetics of troponin C strongly modulate cooperative activation and tension kinetics in cardiac muscle. J. Mol. Cell. Cardiol. 50, 165–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zot A. S., Potter J. D. (1989) Reciprocal coupling between troponin C and myosin cross-bridge attachment. Biochemistry 28, 6751–6756 [DOI] [PubMed] [Google Scholar]
- 41. Wang Y., Xu Y., Guth K., Kerrick W. G. (1999) Troponin C regulates the rate constant for the dissociation of force-generating myosin cross-bridges in cardiac muscle. J. Muscle Res. Cell Motil. 20, 645–653 [DOI] [PubMed] [Google Scholar]
- 42. Güth K., Potter J. D. (1987) Effect of rigor and cycling cross-bridges on the structure of troponin C and on the Ca2+ affinity of the Ca2+-specific regulatory sites in skinned rabbit psoas fibers. J. Biol. Chem. 262, 13627–13635 [PubMed] [Google Scholar]
- 43. Pinto J. R., Veltri T., Sorenson M. M. (2008) Modulation of troponin C affinity for the thin filament by different cross-bridge states in skinned skeletal muscle fibers. Pflugers Arch. 456, 1177–1187 [DOI] [PubMed] [Google Scholar]
- 44. Hofmann P. A., Fuchs F. (1987) Effect of length and cross-bridge attachment on Ca2+ binding to cardiac troponin C. Am. J. Physiol. 253, C90–C96 [DOI] [PubMed] [Google Scholar]
- 45. Martyn D. A., Freitag C. J., Chase P. B., Gordon A. M. (1999) Ca2+ and cross-bridge-induced changes in troponin C in skinned skeletal muscle fibers. Effects of force inhibition. Biophys. J. 76, 1480–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Martyn D. A., Regnier M., Xu D., Gordon A. M. (2001) Ca2+- and cross-bridge-dependent changes in N- and C-terminal structure of troponin C in rat cardiac muscle. Biophys. J. 80, 360–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Swartz D. R., Moss R. L. (1992) Influence of a strong-binding myosin analogue on calcium-sensitive mechanical properties of skinned skeletal muscle fibers. J. Biol. Chem. 267, 20497–20506 [PubMed] [Google Scholar]
- 48. Leavis P. C., Kraft E. L. (1978) Calcium binding to cardiac troponin C. Arch. Biochem. Biophys. 186, 411–415 [DOI] [PubMed] [Google Scholar]
- 49. Johnson J. D., Collins J. H., Potter J. D. (1978) Dansylaziridine-labeled troponin C. A fluorescent probe of Ca2+ binding to the Ca2+-specific regulatory sites. J. Biol. Chem. 253, 6451–6458 [PubMed] [Google Scholar]